[Fe2L3]4+ Cylinders Derived from Bis(bidentate) 2-Pyridyl-1,2,3-triazole “Click” Ligands: Synthesis, Structures and Exploration of Biological Activity


Abstract
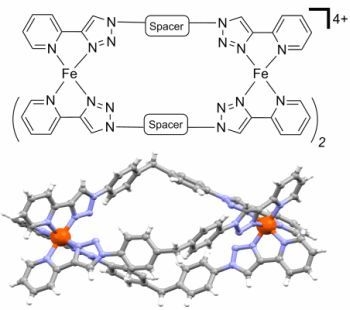
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of Model Iron(II) Complexes
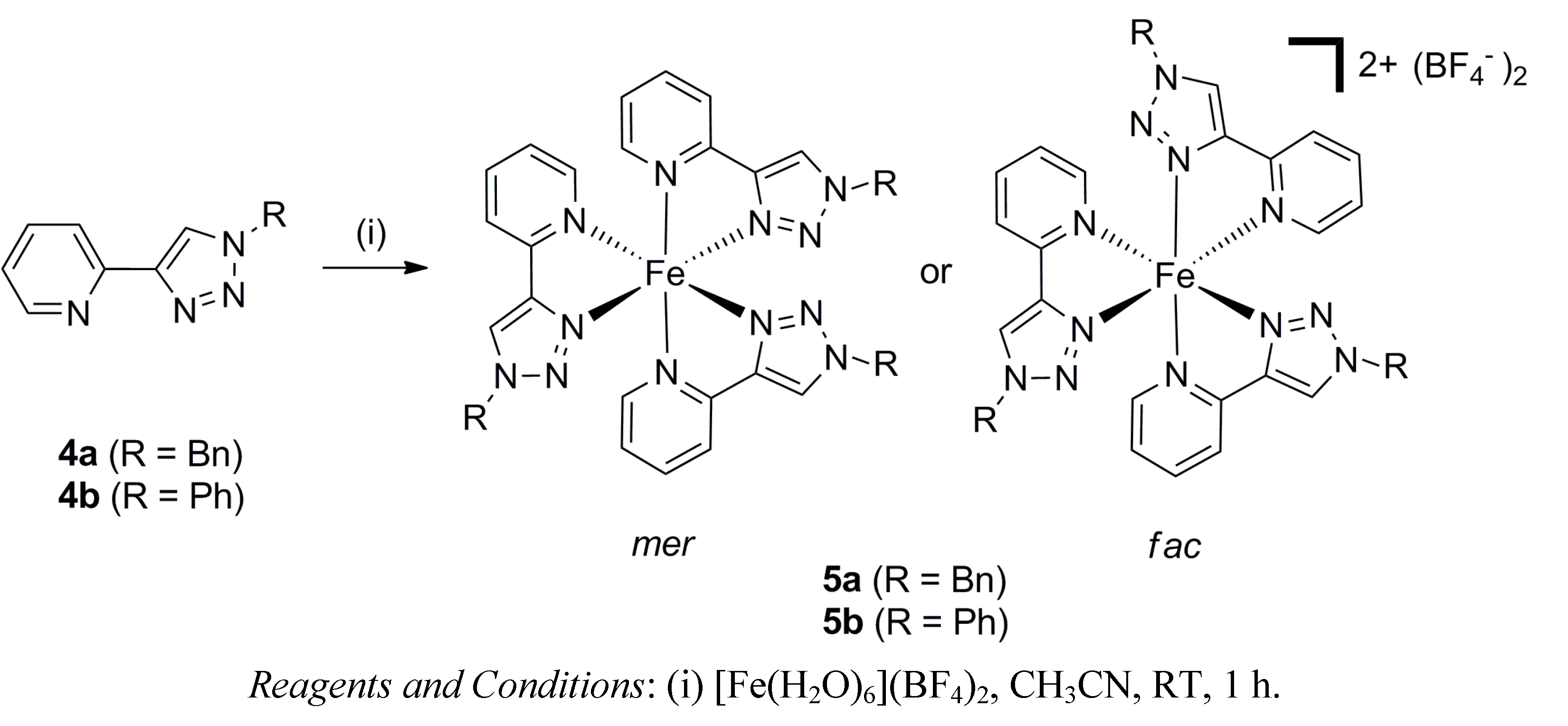
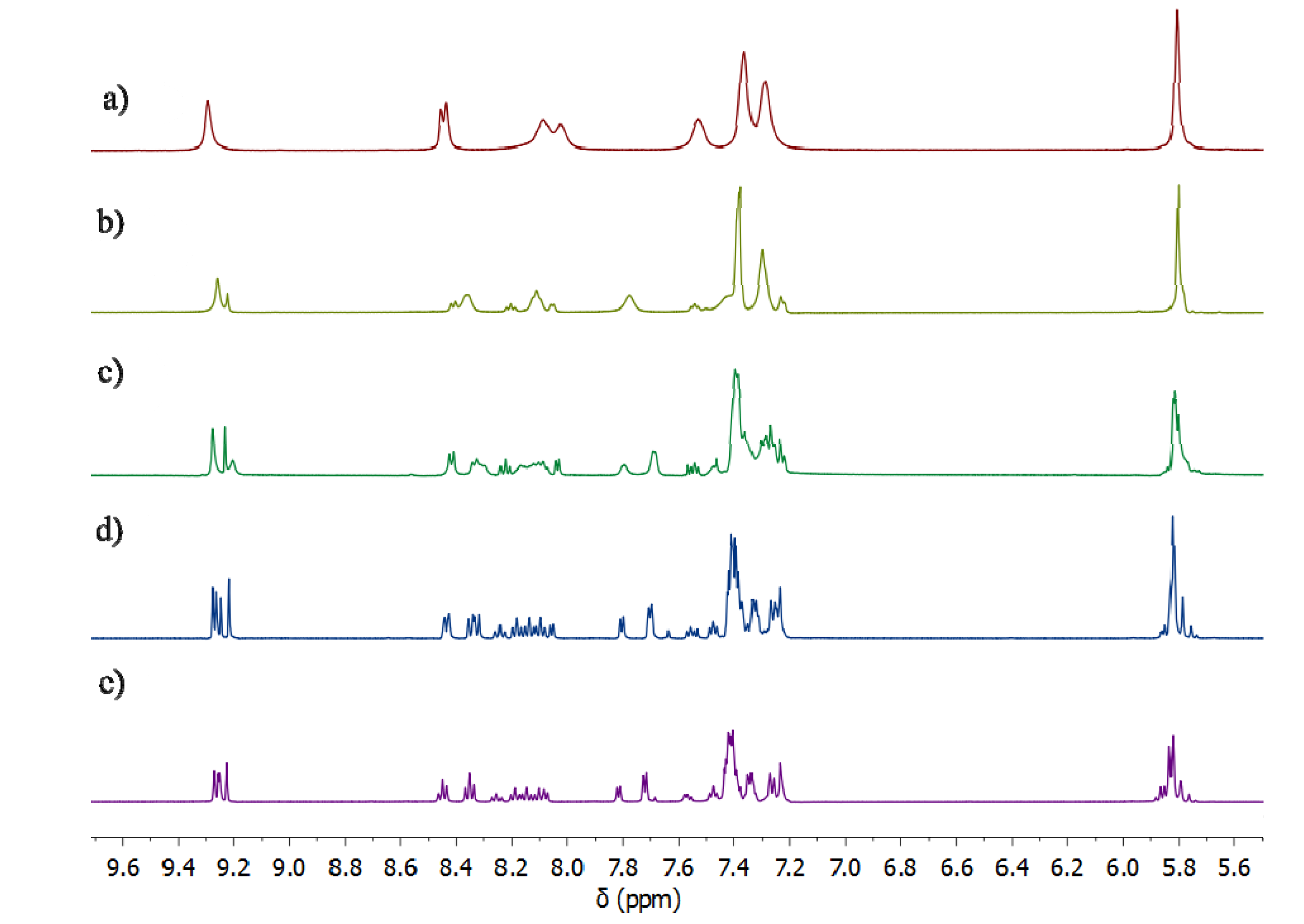
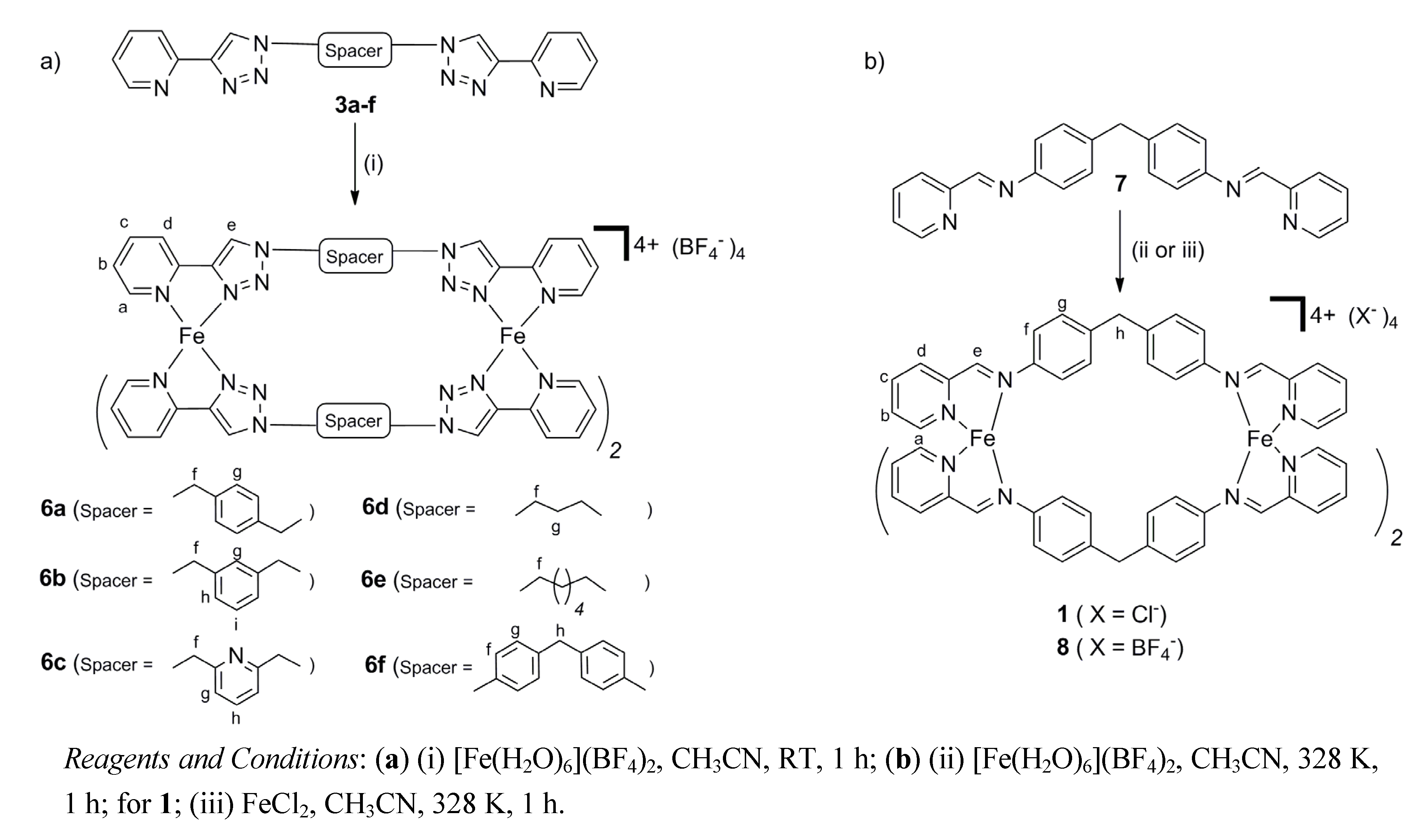
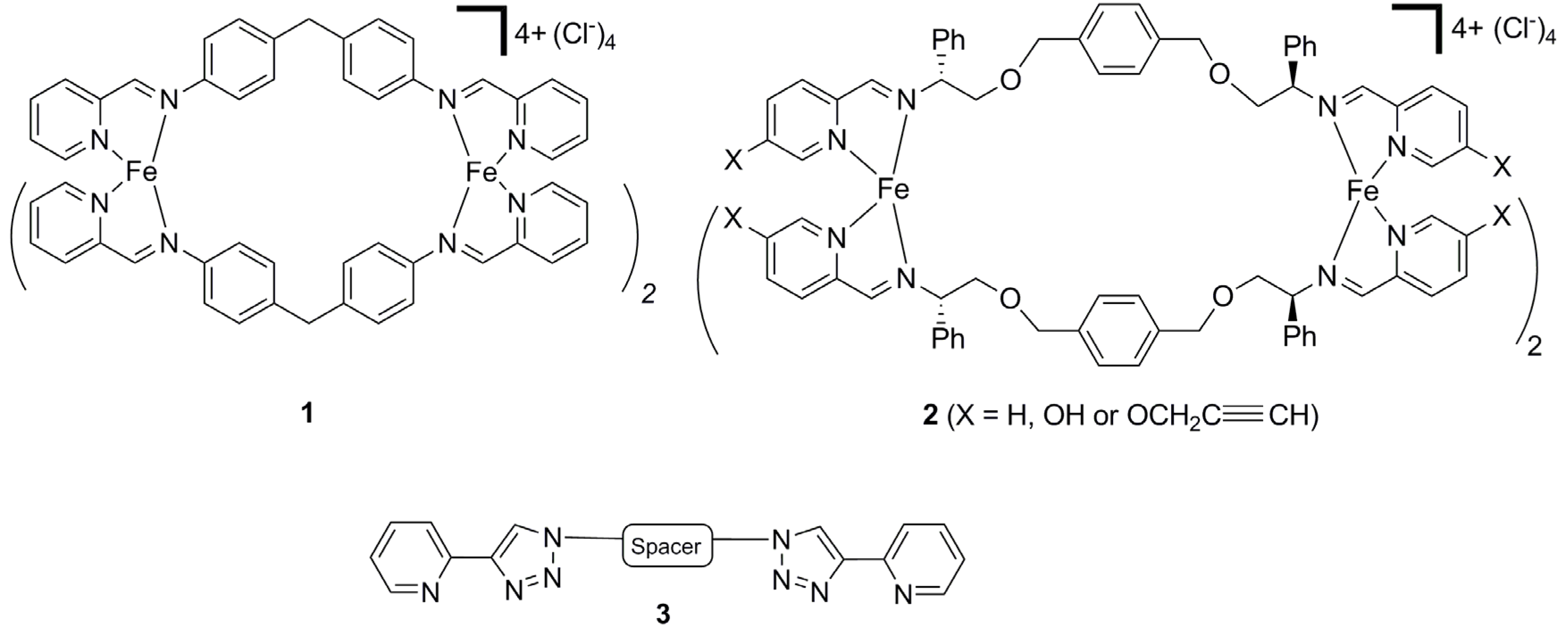
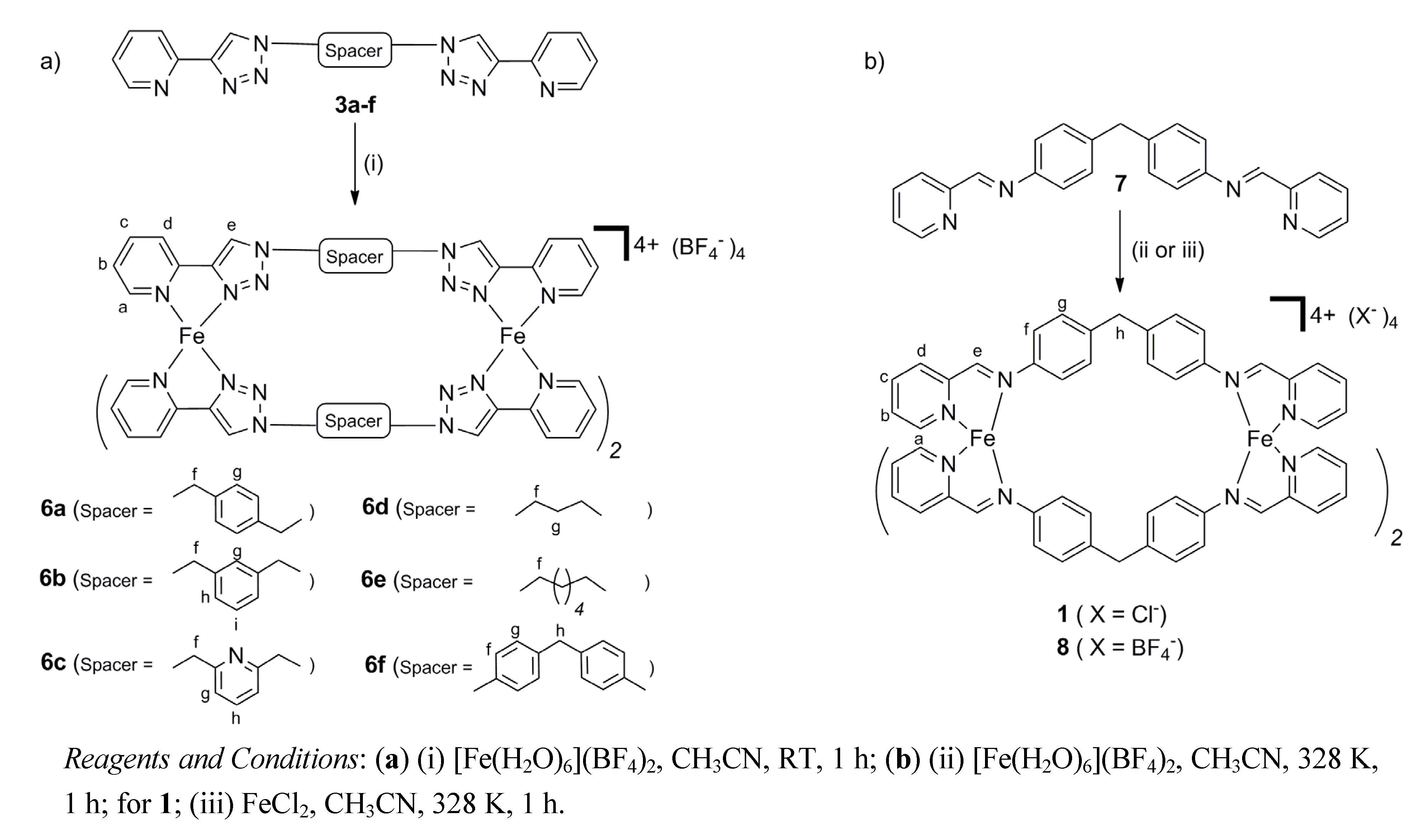
2.2. Synthesis of Iron(II) Metallosupramolecular Cylinders
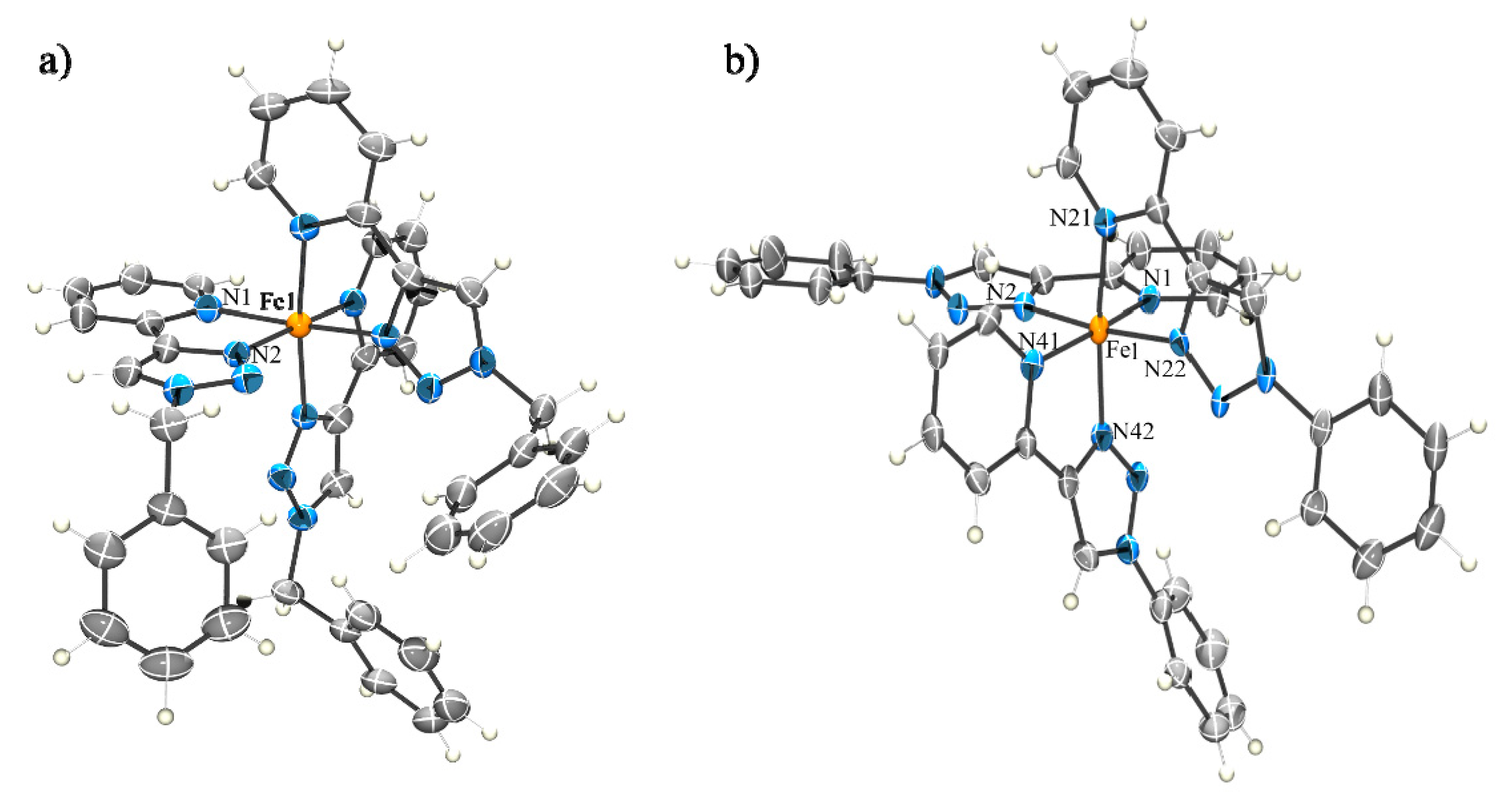
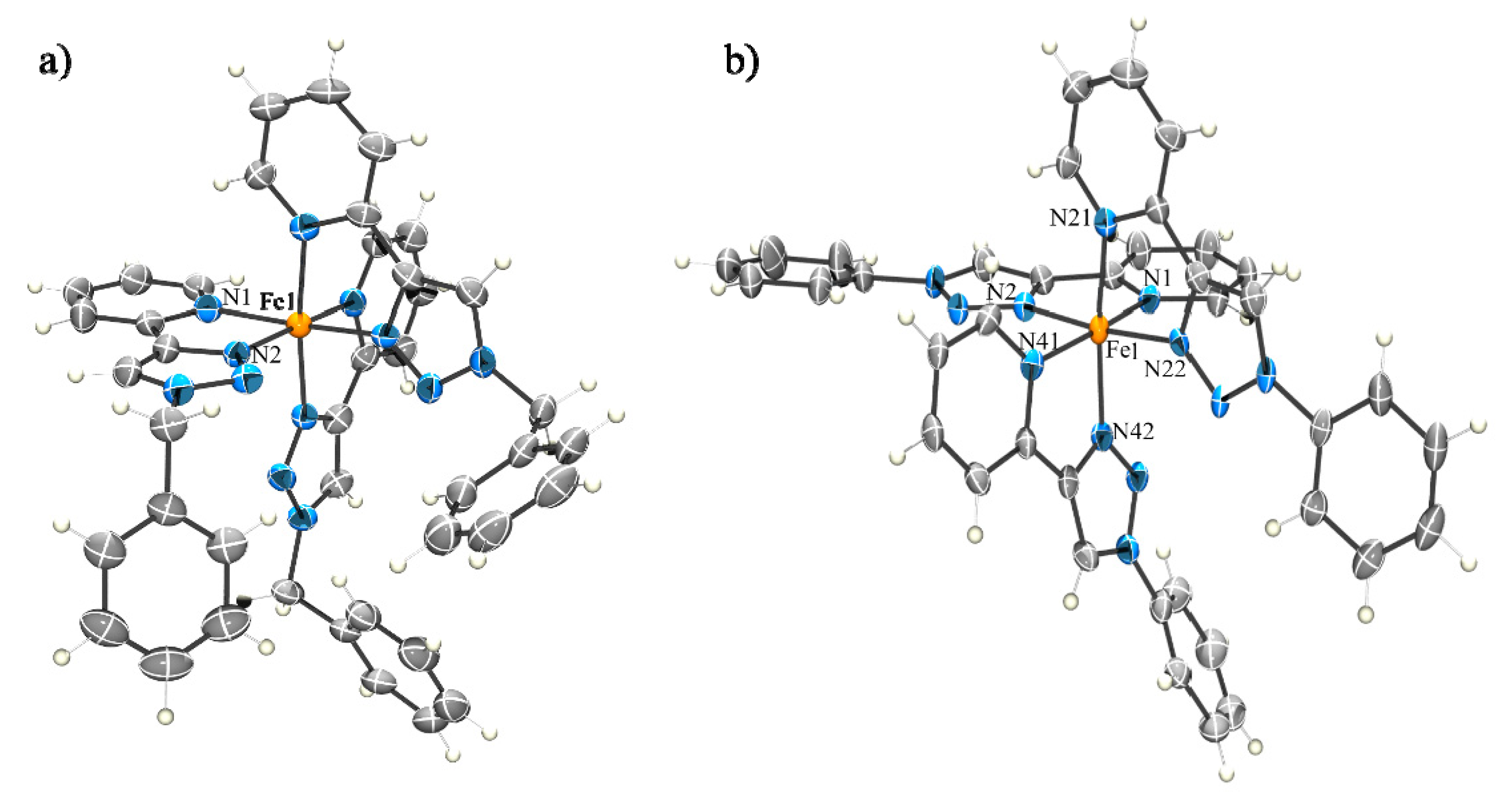
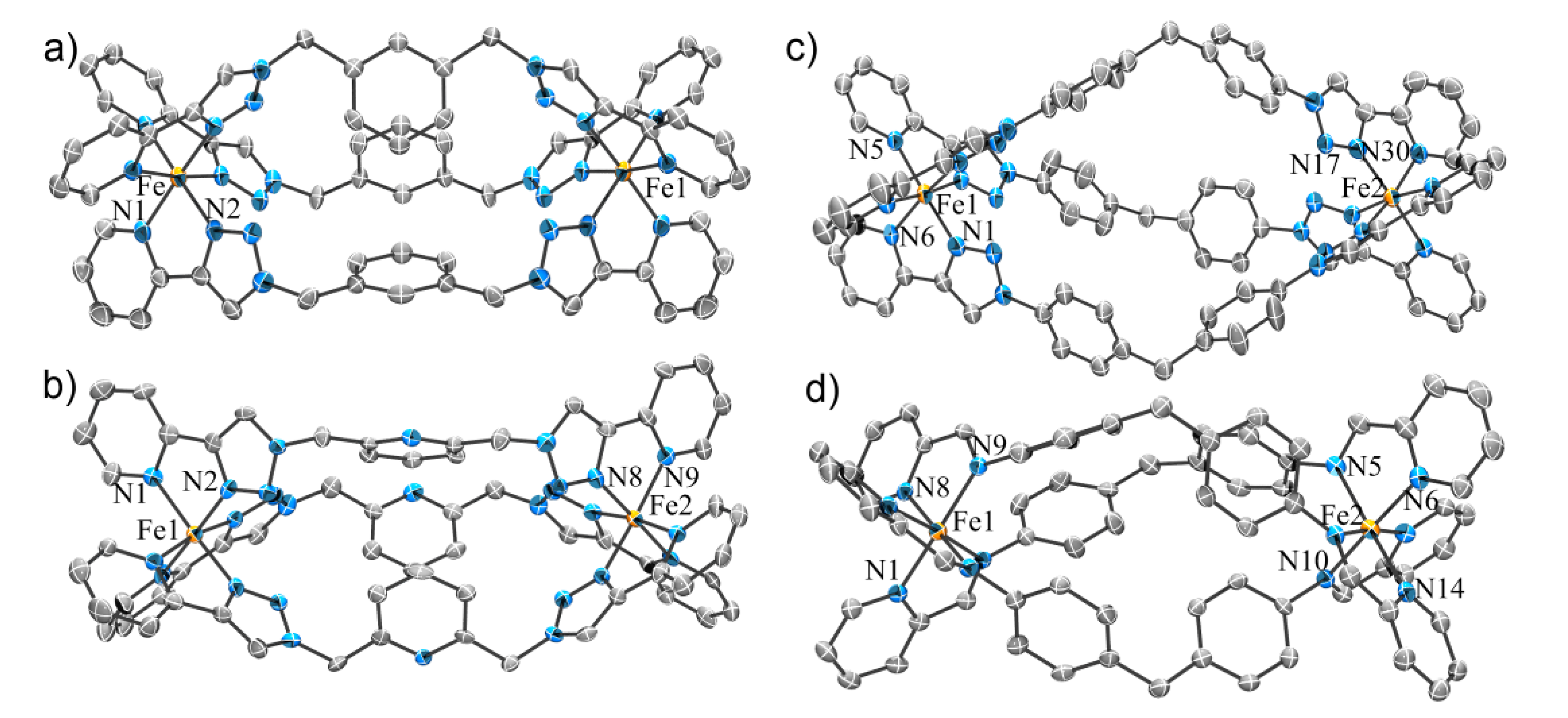
2.2.1. Solid State Structures


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
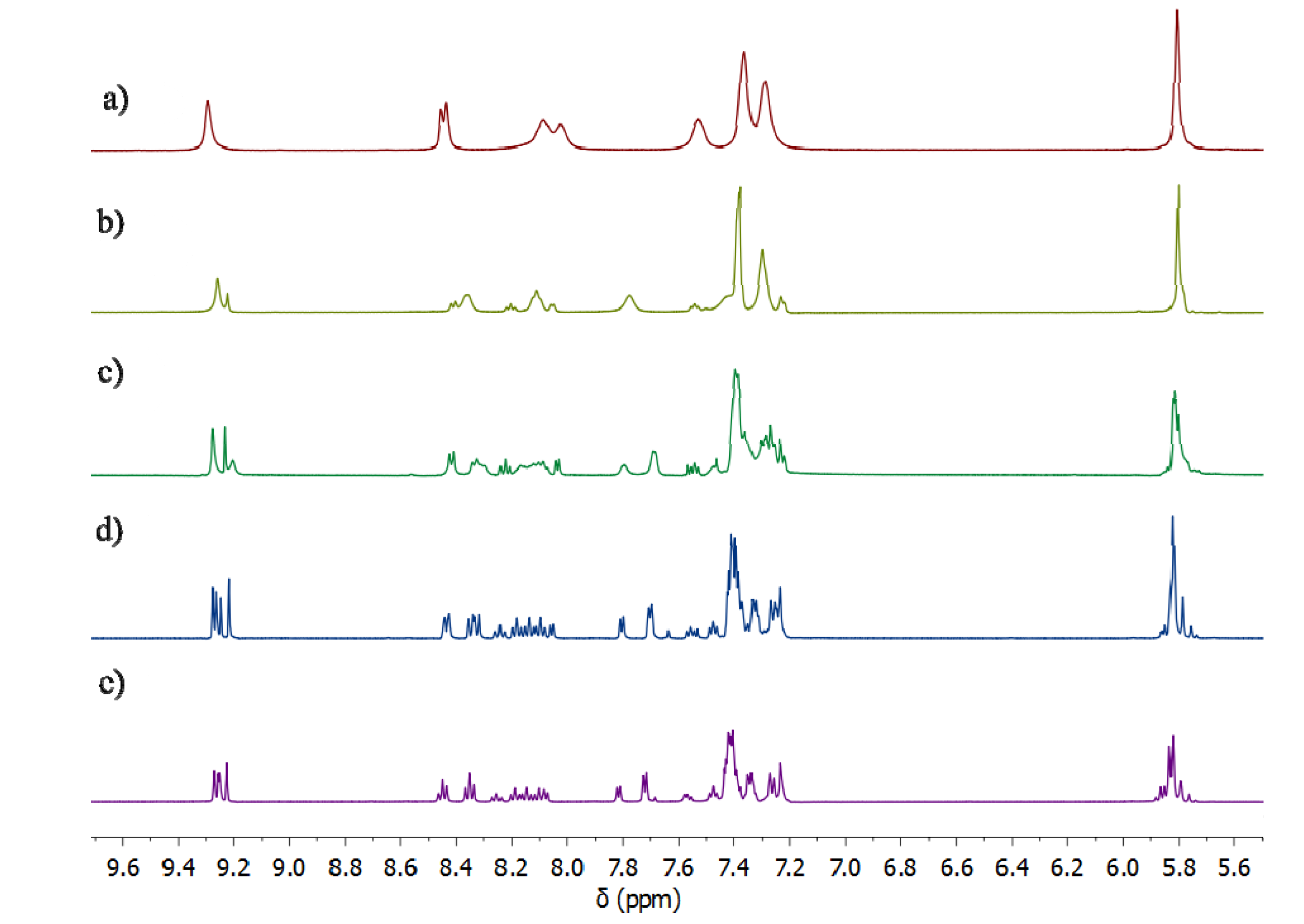
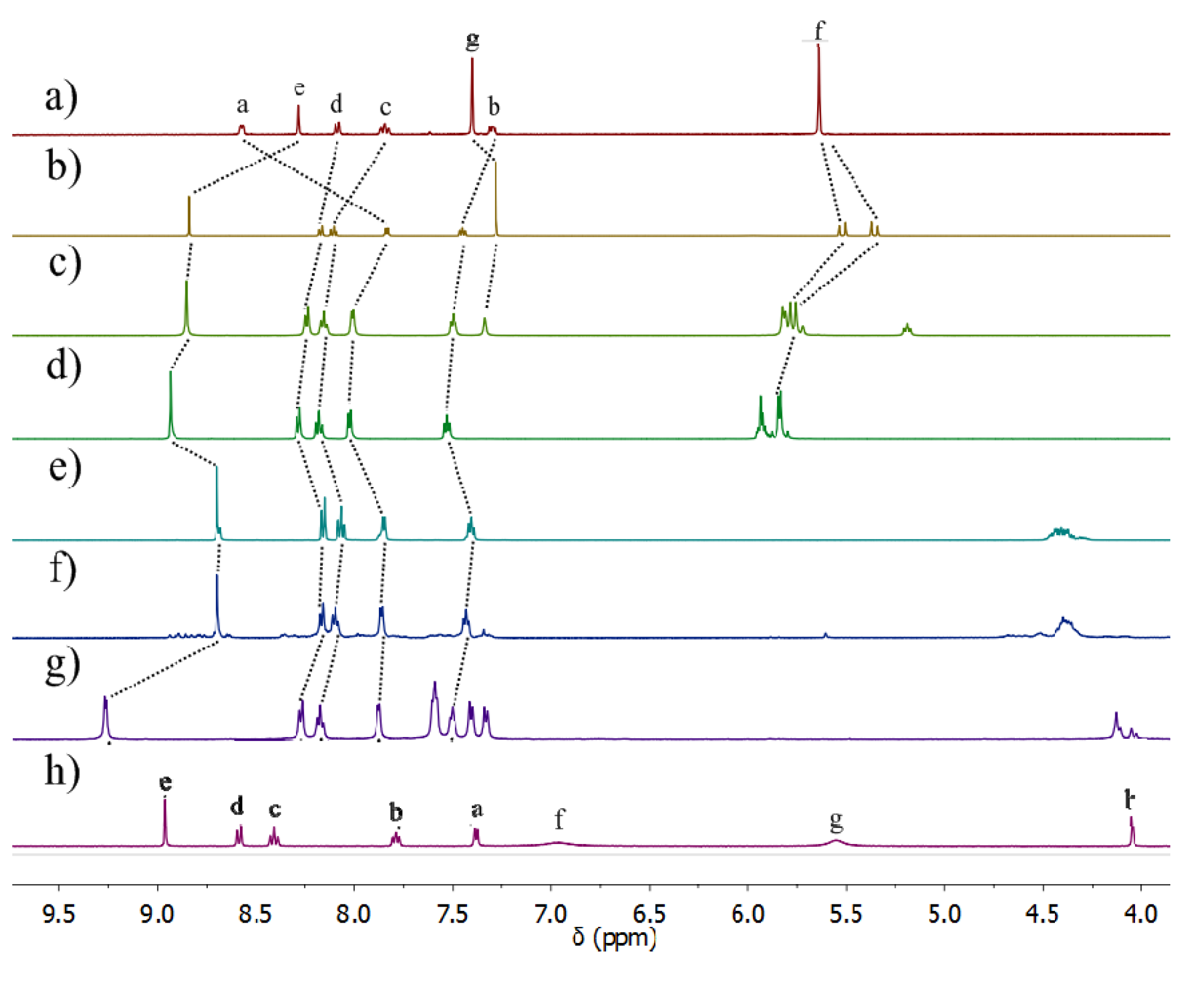
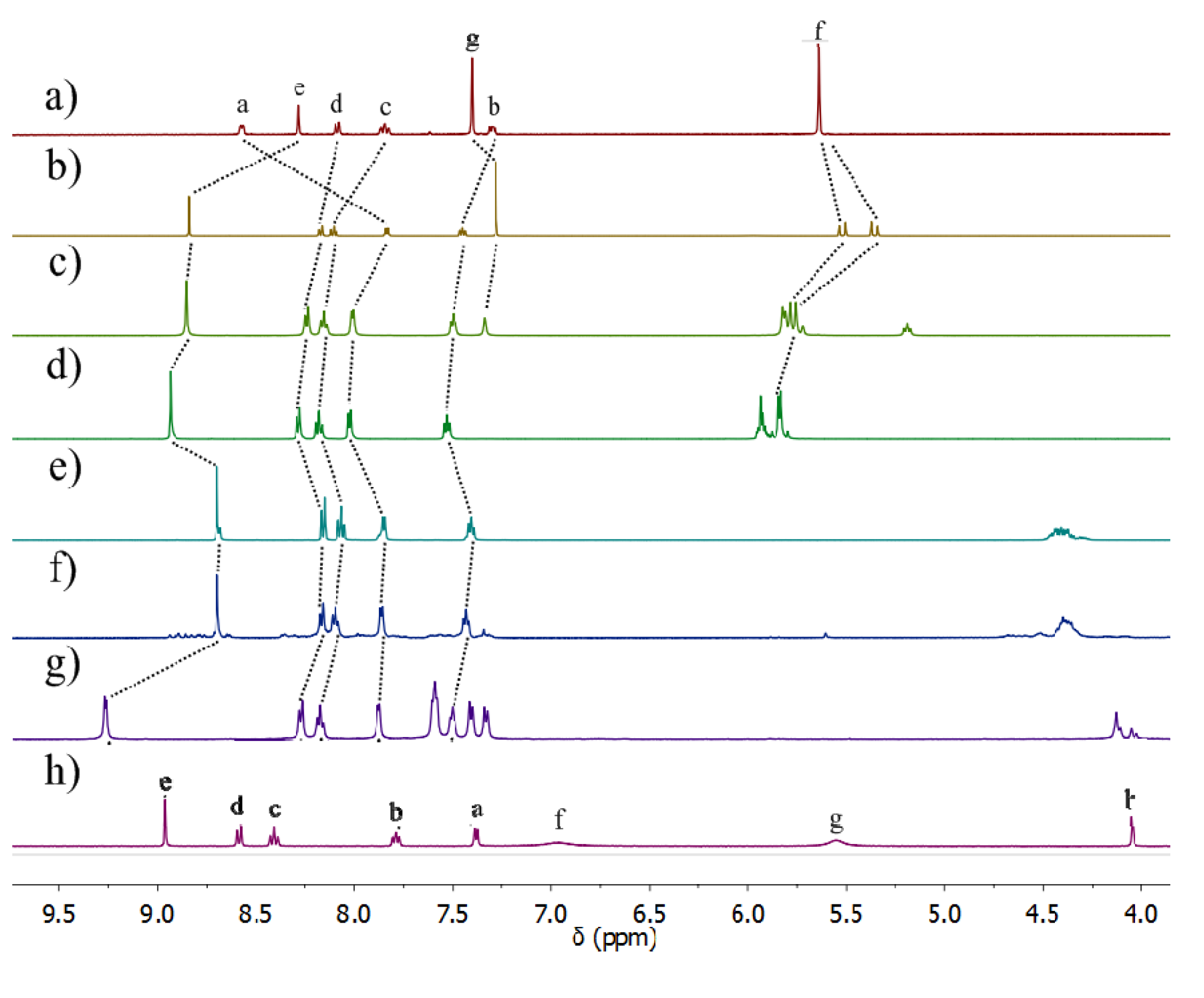
2.2.2. Solution Structures
| Ligand | Diffusion coefficient (×10−10 m2·s−1) | Fe(II) cylinder | Diffusion coefficient (×10−10 m2·s−1) |
|---|---|---|---|
| 7 | 12.9 | 8 | 6.9 |
| 3a | 12.9 | 6a | 4.7 |
| 3b | 12.8 | 6b | 4.7 |
| 3c | 11.9 | 6c | 4.0 |
| 3d | 14.4 | 6d | 3.9 |
| 3e | 13.3 | 6e | 4.9 |
| 3f | 9.6 | 6f | 3.0 |
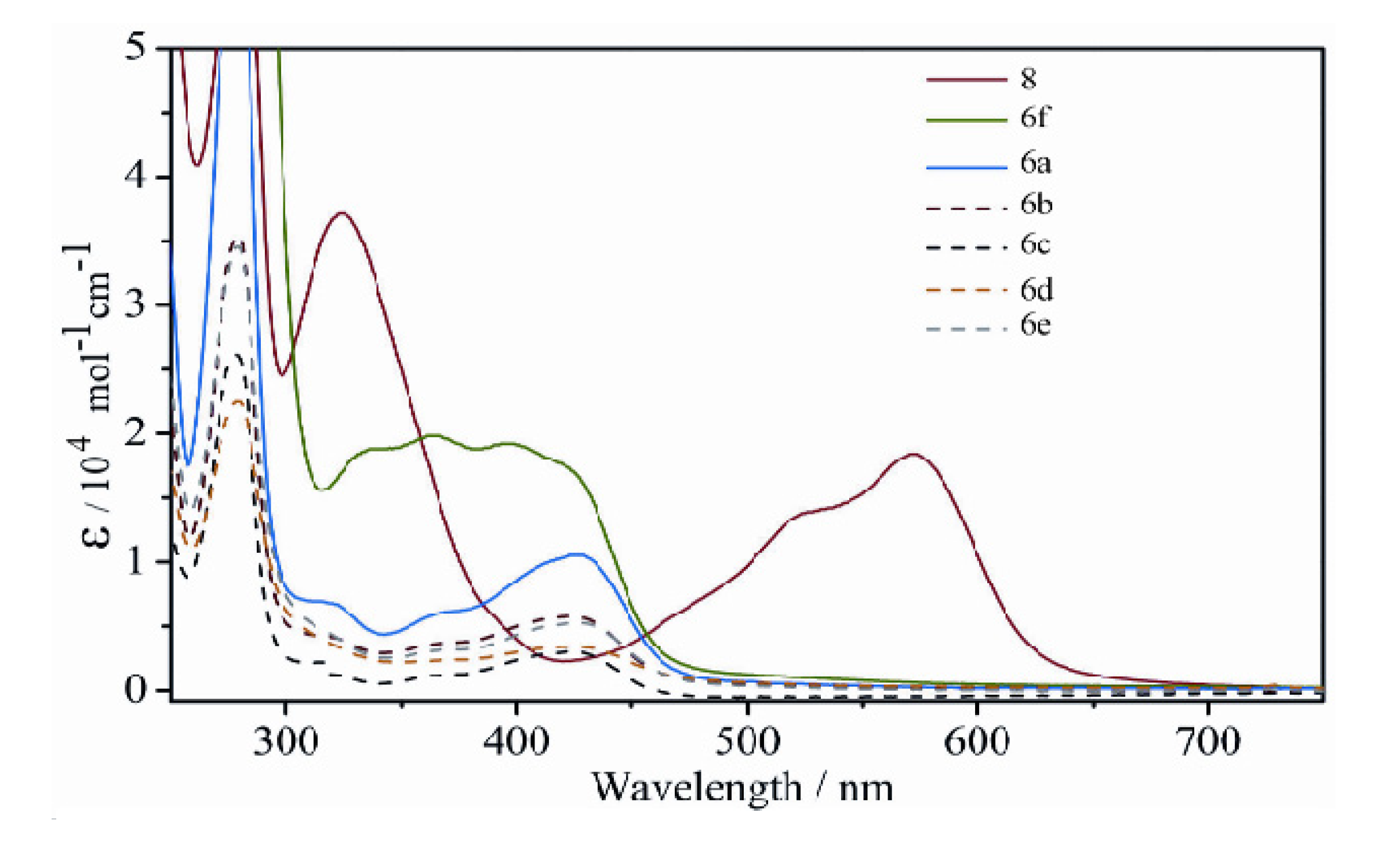
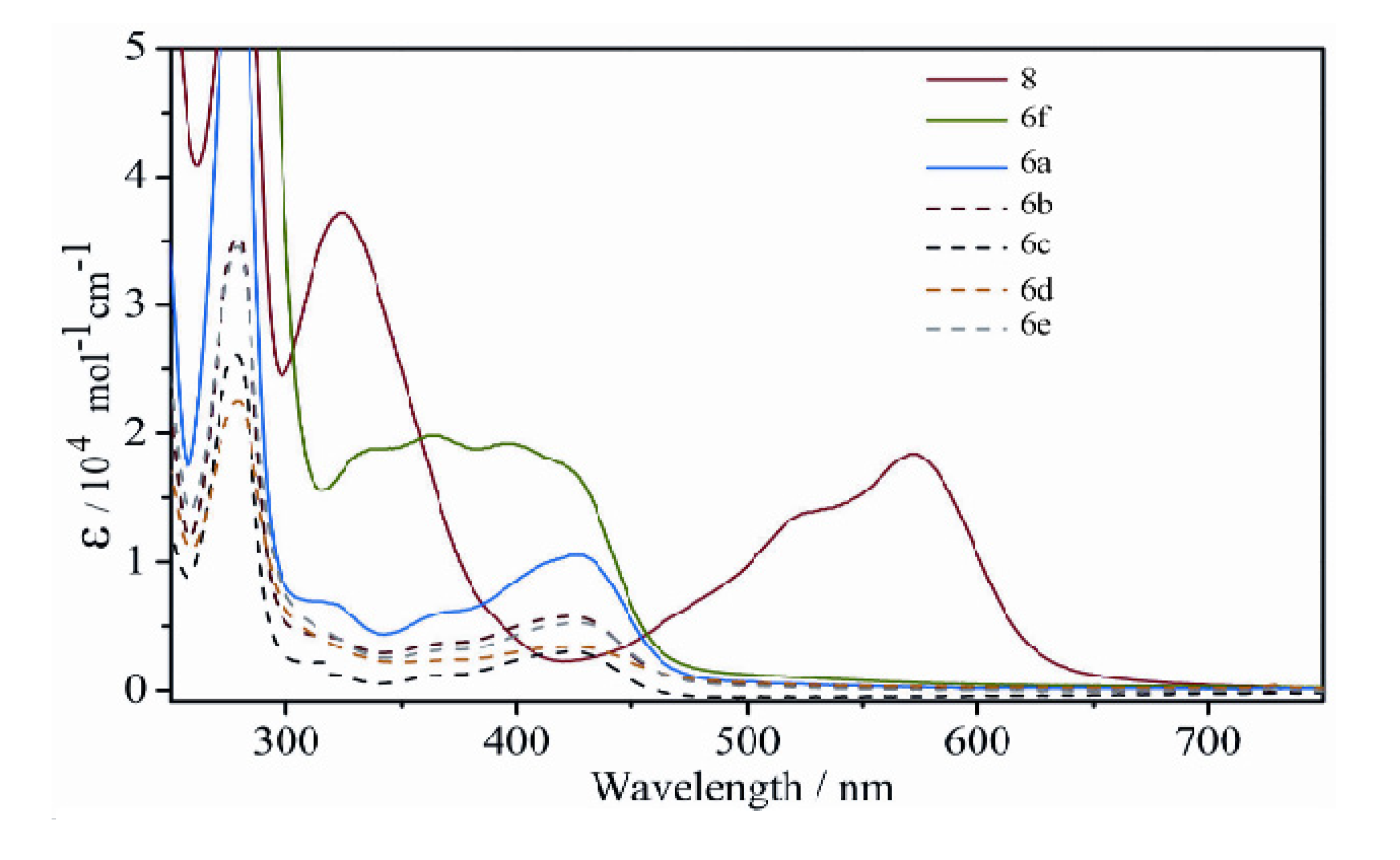
2.2.3. Electronic Spectra
2.3. Molecular Modeling (Docking) and Biological Activity Studies
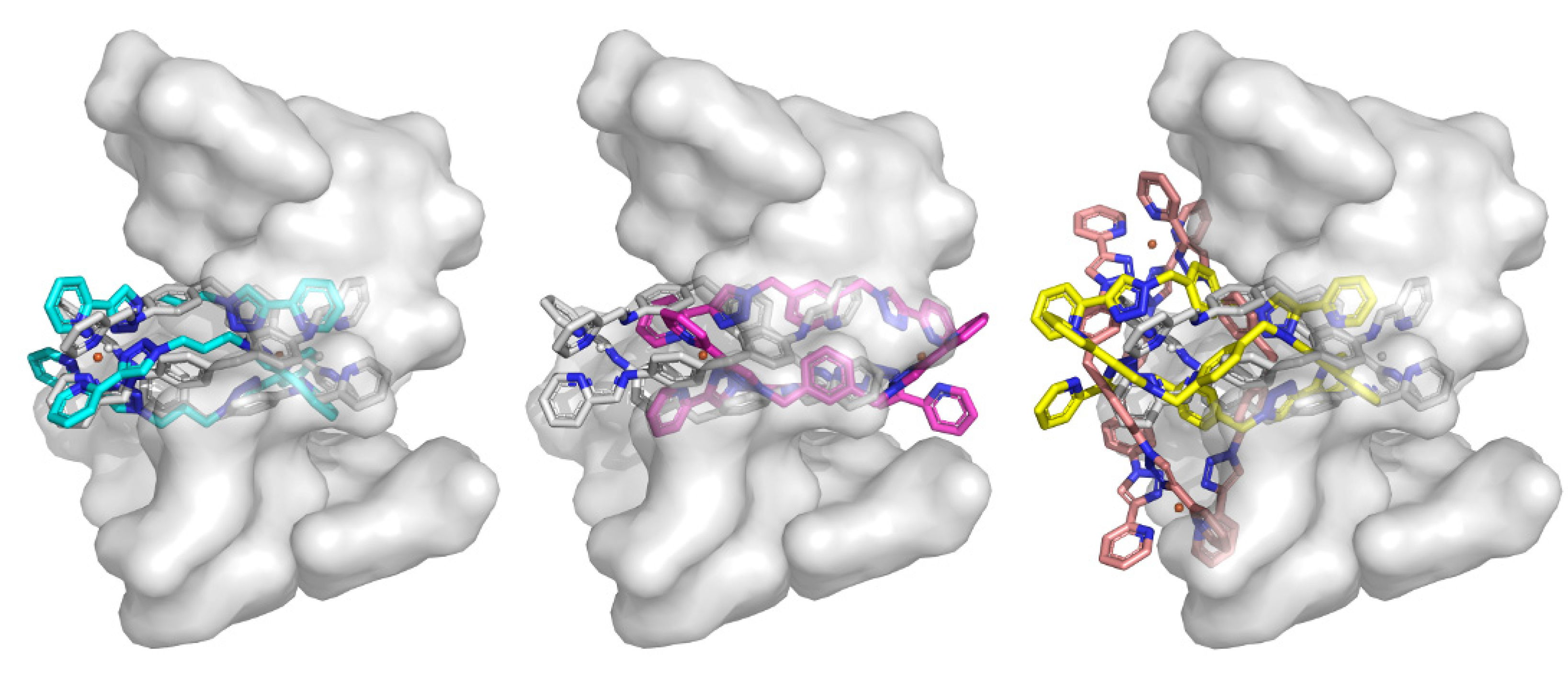
2.3.1. Duplex Modeling
2.3.2. Triplex Modeling
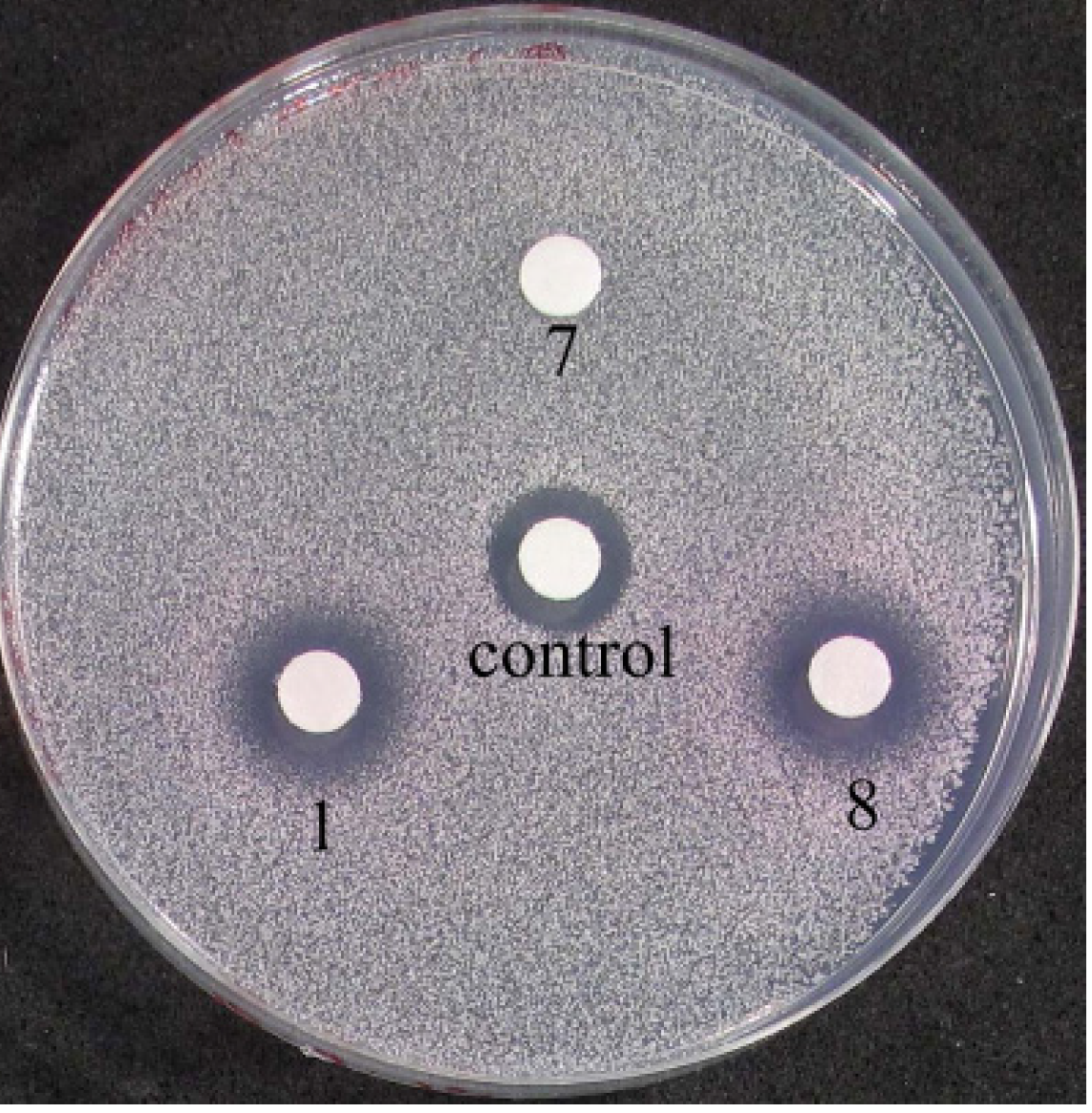
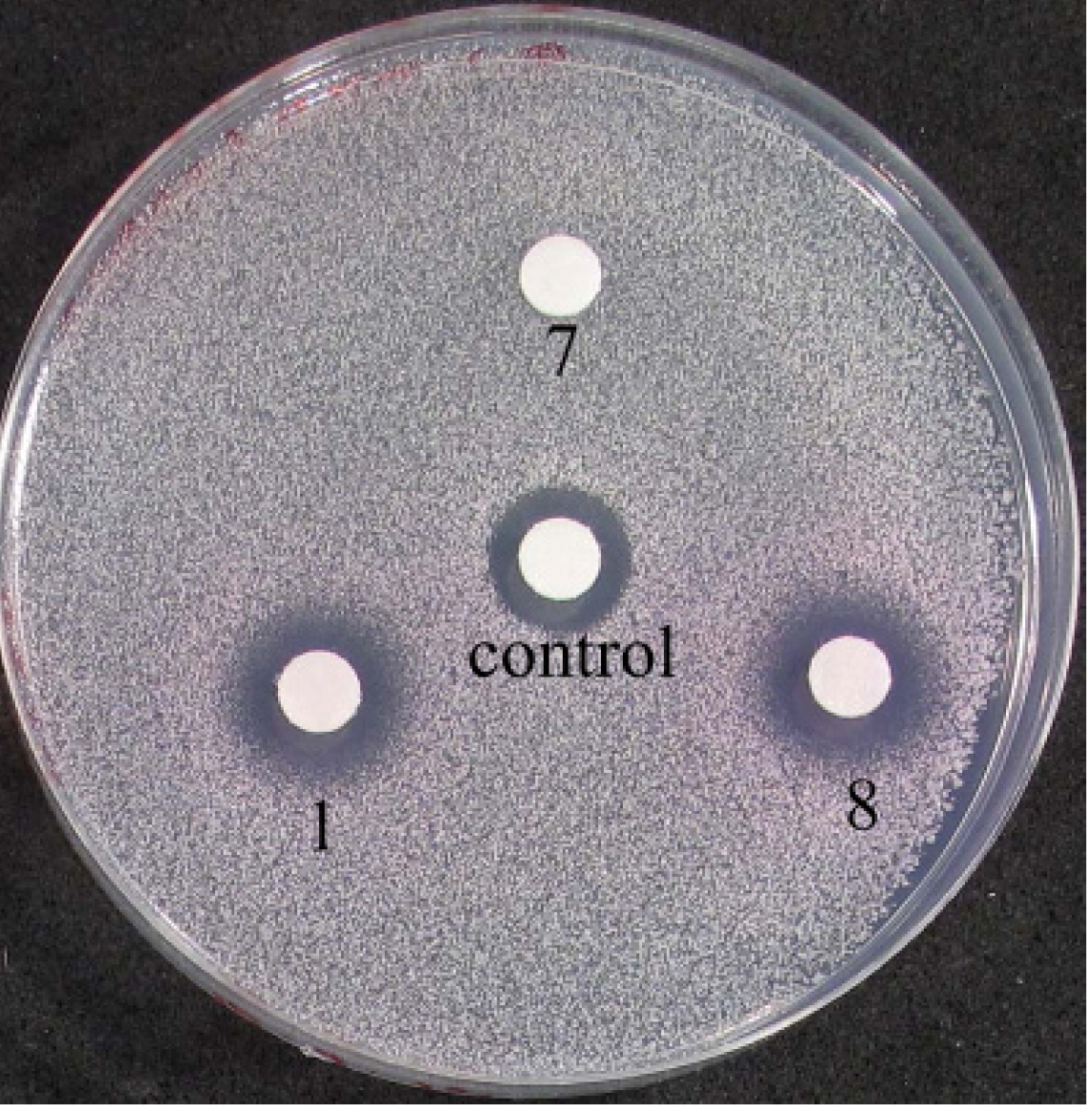
2.3.3. Biological Testing on Yeast Cells
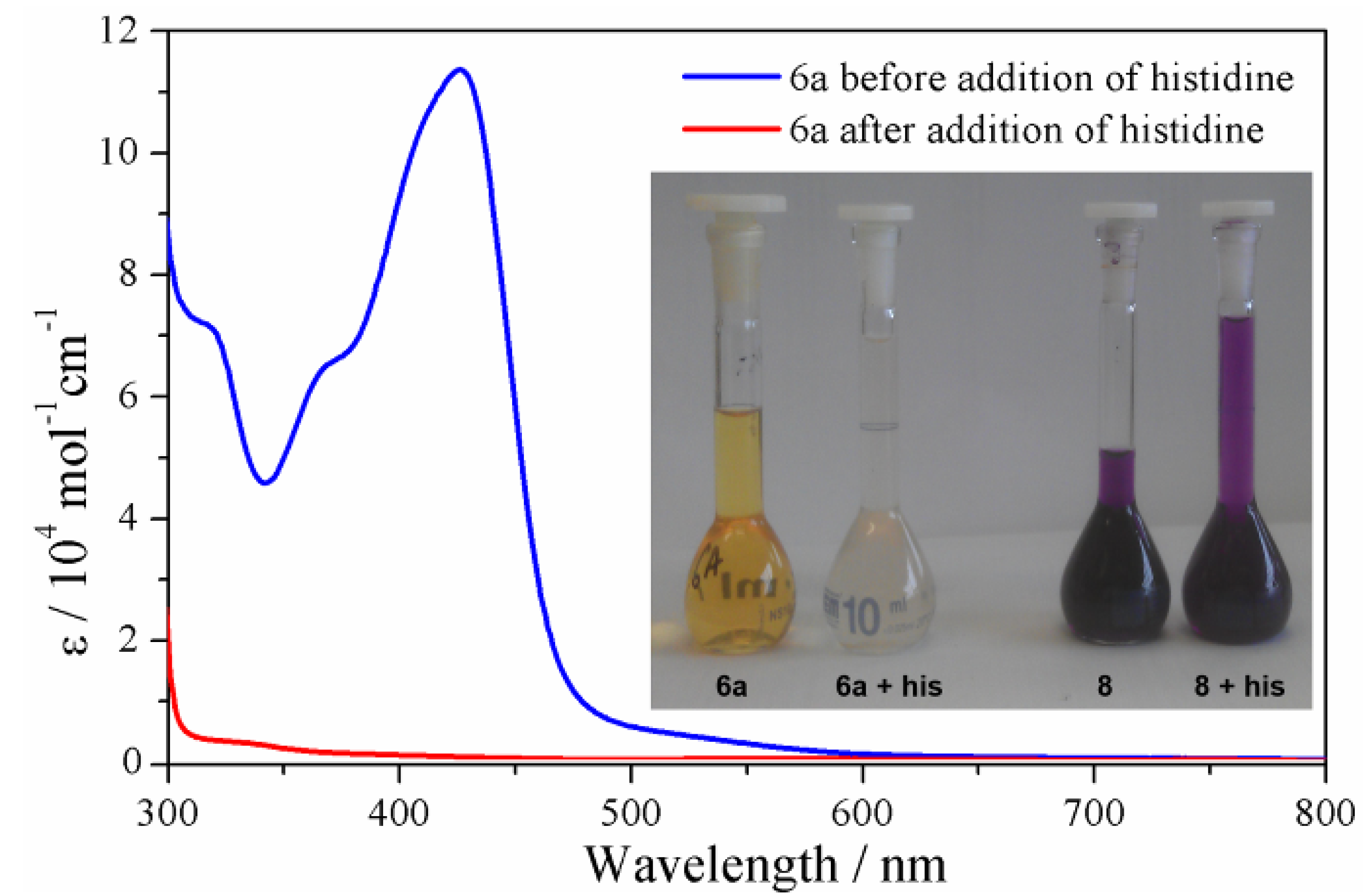
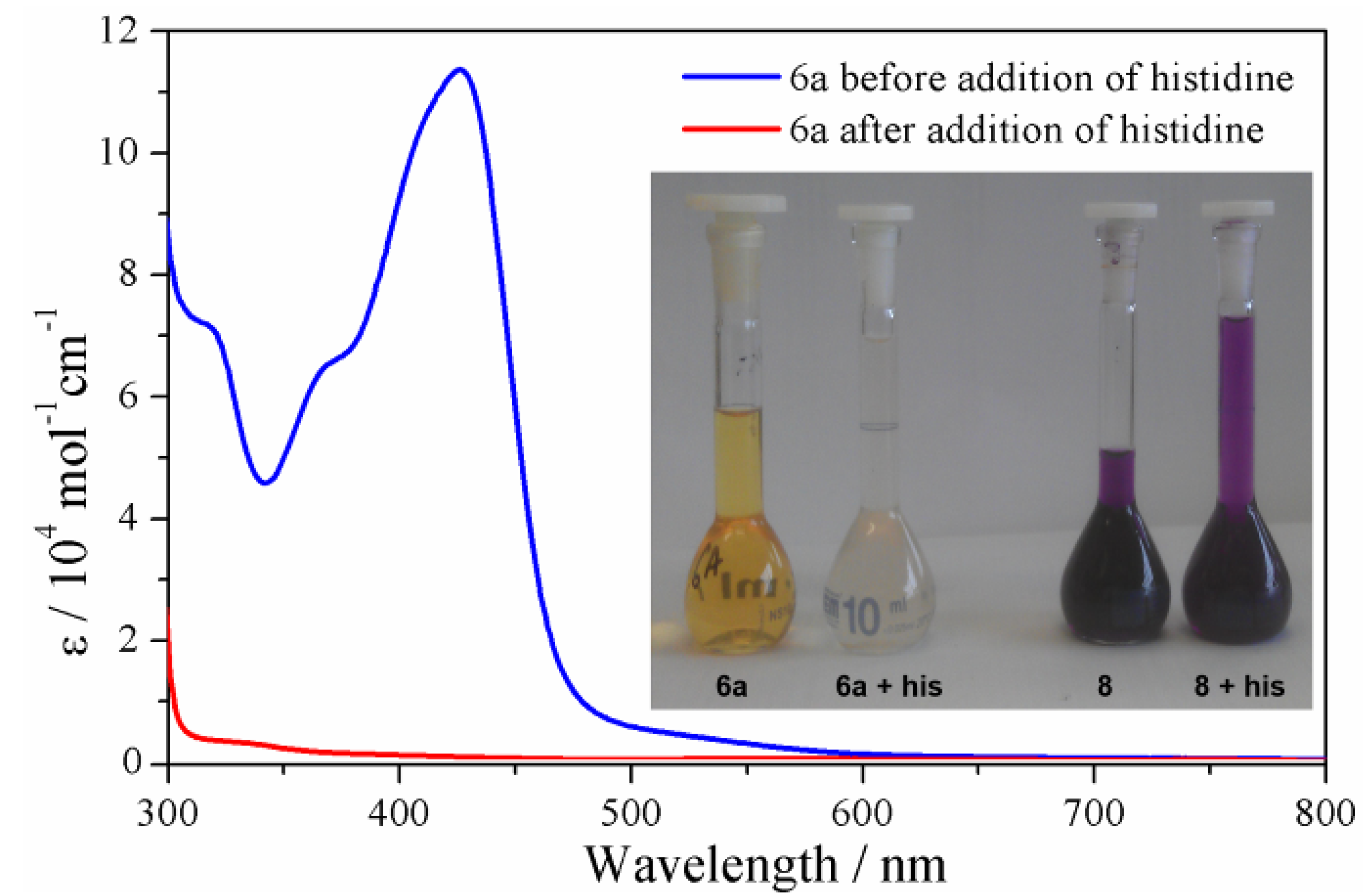
2.3.4. Solution Stability Studies of the Iron(II) Metallosupramolecular Architectures
3. Experimental
3.1. General Information
3.2. General Method for the Synthesis of the Fe(II) Model Complexes 5a–b
3.3. General Method for the Synthesis of the Fe(II) Cylinders 6a–f
3.4. Synthesis of the Fe(II) Pyridylimine Cylinder 8
3.5. General Method for the Molecular Docking Studies
3.6. Yeast Agarose Diffusion Assays
3.7. General X-ray Experimental Section
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Miyake, H.; Tsukube, H. Coordination chemistry strategies for dynamic helicates: Time-programmable chirality switching with labile and inert metal helicates. Chem. Soc. Rev. 2012, 41, 6977–6991. [Google Scholar] [CrossRef]
- Howson, S.E.; Scott, P. Approaches to the synthesis of optically pure helicates. Dalton Trans. 2011, 40, 10268–10277. [Google Scholar] [CrossRef]
- Albrecht, M.; Froehlich, R. Symmetry driven self-assembly of metallo-supramolecular architectures. Bull. Chem. Soc. Jpn. 2007, 80, 797–808. [Google Scholar]
- Piguet, C.; Borkovec, M.; Hamacek, J.; Zeckert, K. Strict self-assembly of polymetallic helicates: the concepts behind the semantics. Coord. Chem. Rev. 2005, 249, 705–726. [Google Scholar] [CrossRef]
- Hannon, M.J.; Childs, L.J. Helices and Helicates. Beautiful supramolecular motifs with emerging applications. Supramol. Chem. 2004, 16, 7–22. [Google Scholar] [CrossRef]
- Albrecht, M. “Let’s Twist Again”-double-stranded, triple-stranded, and circular helicates. Chem. Rev. 2001, 101, 3457–3497. [Google Scholar] [CrossRef]
- Archer, R.J.; Hawes, C.S.; Jameson, G.N.L.; McKee, V.; Moubaraki, B.; Chilton, N.F.; Murray, K.S.; Schmitt, W.; Kruger, P.E. Partial spin crossover behaviour in a dinuclear iron(II) triple helicate. Dalton Trans. 2011, 40, 12368–12373. [Google Scholar] [CrossRef]
- Pelleteret, D.; Clerac, R.; Mathoniere, C.; Harte, E.; Schmitt, W.; Kruger, P.E. Asymmetric spin crossover behavior and evidence of light-induced excited spin state trapping in a dinuclear iron(II) helicate. Chem. Commun. 2009, 221–223. [Google Scholar]
- Telfer, S.G.; Bocquet, B.; Williams, A.F. Thermal spin crossover in binuclear iron(II) helicates: Negative cooperativity and a mixed spin state in solution. Inorg. Chem. 2001, 40, 4818–4820. [Google Scholar] [CrossRef]
- Lincheneau, C.; Peacock, R.D.; Gunnlaugsson, T. Europium directed synthesis of enantiomerically pure dimetallic luminescent “squeezed” triple-stranded helicates; solution studies. Chem. Asian J. 2010, 5, 500–504. [Google Scholar] [CrossRef]
- Stomeo, F.; Lincheneau, C.; Leonard, J.P.; O'Brien, J.E.; Peacock, R.D.; McCoy, C.P.; Gunnlaugsson, T. Metal-directed synthesis of enantiomerically pure dimetallic lanthanide luminescent triple-stranded helicates. J. Am. Chem. Soc. 2009, 131, 9636–9637. [Google Scholar] [CrossRef]
- Bünzli, J.-C.G.; Chauvin, A.-S.; Vandevyver, C.D.B.; Bo, S.; Comby, S. Lanthanide bimetallic helicates forin vitroimaging and sensing. Ann. N.Y. Acad. Sci. 2008, 1130, 97–105. [Google Scholar]
- Vandevyver, C.D.B.; Chauvin, A.-S.; Comby, S.; Bunzli, J.-C.G. Luminescent lanthanide bimetallic triple-stranded helicates as potential cellular imaging probes. Chem. Commun. 2007, 1716–1718. [Google Scholar]
- Sham, K.-C.; Yeung, H.-L.; Yiu, S.-M.; Lau, T.-C.; Kwong, H.-L. New binuclear double-stranded manganese helicates as catalysts for alkene epoxidation. Dalton Trans. 2010, 39, 9469–9471. [Google Scholar] [CrossRef]
- Ayme, J.-F.; Beves, J.E.; Leigh, D.A.; McBurney, R.T.; Rissanen, K.; Schultz, D. Pentameric circular iron(II) double helicates and a molecular pentafoil knot. J. Am. Chem. Soc. 2012, 134, 9488–9497. [Google Scholar]
- Wenzel, M.; Knapp, Q.W.; Plieger, P.G. A bis-salicylaldoximato-copper(II) receptor for selective sulfate uptake. Chem. Commun. 2011, 47, 499–501. [Google Scholar] [CrossRef]
- Stevens, J.R.; Plieger, P.G. Anion-driven conformation control and enhanced sulfate binding utilising aryl linked salicylaldoxime dicopper helicates. Dalton Trans. 2011, 40, 12235–12241. [Google Scholar] [CrossRef]
- Wenzel, M.; Bruere, S.R.; Knapp, Q.W.; Tasker, P.A.; Plieger, P.G. Zwitterionic dicopper helicates: anion encapsulation and binding studies. Dalton Trans. 2010, 39, 2936–2941. [Google Scholar] [CrossRef]
- Wenzel, M.; Jameson, G.B.; Ferguson, L.A.; Knapp, Q.W.; Forgan, R.S.; White, F.J.; Parsons, S.; Tasker, P.A.; Plieger, P.G. Anion-induced contraction of helical receptors. Chem. Commun. 2009, 3606–3608. [Google Scholar]
- Goetz, S.; Kruger, P.E. A new twist in anion binding: metallo-helicate hosts for anionic guests. Dalton Trans. 2006, 1277–1284. [Google Scholar] [CrossRef]
- Ayme, J.-F.; Beves, J.E.; Leigh, D.A.; McBurney, R.T.; Rissanen, K.; Schultz, D. A synthetic molecular pentafoil knot. Nat. Chem. 2012, 4, 15–20. [Google Scholar]
- Hannon, M.J.; Painting, C.L.; Jackson, A.; Hamblin, J.; Errington, W. An inexpensive approach to supramolecular architecture. Chem. Commun. 1997, 1807–1808. [Google Scholar]
- Hannon, M.J. Supramolecular DNA recognition. Chem. Soc. Rev. 2007, 36, 280–295. [Google Scholar] [CrossRef]
- Hannon, M.J. Metal-based anticancer drugs: from a past anchored in platinum chemistry to a post-genomic future of diverse chemistry and biology. Pure Appl. Chem. 2007, 79, 2243–2261. [Google Scholar] [CrossRef]
- Moldrheim, E.; Hannon, M.J.; Meistermann, I.; Rodger, A.; Sletten, E. Interaction between a DNA oligonucleotide and a dinuclear iron(II) supramolecular cylinder; an NMR and molecular dynamics study. J. Biol. Inorg. Chem. 2002, 7, 770–780. [Google Scholar] [CrossRef]
- Meistermann, I.; Moreno, V.; Prieto, M.J.; Moldrheim, E.; Sletten, E.; Khalid, S.; Rodger, P.M.; Peberdy, J.C.; Isaac, C.J.; Rodger, A.; et al. Intramolecular DNA coiling mediated by metallo-supramolecular cylinders: differential binding of P and M helical enantiomers. Proc. Natl. Acad. Sci. USA 2002, 99, 5069–5074. [Google Scholar] [CrossRef]
- Hannon, M.J.; Moreno, V.; Prieto, M.J.; Moldrheim, E.; Sletten, E.; Meistermann, I.; Isaac, C.J.; Sanders, K.J.; Rodger, A. Intramolecular DNA coiling mediated by a metallo-supramolecular cylinder. Angew. Chem. Int. Ed. 2001, 40, 880–884. [Google Scholar]
- Cerasino, L.; Hannon, M.J.; Sletten, E. DNA three-way junction with a dinuclear iron(II) supramolecular helicate at the center: A NMR structural study. Inorg. Chem. 2007, 46, 6245–6251. [Google Scholar] [CrossRef]
- Oleksi, A.; Blanco, A.G.; Boer, R.; Uson, I.; Aymami, J.; Rodger, A.; Hannon, M.J.; Coll, M. Molecular recognition of a three-way DNA junction by a metallosupramolecular helicate. Angew. Chem. Int. Ed. 2006, 45, 1227–1231. [Google Scholar] [CrossRef]
- Hotze, A.C.G.; Hodges, N.J.; Hayden, R.E.; Sanchez-Cano, C.; Paines, C.; Male, N.; Tse, M.-K.; Bunce, C.M.; Chipman, J.K.; Hannon, M.J. Supramolecular iron cylinder with unprecedented DNA binding is a potent cytostatic and apoptotic agent without exhibiting genotoxicity. Chem. Biol. 2008, 15, 1258–1267. [Google Scholar] [CrossRef]
- Richards, A.D.; Rodger, A.; Hannon, M.J.; Bolhuis, A. Antimicrobial activity of an iron triple helicate. Int. J. Antimicrob. Agents 2009, 33, 469–472. [Google Scholar] [CrossRef]
- Howson, S.E.; Bolhuis, A.; Brabec, V.; Clarkson, G.J.; Malina, J.; Rodger, A.; Scott, P. Optically pure, water-stable metallo-helical ‘flexicate’ assemblies with antibiotic activity. Nat. Chem. 2012, 4, 31–36. [Google Scholar]
- Buckley, B.R.; Heaney, H. Mechanistic investigations of copper(I)-catalysed alkyne-azide cycloaddition reactions. Top. Heterocycl. Chem. 2012, 28, 1–29. [Google Scholar] [CrossRef]
- Liang, L.; Astruc, D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Tornoe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Meldal, M.; Tornoe, C.W. Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Wu, P.; Fokin, V.V. Catalytic azide-alkyne cycloaddition: reactivity and applications. Aldrichimica Acta 2007, 40, 7–17. [Google Scholar]
- Zheng, T.; Rouhanifard, S.H.; Jalloh, A.S.; Wu, P. Click triazoles for bioconjugation. Top. Heterocycl. Chem. 2012, 28, 163–183. [Google Scholar] [CrossRef]
- Agalave, S.G.; Maujan, S.R.; Pore, V.S. Click chemistry: 1,2,3-Triazoles as pharmacophores. Chem. Asian J. 2011, 6, 2696–2718. [Google Scholar] [CrossRef]
- Holub, J.M.; Kirshenbaum, K. Tricks with clicks: modification of peptidomimetic oligomers via copper-catalyzed azide-alkyne [3 + 2] cycloaddition. Chem. Soc. Rev. 2010, 39, 1325–1337. [Google Scholar] [CrossRef]
- Aragao-Leoneti, V.; Campo, V.L.; Gomes, A.S.; Field, R.A.; Carvalho, I. Application of copper(I)-catalyzed azide/alkyne cycloaddition (CuAAC) click chemistry' in carbohydrate drug and neoglycopolymer synthesis. Tetrahedron 2010, 66, 9475–9492. [Google Scholar] [CrossRef]
- Amblard, F.; Cho, J.H.; Schinazi, R.F. Cu(I)-catalyzed huisgen azide-alkyne 1,3-dipolar cycloaddition reaction in nucleoside, nucleotide, and oligonucleotide chemistry. Chem. Rev. 2009, 109, 4207–4220. [Google Scholar] [CrossRef]
- Li, N.; Binder, W.H. Click-chemistry for nanoparticle-modification. J. Mater. Chem. 2011, 21, 16717–16734. [Google Scholar] [CrossRef]
- Fahrenbach, A.C.; Stoddart, J.F. Reactions under the click chemistry philosophy employed in supramolecular and mechanostereochemical systems. Chem. Asian J. 2011, 6, 2660–2669. [Google Scholar] [CrossRef]
- Qin, A.; Lam, J.W.Y.; Tang, B.Z. Click polymerization. Chem. Soc. Rev. 2010, 39, 2522–2544. [Google Scholar] [CrossRef]
- McDonald, K.P.; Hua, Y.; Flood, A.H. 1,2,3-Triazoles and the expanding utility of charge neutral CH···anion interactions. Top. Heterocycl. Chem. 2010, 24, 341–366. [Google Scholar] [CrossRef]
- Crowley, J.D.; Goldup, S.M.; Lee, A.-L.; Leigh, D.A.; McBurney, R.T. Active metal template synthesis of rotaxanes, catenanes and molecular shuttles. Chem. Soc. Rev. 2009, 38, 1530–1541. [Google Scholar] [CrossRef]
- Crowley, J.D.; McMorran, D.A. “Click-triazole” coordination chemistry: Exploiting 1,4-disubstituted-1,2,3-triazoles as ligands. Top. Heterocycl. Chem. 2012, 28, 31–83. [Google Scholar] [CrossRef]
- Crowley, J.D.; Lee, A.-L.; Kilpin, K.J. 1,3,4-Trisubstituted-1,2,3-triazol-5-ylidene click carbene ligands. Synthesis, catalysis, and self-assembly. Aust. J. Chem. 2011, 64, 1118–1132. [Google Scholar] [CrossRef]
- Schweinfurth, D.; Deibel, N.; Weisser, F.; Sarkar, B. Getting new ligands with a click. Nachr. Chem. 2011, 59, 937–941. [Google Scholar] [CrossRef]
- Struthers, H.; Mindt, T.L.; Schibli, R. Metal chelating systems synthesized using the copper(I) catalyzed azide-alkyne cycloaddition. Dalton Trans. 2010, 39, 675–696. [Google Scholar] [CrossRef]
- Scott, S.O.; Gavey, E.L.; Lind, S.J.; Gordon, K.C.; Crowley, J.D. Self-assembled palladium(II) “click” cages: Synthesis, structural modification and stability. Dalton Trans. 2011, 40, 12117–12124. [Google Scholar]
- Kilpin, K.J.; Paul, U.S.D.; Lee, A.-L.; Crowley, J.D. Gold(I) “click” 1,2,3-triazolylidenes: Synthesis, self-assembly and catalysis. Chem. Commun. 2011, 47, 328–330. [Google Scholar] [CrossRef]
- Gower, M.L.; Crowley, J.D. Self-assembly of silver(I) metallomacrocycles using unsupported 1,4-substituted-1,2,3-triazole “click” ligands. Dalton Trans. 2010, 39, 2371–2378. [Google Scholar] [CrossRef]
- Crowley, J.D.; Gavey, E.L. Use of di-1,4-substituted-1,2,3-triazole “click” ligands to self-assemble dipalladium(II) coordinatively saturated, quadruply stranded helicate cages. Dalton Trans. 2010, 39, 4035–4037. [Google Scholar] [CrossRef]
- Crowley, J.D.; Bandeen, P.H. A multicomponent CuAAC “click” approach to a library of hybrid polydentate 2-pyridyl-1,2,3-triazole ligands: new building blocks for the generation of metallosupramolecular architectures. Dalton Trans. 2010, 39, 612–623. [Google Scholar] [CrossRef]
- Garcia, L.; Maisonneuve, S.; Xie, J.; Guillot, R.; Dorlet, P.; Riviere, E.; Desmadril, M.; Lambert, F.; Policar, C. Sugars to Control Ligand Shape in Metal Complexes: Conformationally Constrained Glycoligands with a Predetermination of Stereochemistry and a Structural Control. Inorg. Chem. 2010, 49, 7282–7288. [Google Scholar]
- Najar, A.M.; Tidmarsh, I.S.; Ward, M.D. Lead(II) complexes of bis- and tris-bidentate compartmental ligands based on pyridyl-pyrazole and pyridyl-triazole fragments: coordination networks and a discrete dimeric box. CrystEngComm 2010, 12, 3642–3650. [Google Scholar]
- The biological properties of “click” complexes have received considerable recent attention for selected examples see ref 61–69.
- Yano, S.; Ohi, H.; Ashizaki, M.; Obata, M.; Mikata, Y.; Tanaka, R.; Nishioka, T.; Kinoshita, I.; Sugai, Y.; Okura, I.; et al. Syntheses, Characterization, and Antitumor Activities of Platinum(II) and Palladium(II) Complexes with Sugar-Conjugated Triazole Ligands. Chem. Biodivers. 2012, 9, 1903–1915. [Google Scholar] [CrossRef]
- Clede, S.; Lambert, F.; Sandt, C.; Gueroui, Z.; Refregiers, M.; Plamont, M.-A.; Dumas, P.; Vessieres, A.; Policar, C. A rhenium tris-carbonyl derivative as a single core multimodal probe for imaging (SCoMPI) combining infrared and luminescent properties. Chem. Commun. 2012, 48, 7729–7731. [Google Scholar] [CrossRef]
- Seridi, A.; Wolff, M.; Boulay, A.; Saffon, N.; Coulais, Y.; Picard, C.; Machura, B.; Benoist, E. Rhenium(I) and technetium(I) complexes of a novel pyridyltriazole-based ligand containing an arylpiperazine pharmacophore: Synthesis, crystal structures, computational studies and radiochemistry. Inorg. Chem. Commun. 2011, 14, 238–242. [Google Scholar] [CrossRef]
- Benoist, E.; Coulais, Y.; Almant, M.; Kovensky, J.; Moreau, V.; Lesur, D.; Artigau, M.; Picard, C.; Galaup, C.; Gouin, S.G. A Click procedure with heterogeneous copper to tether technetium-99m chelating agents and rhenium complexes. Evaluation of the chelating properties and biodistribution of the new radiolabelled glucose conjugates. Carbohydr. Res. 2011, 346, 26–34. [Google Scholar] [CrossRef]
- Urankar, D.; Pevec, A.; Kosmrlj, J. Synthesis and characterization of platinum(II) complexes with a diazenecarboxamide-appended picolyl-triazole ligand. Eur. J. Inorg. Chem. 2011, 1921–1929. [Google Scholar] [CrossRef]
- Bratsos, I.; Urankar, D.; Zangrando, E.; Genova-Kalou, P.; Kosmrlj, J.; Alessio, E.; Turel, I. 1-(2-Picolyl)-substituted 1,2,3-triazole as novel chelating ligand for the preparation of ruthenium complexes with potential anticancer activity. Dalton Trans. 2011, 40, 5188–5199. [Google Scholar] [CrossRef]
- Chevry, A.; Teyssot, M.-L.; Maisonial, A.; Lemoine, P.; Viossat, B.; Traikia, M.; Aitken, D.J.; Alves, G.; Morel, L.; Nauton, L.; et al. Click chelators - The behavior of platinum and palladium complexes in the presence of guanosine and DNA. Eur. J. Inorg. Chem. 2010, 3513–3519. [Google Scholar]
- Obata, M.; Kitamura, A.; Mori, A.; Kameyama, C.; Czaplewska, J.A.; Tanaka, R.; Kinoshita, I.; Kusumoto, T.; Hashimoto, H.; Harada, M.; et al. Syntheses, structural characterization and photophysical properties of 4-(2-pyridyl)-1,2,3-triazole rhenium(I) complexes. Dalton Trans. 2008, 3292–3300. [Google Scholar]
- Maisonial, A.; Serafin, P.; Traikia, M.; Debiton, E.; Thery, V.; Aitken, D.J.; Lemoine, P.; Viossat, B.; Gautier, A. Click chelators for platinum-based anticancer drugs. Eur. J. Inorg. Chem. 2008, 298–305. [Google Scholar]
- Kilpin, K.J.; Gavey, E.L.; McAdam, C.J.; Anderson, C.B.; Lind, S.J.; Keep, C.C.; Gordon, K.C.; Crowley, J.D. Palladium(II) complexes of readily functionalized bidentate 2-pyridyl-1,2,3-triazole “Click” ligands: A synthetic, structural, spectroscopic, and computational study. Inorg. Chem. 2011, 50, 6334–6346. [Google Scholar] [CrossRef]
- Crowley, J.D.; Bandeen, P.H.; Hanton, L.R. A one pot multi-component CuAAC “click” approach to bidentate and tridentate pyridyl-1,2,3-triazole ligands: synthesis, x-ray structures and copper(II) and silver(I) complexes. Polyhedron 2010, 29, 70–83. [Google Scholar] [CrossRef]
- The related bis(2,6-bis(1,2,3-triazol-4-yl)pyridine)) Fe(II) complexes had been reported by the Flood group in 2007 see ref. [73].
- Li, Y.; Huffman, J.C.; Flood, A.H. Can terdentate 2,6-bis(1,2,3-triazol-4-yl)pyridines form stable coordination compounds? Chem. Commun. 2007, 2692–2694. [Google Scholar]
- Fletcher, J.T.; Bumgarner, B.J.; Engels, N.D.; Skoglund, D.A. Multidentate 1,2,3-triazole-containing chelators from tandem deprotection/click reactions of (trimethylsilyl)alkynes and comparison of their ruthenium(II) complexes. Organometallics 2008, 27, 5430–5433. [Google Scholar]
- Happ, B.; Friebe, C.; Winter, A.; Hager, M.D.; Hoogenboom, R.; Schubert, U.S. 2-(1H-1,2,3-Triazol-4-yl)-pyridine ligands as alternatives to 2,2′-bipyridines in ruthenium(II) complexes. Chem. Asian J. 2009, 4, 154–163. [Google Scholar] [CrossRef]
- Howson, S.E.; Allan, L.E.N.; Chmel, N.P.; Clarkson, G.J.; van Gorkum, R.; Scott, P. Self-assembling optically pure Fe(A-B)3 chelates. Chem. Commun. 2009, 1727–1729. [Google Scholar]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Stevenson, K.A.; Melan, C.F.C.; Fleischel, O.; Wang, R.; Petitjean, A. Solid-state self-assembly of triazolylpyridine-based helicates and mesocate: control of the metal-metal distances. Cryst. Growth Des. 2012, 12, 5169–5173. [Google Scholar] [CrossRef]
- Albrecht, M.; Riether, C. Self assembly of a triple-stranded meso-helicate from two iron(II) ions and three [CH2]3-bridged bis(2,2'-bipyridine) ligands. Chem. Ber. 1996, 129, 829–832. [Google Scholar] [CrossRef]
- Albrecht, M.; Kotila, S. Formation of a “meso-helicate” by self-assembly of three bis(catecholate) ligands and two titanium(IV) ions. Angew. Chem. Int. Ed. 1995, 34, 2134–2137. [Google Scholar] [CrossRef]
- Kerckhoffs, J.M.C.A.; Peberdy, J.C.; Meistermann, I.; Childs, L.J.; Isaac, C.J.; Pearmund, C.R.; Reudegger, V.; Khalid, S.; Alcock, N.W.; Hannon, M.J.; et al. Enantiomeric resolution of supramolecular helicates with different surface topographies. Dalton Trans. 2007, 734–742. [Google Scholar]
- Anderson, C.B.; Elliott, A.B.S.; Lewis, J.E.M.; McAdam, C.J.; Gordon, K.C.; Crowley, J.D. fac-Re(CO)3 complexes of 2,6-bis(4-substituted-1,2,3-triazol-1-ylmethyl)pyridine “click” ligands: Synthesis, characterisation and photophysical properties. Dalton Trans. 2012, 41, 14625–14632. [Google Scholar] [CrossRef]
- Brotherton, W.S.; Guha, P.M.; Phan, H.; Clark, R.J.; Shatruk, M.; Zhu, L. Tridentate complexes of 2,6-bis(4-substituted-1,2,3-triazol-1-ylmethyl)pyridine and its organic azide precursors: an application of the copper(II) acetate-accelerated azide-alkyne cycloaddition. Dalton Trans. 2011, 40, 3655–3665. [Google Scholar] [CrossRef]
- Kilpin, K.J.; Crowley, J.D. Palladium(II) and platinum(II) complexes of bidentate 2-pyridyl-1,2,3-triazole “click” ligands: Synthesis, properties and X-ray structures. Polyhedron 2010, 29, 3111–3117. [Google Scholar] [CrossRef]
- Guha, P.M.; Phan, H.; Kinyon, J.S.; Brotherton, W.S.; Sreenath, K.; Simmons, J.T.; Wang, Z.; Clark, R.J.; Dalal, N.S.; Shatruk, M.; et al. Structurally diverse copper(II) complexes of polyaza ligands containing 1,2,3-triazoles: Site selectivity and magnetic properties. Inorg. Chem. 2012, 51, 3465–3477. [Google Scholar] [CrossRef]
- Young, M.C.; Johnson, A.M.; Gamboa, A.S.; Hooley, R.J. Achiral endohedral functionality provides stereochemical control in Fe(II)-based self-assemblies. Chem. Commun. 2013, 49, 1627–1629. [Google Scholar] [CrossRef]
- Sakamoto, S.; Fujita, M.; Kim, K.; Yamaguchi, K. Characterization of self-assembling nano-sized structures by means of coldspray ionization mass spectrometry. Tetrahedron 2000, 56, 955–964. [Google Scholar] [CrossRef]
- Miras, H.N.; Wilson, E.F.; Cronin, L. Unravelling the complexities of inorganic and supramolecular self-assembly in solution with electrospray and cryospray mass spectrometry. Chem. Commun. 2009, 1297–1311. [Google Scholar] [CrossRef]
- Hawes, C.S.; Fitchett, C.M.; Kruger, P.E. Synthesis of an Fe(II) dinuclear triple helicate from a novel -bis-(N-pyrazolyl)pyridine ligand, [Fe2L3]4+: Solution and solid-state studies. Supramol. Chem. 2012, 24, 553–562. [Google Scholar] [CrossRef]
- Welby, C.E.; Grkinic, S.; Zahid, A.; Uppal, B.S.; Gibson, E.A.; Rice, C.R.; Elliott, P.I.P. Synthesis, characterisation and theoretical study of ruthenium 4,4[prime or minute]-bi-1,2,3-triazolyl complexes: fundamental switching of the nature of S1 and T1 states from MLCT to MC. Dalton Trans. 2012, 41, 7637–7646. [Google Scholar] [CrossRef]
- Lamping, E.; Monk, B.C.; Niimi, K.; Holmes, A.R.; Tsao, S.; Tanabe, K.; Niimi, M.; Uehara, Y.; Cannon, R.D. Characterization of Three Classes of Membrane Proteins Involved in Fungal Azole Resistance by Functional Hyperexpression in Saccharomyces cerevisiae. Eukaryotic Cell 2007, 6, 1150–1165. [Google Scholar] [CrossRef]
- Paul, L.E.H.; Therrien, B.; Furrer, J. Investigation of the reactivity between a ruthenium hexacationic prism and biological ligands. Inorg. Chem. 2012, 51, 1057–1067. [Google Scholar] [CrossRef]
- Paul, L.E.H.; Therrien, B.; Furrer, J. Interaction of a ruthenium hexacationic prism with amino acids and biological ligands: ESI mass spectrometry and NMR characterization of the reaction products. J. Biol. Inorg. Chem. 2012, 17, 1053–1062. [Google Scholar] [CrossRef]
- Lavanant, H.; Hecquet, E.; Hoppilliard, Y. Complexes of l-histidine with Fe2+, Co2+, Ni2+, Cu2+, Zn2+ studied by electrospray ionization mass spectrometry. Int. J. Mass Spectrom. 1999, 185–187, 11–23. [Google Scholar] [CrossRef]
- Fleischel, O.; Wu, N.; Petitjean, A. Click-triazole: Coordination of 2-(1,2,3-triazol-4-yl)-pyridine to cations of traditional tetrahedral geometry (Cu(I), Ag(I)). Chem. Commun. 2010, 46, 8454–8456. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Narayana, N.; Weiss, M.A. crystallographic analysis of a sex-specific enhancer element: Sequence-dependent DNA structure, hydration, and dynamics. J. Mol. Biol. 2009, 385, 469–490. [Google Scholar] [CrossRef]
- McPhillips, T.M.; McPhillips, S.E.; Chiu, H.J.; Cohen, A.E.; Deacon, A.M.; Ellis, P.J.; Garman, E.; Gonzalez, A.; Sauter, N.K.; Phizackerley, R.P.; et al. Blu-Ice and the distributed control system: Software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 2002, 9, 401–406. [Google Scholar] [CrossRef]
- Blessing, R.H. An empirical correction for absorption anisotropy? Acta Cryst. Sect. A 1995, 51, 33–38. [Google Scholar] [CrossRef]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A: Found. Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Farrugia, L.J. ORTEP-3 for windows - a version of ORTEP-III with a graphical user interface (GUI). J. Appl. Crystallogr. 1997, 30, 565–565. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 3a–f, 6a–f, and 8 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vellas, S.K.; Lewis, J.E.M.; Shankar, M.; Sagatova, A.; Tyndall, J.D.A.; Monk, B.C.; Fitchett, C.M.; Hanton, L.R.; Crowley, J.D. [Fe2L3]4+ Cylinders Derived from Bis(bidentate) 2-Pyridyl-1,2,3-triazole “Click” Ligands: Synthesis, Structures and Exploration of Biological Activity. Molecules 2013, 18, 6383-6407. https://doi.org/10.3390/molecules18066383
Vellas SK, Lewis JEM, Shankar M, Sagatova A, Tyndall JDA, Monk BC, Fitchett CM, Hanton LR, Crowley JD. [Fe2L3]4+ Cylinders Derived from Bis(bidentate) 2-Pyridyl-1,2,3-triazole “Click” Ligands: Synthesis, Structures and Exploration of Biological Activity. Molecules. 2013; 18(6):6383-6407. https://doi.org/10.3390/molecules18066383
Chicago/Turabian StyleVellas, Sreedhar K., James E. M. Lewis, Madhu Shankar, Alia Sagatova, Joel D. A. Tyndall, Brian C. Monk, Christopher M. Fitchett, Lyall R. Hanton, and James D. Crowley. 2013. "[Fe2L3]4+ Cylinders Derived from Bis(bidentate) 2-Pyridyl-1,2,3-triazole “Click” Ligands: Synthesis, Structures and Exploration of Biological Activity" Molecules 18, no. 6: 6383-6407. https://doi.org/10.3390/molecules18066383