Synthesis and Biological Evaluation of 3-Benzisoxazolyl-4-indolylmaleimides as Potent, Selective Inhibitors of Glycogen Synthase Kinase-3β

Abstract
:1. Introduction
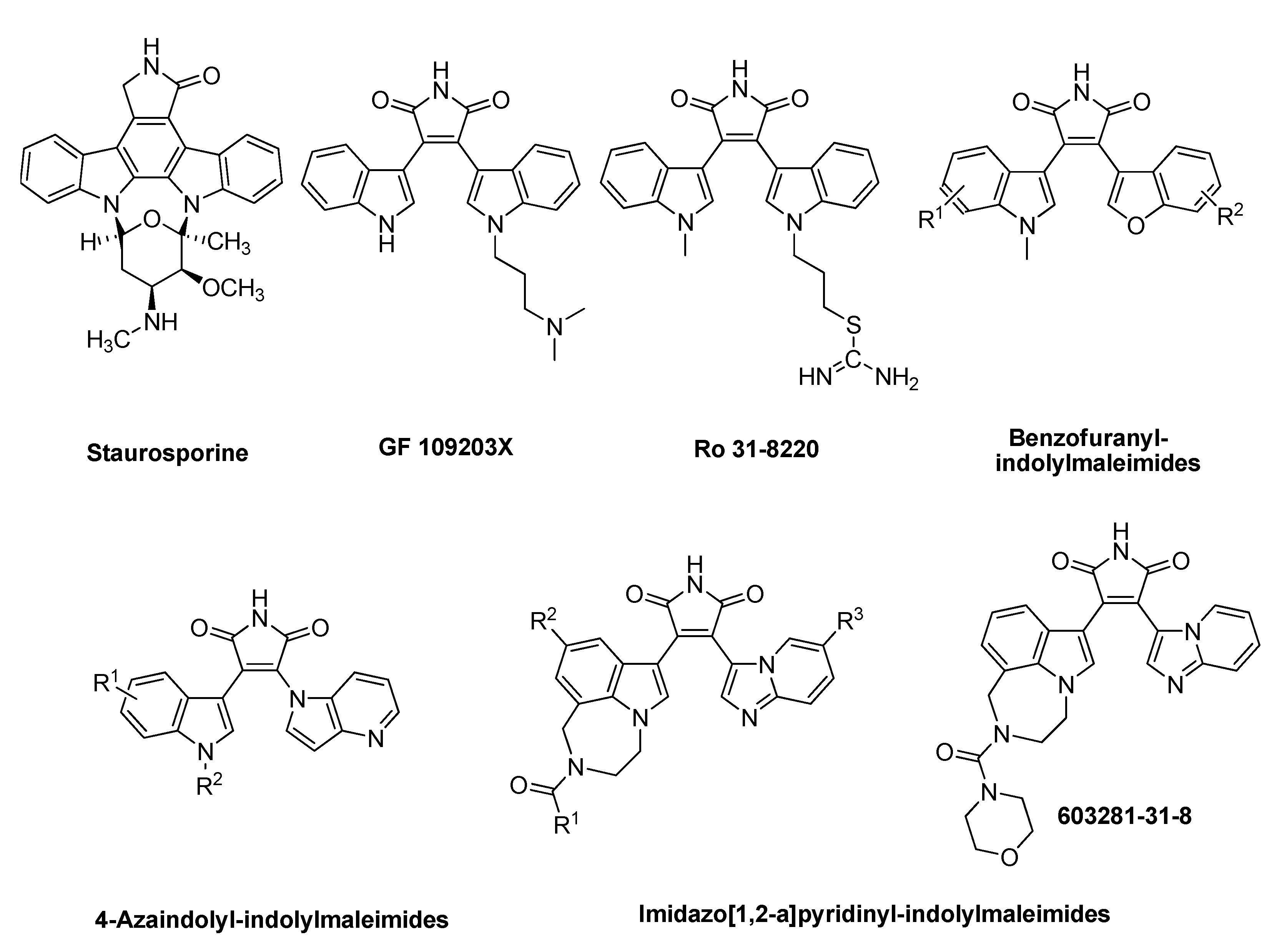
2. Results and Discussion
2.1. Chemistry
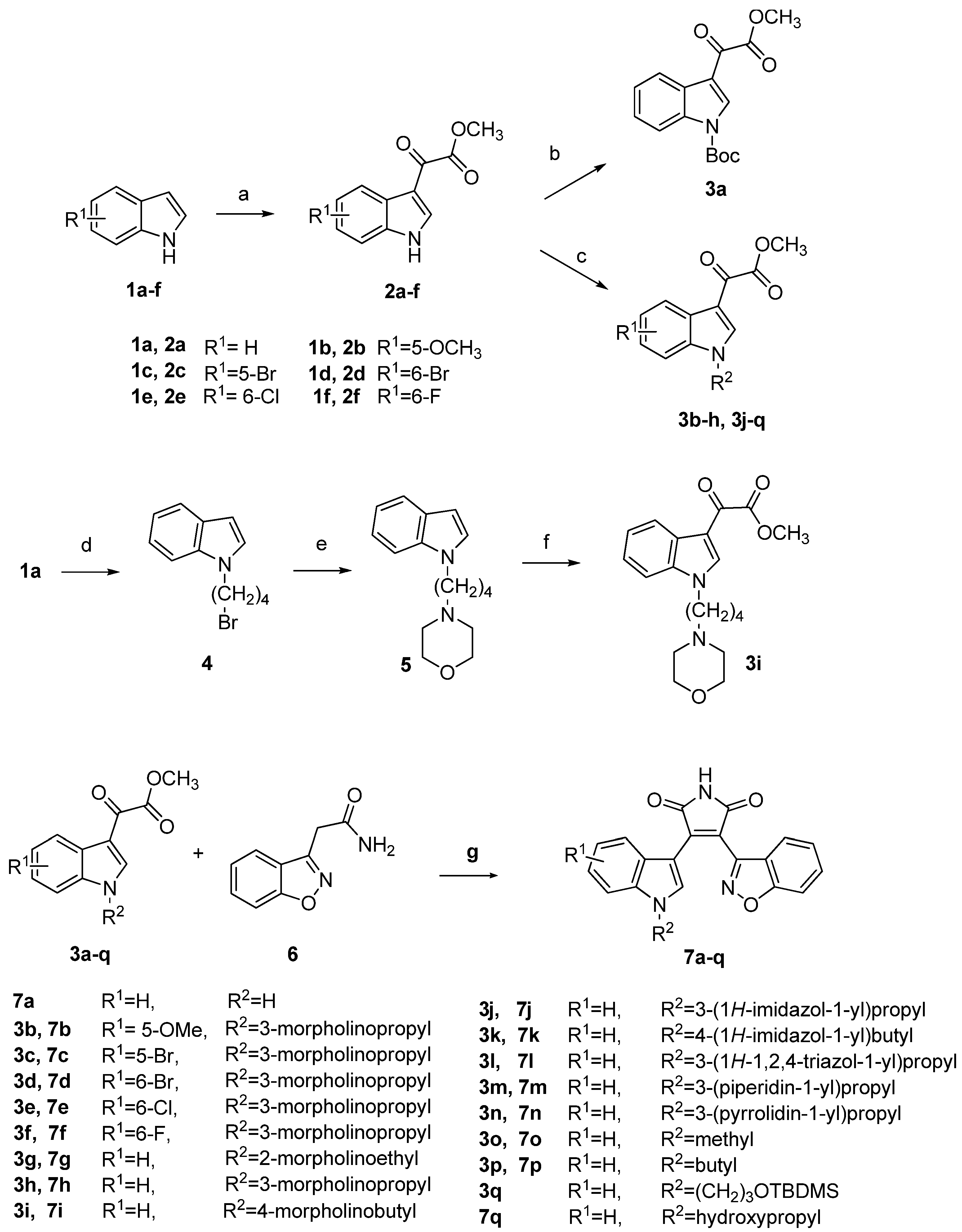
2.2. Biological Activity and Molecular Modeling
2.2.1. Enzymatic Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | R 1 | R 2 | IC50 (nM) ± SE a |
|---|---|---|---|
| Staurosporine | 72.2 ± 3.6 | ||
| 7a | H | H | 332.2 ± 25.7 |
| 7b | 5-OMe | 3-morpholinopropyl | 357.2 ± 17.1 |
| 7c | 5-Br | 3-morpholinopropyl | 10.2 ± 5.9 |
| 7d | 6-Br | 3-morpholinopropyl | 209.6 ± 8.9 |
| 7e | 6-Cl | 3-morpholinopropyl | 510.0 ± 20.1 |
| 7f | 6-F | 3-morpholinopropyl | 126.7 ± 3.2 |
| 7g | H | 2-morpholinoethyl | 615.6 ± 6.1 |
| 7h | H | 3-morpholinopropyl | 137.7 ± 6.0 |
| 7i | H | 4-morpholinobutyl | 410.7 ± 8.2 |
| 7j | H | 3-(1H-imidazol-1-yl)propyl | 0.73 ± 0.02 |
| 7k | H | 4-(1H-imidazol-1-yl)butyl | 89.8 ± 4.5 |
| 7l | H | 3-(1H-1,2,4-triazol-1-yl)propyl | 20.9 ± 5.1 |
| 7m | H | 3-(piperidin-1-yl)propyl | 511.8 ± 15.6 |
| 7n | H | 3-(pyrrolidin-1-yl)propyl | 658.8 ± 15.7 |
| 7o | H | methyl | 22.1 ± 1.5 |
| 7p | H | butyl | 58.7 ± 2.8 |
| 7q | H | hydroxypropyl | 38.9 ± 2.6 |
| Kinases | IC50 (nM) ± SE a or inhibition% at 0.8 μg/mL (~2 μM) | |||
|---|---|---|---|---|
| Staurosporine | 7c | 7j | 7o | |
| GSK-3β | 72.2 ±3.6 | 10.2 ± 5.9 | 0.7 ± 0.02 | 22.1 ± 1.5 |
| PKC-epsilon | 0.88 ± 0.03 | 12.1% | 10.9% | 21.2% |
| JAK2 | 2.26 ± 0.14 | 0.1% | 4.9% | 9.4% |
| BRAF | 14.37 ± 0.86 | 3.4% | 0.8% | 1.1% |
| IKK2 | 1.21 ± 0.13 | 13.1% | 3.5% | 2.9% |
| Drak2 | 25.82 ± 2.11 | 1.67% | 4.47% | 13.76% |
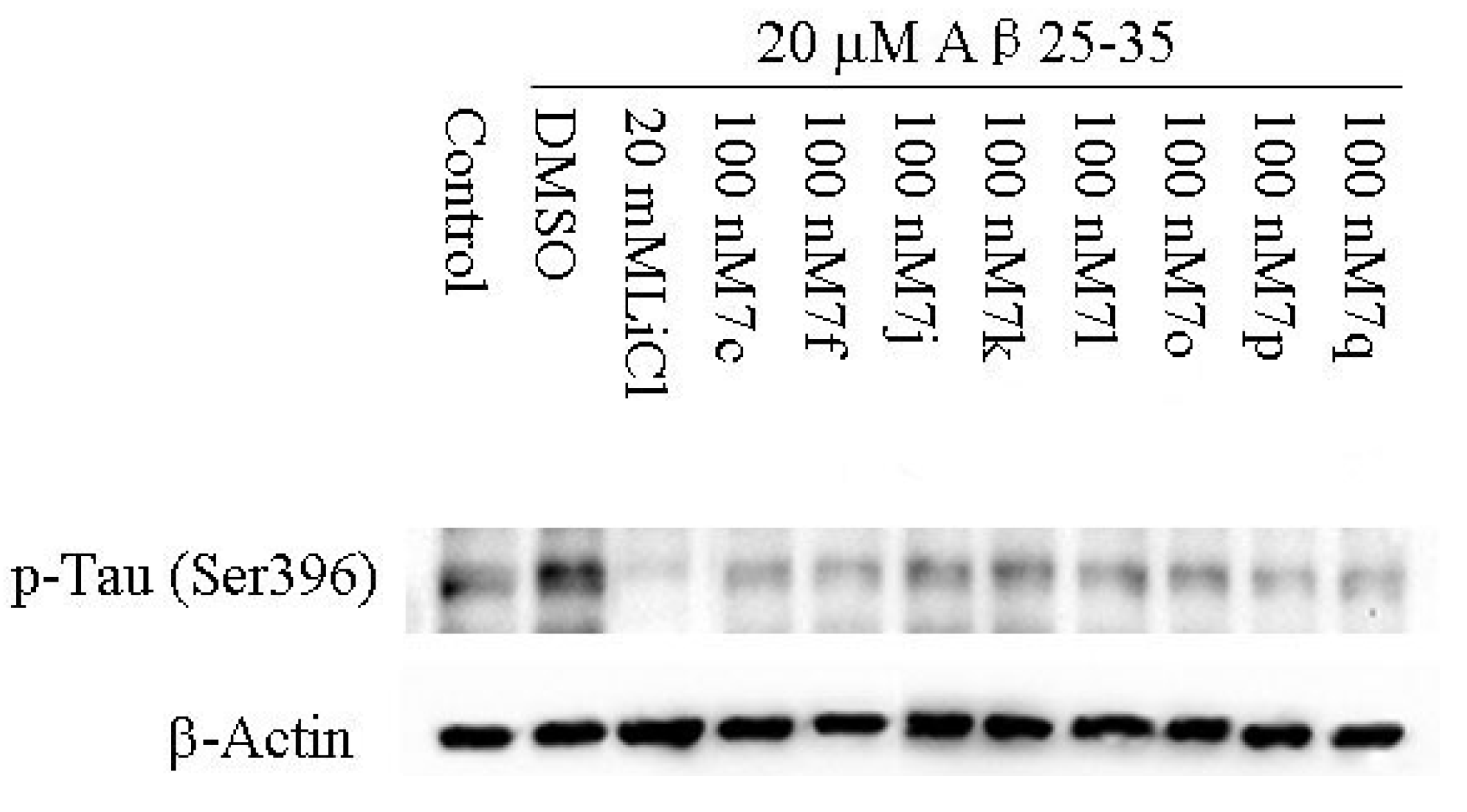
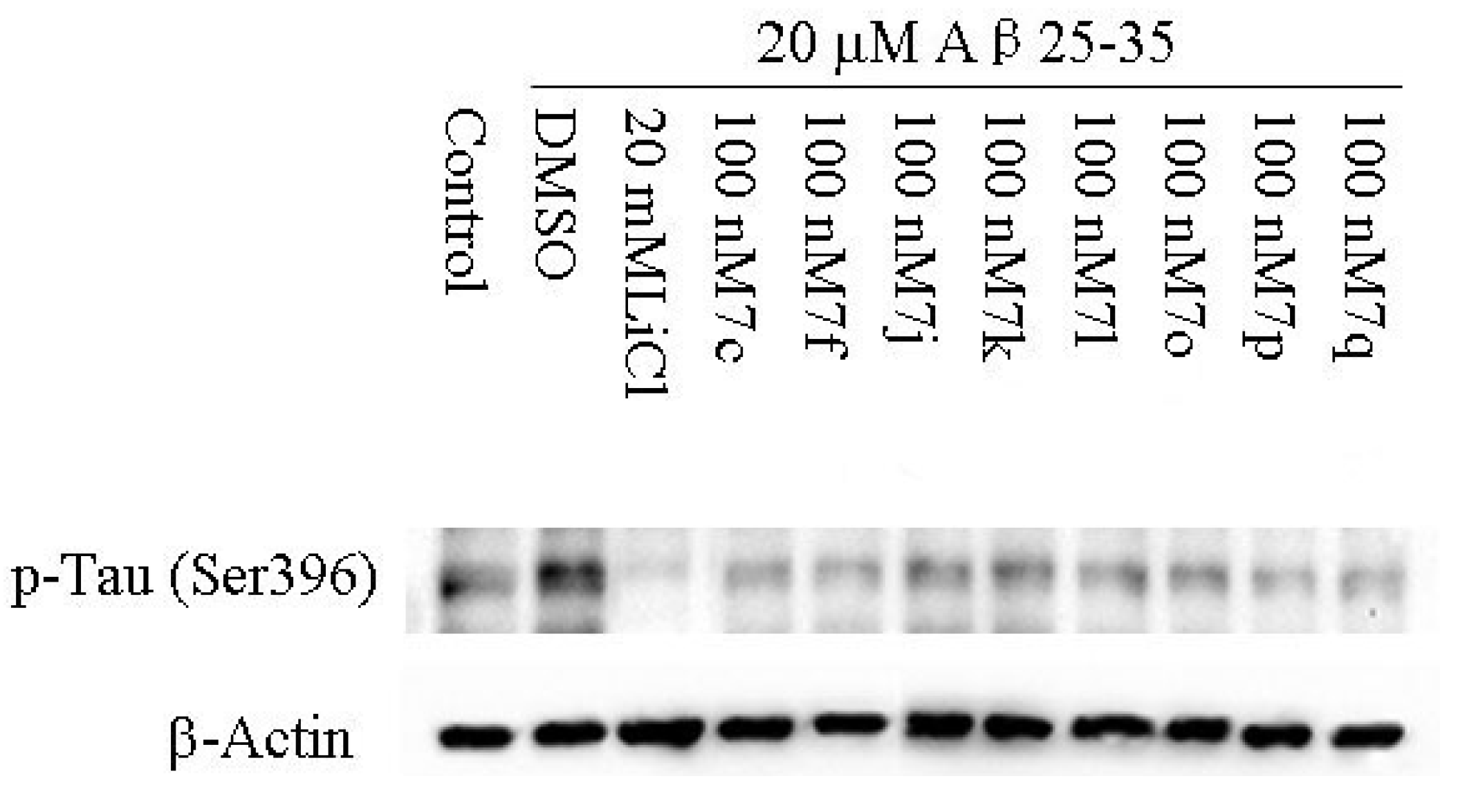
2.2.2. Cellular Activity
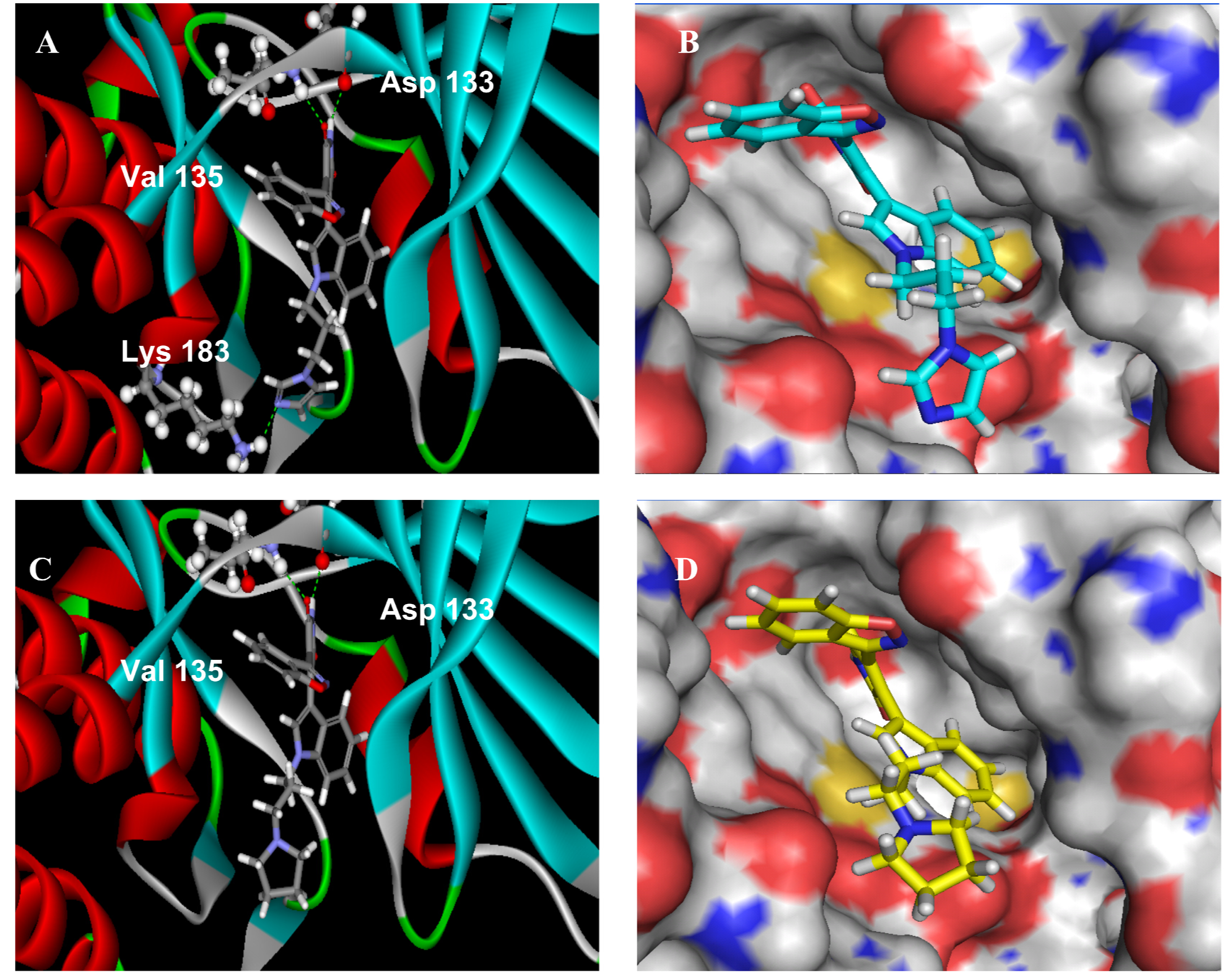
2.2.3. Molecular Modeling

3. Experimental
3.1. General
3.2. General Procedure for the Preparation of 2a–f
3.3. tert-Butyl 3-(2-methoxy-2-oxoacetyl)-1H-indole-1-carboxylate (3a)
3.4. General Procedure for the Preparation of 3b–h, 3j–n and 3q
3.5. 1-(4-Bromobutyl)-1H-indole (4)
3.6. 4-(4-(1H-Indol-1-yl)butyl)morpholine (5)
3.7. Methyl 2-(1-(4-morpholinobutyl)-1H-indol-3-yl)-2-oxoacetate (3i)
3.8. General Procedure for the Preparation of 3o and 3p
3.9. General Procedure for the Preparation of 7a–q
3.10. Biological Activity Assay
3.10.1. Kinase Assay
3.10.2. Cell Culture and Western Blotting
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Embi, N.; Rylatt, D.B.; Cohen, P. Glycogen synthase-3 from rabbit skeletal muscle separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur. J. Biochem. 1980, 107, 519–527. [Google Scholar] [CrossRef]
- Harwood, A.J. Regulation of GSK-3: A cellular multiprocessor. Cell 2001, 105, 821–824. [Google Scholar] [CrossRef]
- Yao, H.; Shaw, P.; Wrong, C.; Wan, D.C. Expression of glycogen synthase kinase-3 isoforms in mouse tissues and their transcription in the brain. J. Chem. Neuroanat. 2002, 23, 291–297. [Google Scholar] [CrossRef]
- Woodgett, J.R. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990, 9, 2431–2438. [Google Scholar]
- Eldar-Finkelman, H. Glycogen synthase kinase 3: An emerging therapeutic target. Trends Mol. Med. 2002, 8, 126–132. [Google Scholar] [CrossRef]
- Doble, B.W.; Woodgett, J.R. GSK-3: Tricks of the trade for a multi-tasking kinase. J. Cell. Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen Synthase Kinase-3 (GSK3): Inflammation, Diseases, and Therapeutics. Neurochem. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef]
- Frame, S.; Cohen, P. GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 2001, 359, 1–16. [Google Scholar] [CrossRef]
- Kim, L.; Kimmel, A.R. GSK3, a master switch regulating cell-fate specification and tumorigenesis. Curr. Opin. Genet. Dev. 2000, 10, 508–514. [Google Scholar] [CrossRef]
- Wagman, A.S.; Nuss, J.M. Current therapies and emerging targets for the treatment of diabetes. Curr. Pharm. Des. 2001, 7, 417–450. [Google Scholar] [CrossRef]
- Cohen, P. The role of protein phosphorylation in human health and disease. Eur. J. Biochem. 2001, 268, 5001–5010. [Google Scholar] [CrossRef]
- Sasaki, C.; Hayashi, T.; Zhang, W.R.; Warita, H.; Manabe, Y.; Sakai, K.; Abe, K. Different expression of glycogen synthase kinase-3β between young and old rat brains after transient middle cerebral artery occlusion. Neurol. Res. 2001, 23, 588–592. [Google Scholar] [CrossRef]
- Castro, A.; Martinez, A. Inhibition of tau phosphorylation: A new therapeutic strategy for the treatment of Alzheimer’s disease and other neurodegenerative disorders. Exp. Opin. Ther. Pat. 2000, 10, 1519–1527. [Google Scholar] [CrossRef]
- Bertrand, J.A.; Thieffine, S.; Vulpetti1, A.; Cristiani, C.; Valsasina, B.; Knapp, S.; Kalisz, H.M.; Flocco, M. Structural Characterization of the GSK-3β Active Site Using Selective and Non-selective ATP-mimetic Inhibitors. J. Mol. Biol. 2003, 333, 393–407. [Google Scholar] [CrossRef]
- Hers, I.; Tavare, J.M.; Denton, R.M. The protein kinase C inhibitors bisindolylmaleimide I (GF 109203x) and IX (Ro 31–8220) are potent inhibitors of glycogen synthase kinase-3 activity. FEBS Lett. 1999, 460, 433–436. [Google Scholar] [CrossRef]
- Zhang, H.-C.; White, K.B.; Ye, H.; McComsey, D.F.; Derian, C.K.; Addo, M.F.; Andrade-Gordon, P.; Eckardt, A.J.; Conway, B.R.; Westover, L.; et al. Macrocyclic bisindolylmaleimides as inhibitors of protein kinase C and glycogen synthase kinase-3. Bioorg. Med. Chem. Lett. 2003, 13, 3049–3053. [Google Scholar] [CrossRef]
- Gani, O.A.; Engh, R.A. Protein kinase inhibition of clinically important staurosporine analogues. Nat. Prod. Rep. 2010, 27, 489–498. [Google Scholar] [CrossRef]
- Pajak, B.; Orzechowska, S.; Gajkowska, B.; Orzechowski, A. Bisindolylmaleimides in anti-cancer therapy-more than PKC inhibitors. Adv. Med. Sci. 2008, 5321–5331. [Google Scholar]
- Engler, T.A.; Henry, J.R.; Malhotra, S.; Cunningham, B.; Furness, K.; Brozinick, J.; Burkholder, T.P.; Clay, M.P.; Clayton, J.; Diefenbacher, C.; et al. Substituted 3-Imidazo[1,2-a]pyridin-3-yl-4-(1,2,3,4-tetrahydro-[1,4]diazepino-[6,7,1-hi]indol-7- yl) pyrrole-2,5-diones as Highly Selective and Potent Inhibitors of Glycogen Synthase Kinase-3. J. Med. Chem. 2004, 47, 3934–3937. [Google Scholar] [CrossRef]
- Ye, Q.; Xu, G.; Lv, D.; Cheng, Z.; Li, J.; Hu, Y. Synthesis and biological evaluation of novel 4-azaindolyl-indolyl-maleimides as glycogen synthase kinase-3β (GSK-3β) inhibitors. Bioorg. Med. Chem. 2009, 17, 4302–4312. [Google Scholar] [CrossRef]
- Gaisina, I.N.; Gallier, F.; Ougolkov, A.V.; Kim, K.H.; Kurome, T.; Guo, S.; Holzle, D.; Luchini, D.N.; Blond, S.Y.; Billadeau, D.D.; et al. From a Natural Product Lead to the Identification of Potent and Selective Benzofuran-3- yl-(indol-3-yl)maleimides as Glycogen Synthase Kinase 3β Inhibitors That Suppress Proliferation and Survival of Pancreatic Cancer Cells. J. Med. Chem. 2009, 52, 1853–1863. [Google Scholar] [CrossRef]
- Uno, H.; Kurokawa, M.; Nishimura, H. Studies on 3-substituted 1, 2-benzisoxazole derivatives. II. The catalytic reductions of 1,2-benzisoxazole-3-acetamide oxime and related compounds. Chem. Pharm. Bull. 1976, 24, 632–643. [Google Scholar] [CrossRef]
- Gong, E.J.; Park, H.R.; Kim, M.E.; Piao, S.; Lee, E.; Jo, D.G.; Chung, H.Y.; Ha, N.C.; Mattson, M.P.; Lee, J. Morin attenuates tau hyperphosphorylation by inhibiting GSK3β. Neurobiol. Dis. 2011, 24, 223–230. [Google Scholar]
- Sun, Z.K.; Yang, H.Q.; Pan, J.; Zhen, H.; Wang, Z.Q.; Chen, S.D.; Ding, J.Q. Protective effects of erythropoietin on tau phosphorylation induced by β-amyloid. J. Neurosci. Res. 2008, 86, 3018–3027. [Google Scholar] [CrossRef]
- Gong, L.; Hirschfeld, D.; Tan, Y.C.; Hogg, J.H.; Peltz, G.; Avnur, Z.; Dunten, P. Discovery of potent and bioavailable GSK-3β inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1693–1696. [Google Scholar] [CrossRef]
- Liu, X.H.; Pan, L.; Ma, Y.; Weng, J.Q.; Tan, C.X.; Li, Y.H.; Shi, Y.X.; Li, B.J.; Li, Z.M.; Zhang, Y.G. Design, Synthesis, Biological Activities, and 3D-QSAR of New N,N'-Diacylhydrazines Containing 2-(2,4-dichlorophenoxy)propane Moiety. Chem. Biol. Drug Des. 2011, 78, 689–694. [Google Scholar] [CrossRef]
- Liu, X.H.; Pan, L.; Tan, C.X.; Weng, J.Q.; Wang, B.L.; Li, Z.M. Synthesis, crystal structure, bioactivity and DFT calculation of new oxime ester derivatives containing cyclopropane moiety. Pestic. Biochem. Physiol. 2011, 101, 143–147. [Google Scholar] [CrossRef]
- Liu, X.H.; Tan, C.X.; Weng, J.Q. Synthesis, Dimeric Crystal Structure, and Fungicidal Activity of 1-(4-Methylphenyl)-2-(5-((3,5-Dimethyl-1H-Pyrazol-1-yl)methyl)-4-Phenyl-4H-1,2,4-Trizol-3-ylthio)Ethanone. Phosphorus Sulfur Silicon Relat. Elem. 2011, 186, 558–564. [Google Scholar] [CrossRef]
- Liu, X.H.; Pan, L.; Weng, J.Q.; Tan, C.X.; Li, Y.H.; Wang, B.L.; Li, Z.M. Synthesis, structure, and biological activity of novel (oxdi/tri)azoles derivatives containing 1,2,3-thiadiazole or methyl moiety. Mol. Divers. 2012, 16, 251–260. [Google Scholar] [CrossRef]
- Tan, C.X.; Shi, Y.X.; Weng, J.Q.; Liu, X.H.; Zhao, W.G.; Li, B.J. Synthesis and Antifungal Activity of Novel 1,2,4-Triazole Derivatives Containing 1,2,3-Thiadiazole Moiety. J. Heterocycl. Chem. 2013. [Google Scholar] [CrossRef]
- Liu, X.H.; Weng, J.Q.; Wang, B.L.; Li, Y.H.; Tan, C.X.; Li, Z.M. Microwave-assisted Synthesis and Biological Activity Study of Novel Fluorinated 1,2,4-Triazole Derivatives. Res. Chem. Intermed. 2013. [Google Scholar] [CrossRef]
- Tan, C.X.; Shi, Y.X.; Weng, J.Q.; Liu, X.H.; Li, B.J.; Zhao, W.G. Synthesis and Antifungal Activity of 1,2,4-triazole Derivatives Containing Cyclopropane Moiety. Lett. Drug Des. Discov. 2012, 9, 431–435. [Google Scholar] [CrossRef]
- Liu, X.H.; Weng, J.Q.; Tan, C.X. Synthesis, Crystal Structure and Fungicidal Activity of 5-(4-cyclopropyl-5-((3-fluorobenzyl)thio)-4H-1,2,4-triazol-3-yl)-4-methyl-1,2,3-thiadiazole. J. Chem. 2013, 2013, 306361. [Google Scholar] [CrossRef]
- Weng, J.Q.; Wang, L.; Liu, X.H. Synthesis, Crystal Structure and Herbicidal Activity of a 1,2,4-triazol-5(4H)-one Derivative. J. Chem. Soc. Pakistan 2012, 34, 1248–1252. [Google Scholar]
- Liu, X.H.; Zhao, W.G.; Wang, B.L.; Li, Z.M. Synthesis, Bioactivity and DFT Structure-Activity Relationship Study of Novel 1,2,3-Thiadiazole Derivatives. Res. Chem. Intermed. 2012, 38, 1999–2008. [Google Scholar] [CrossRef]
- Tong, J.Y.; Shi, Y.X.; Liu, X.H.; Sun, N.B.; Li, B.J. Synthesis and Fungicidal Activity of 1,2,4-Triazole Derivatives Containing 2-Fluorophenyl Moiety. Chin. J. Org. Chem. 2012, 32, 2373–2377. [Google Scholar] [CrossRef]
- Tong, J.Y.; Wu, H.K.; Sun, N.B.; Liu, X.H. Synthesis, Crystal Structure and Biological Activity of a New 1,2,4-Triazole Derivative. Chin. J. Struct. Chem. 2013, 32, 607–611. [Google Scholar]
- Sun, N.B.; Liu, X.H.; Weng, J.Q.; Tan, C.X. An Unexpected Product N-(3-((2-fluorobenzyl)thio)-5-methyl-4H- 1,2,4-triazol-4-yl)acetimidamide: Synthesis and Structure Analysis. J. Chem. Soc. Pakistan 2013, 35, 499–502. [Google Scholar]
- Cui, C.; Wang, Z.P.; Du, X.j.; Wang, L.Z.; Yu, S.J.; Liu, X.H.; Li, Z.M.; Zhao, W.G. Synthesis and Antiviral Activity of Hydrogenated Ferulic Acid Derivatives. J. Chem. 2013, 2013, 269434. [Google Scholar]
- Wu, R.; Zhu, C.; Du, X.J.; Xiong, L.X.; Yu, S.J.; Liu, X.H.; Li, Z.M.; Zhao, W.G. Synthesis, crystal structure and larvicidal activity of novel diamide derivatives against Culex. pipiens. Chem. Cent. J. 2012, 9, 99. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ye, Q.; Li, M.; Zhou, Y.; Pang, T.; Xu, L.; Cao, J.; Han, L.; Li, Y.; Wang, W.; Gao, J.; et al. Synthesis and Biological Evaluation of 3-Benzisoxazolyl-4-indolylmaleimides as Potent, Selective Inhibitors of Glycogen Synthase Kinase-3β. Molecules 2013, 18, 5498-5516. https://doi.org/10.3390/molecules18055498
Ye Q, Li M, Zhou Y, Pang T, Xu L, Cao J, Han L, Li Y, Wang W, Gao J, et al. Synthesis and Biological Evaluation of 3-Benzisoxazolyl-4-indolylmaleimides as Potent, Selective Inhibitors of Glycogen Synthase Kinase-3β. Molecules. 2013; 18(5):5498-5516. https://doi.org/10.3390/molecules18055498
Chicago/Turabian StyleYe, Qing, Meng Li, Yubo Zhou, Tao Pang, Lei Xu, Jiayi Cao, Liang Han, Yujin Li, Weisi Wang, Jianrong Gao, and et al. 2013. "Synthesis and Biological Evaluation of 3-Benzisoxazolyl-4-indolylmaleimides as Potent, Selective Inhibitors of Glycogen Synthase Kinase-3β" Molecules 18, no. 5: 5498-5516. https://doi.org/10.3390/molecules18055498