Water Complexes of Cytochrome P450: Insights from Energy Decomposition Analysis

Abstract
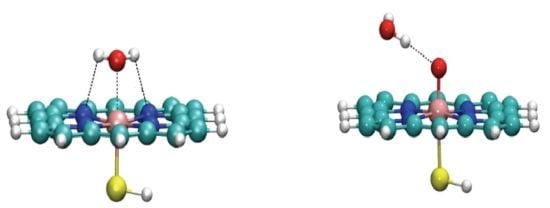
:
1. Introduction

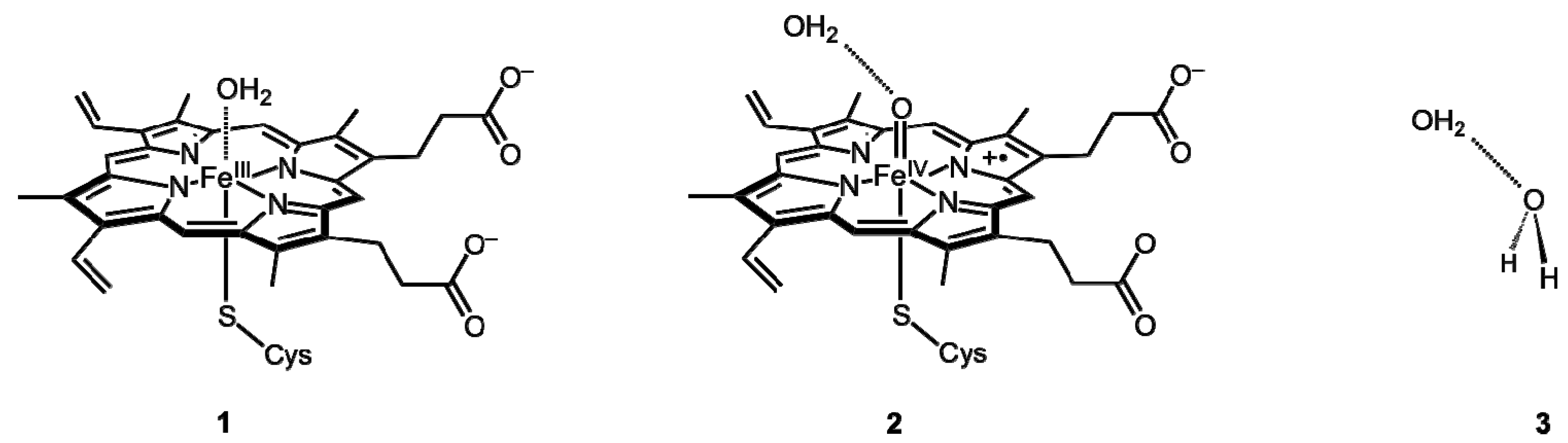
2. Results and Discussion
2.1. LMOEDA of Complexes 1–3


{kind=link}
{kind=link}
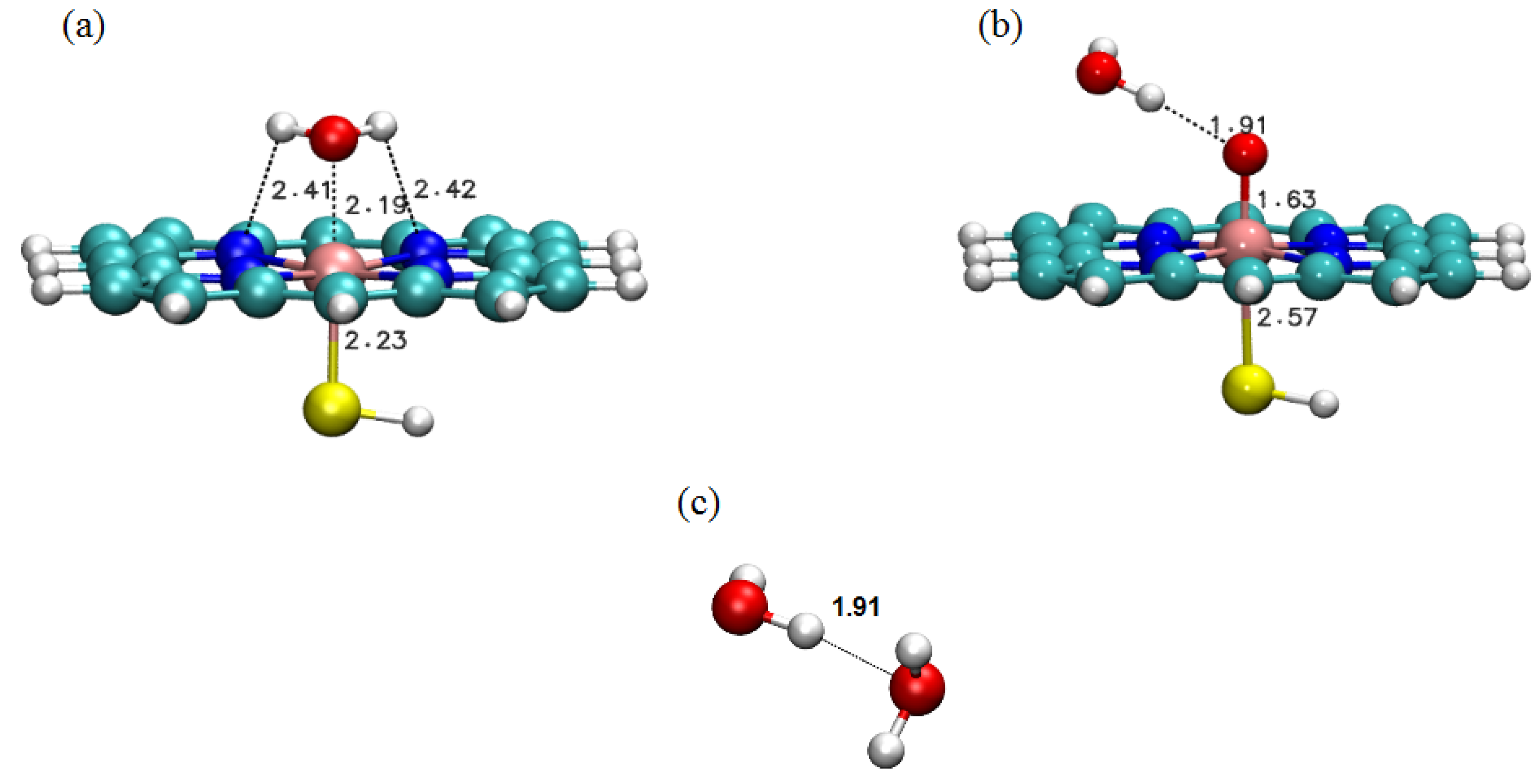{kind=link}
| 1 | 2 | 3 | |
|---|---|---|---|
| Electrostatic | −33.30 | −8.34 | −8.97 |
| Exchange | −22.30 | −2.67 | −3.01 |
| Repulsion | +72.40 | +14.12 | +12.39 |
| Polarization | −21.96 | −7.99 | −5.53 |
| Dispersion | −8.91 | −3.54 | −2.58 |
| Total | −14.07 | −8.41 | −7.70 |
2.2. ADF-EDA of Complexes 1 and 3
| 1 | 3 | |
|---|---|---|
| Electrostatic | −40.28 | −9.14 |
| Pauli repulsion | +50.86 | +8.59 |
| Orbital interaction | −24.96 | −4.21 |
| Total | −14.38 | −4.75 |
2.3. Spin and Charge Distributions
| Group | Charge | Spin | ||
|---|---|---|---|---|
| In fragments | In complex | In fragments | In complex | |
| Ha | +0.39 | +0.45 | 0.00 | 0.00 |
| O | −0.77 | −0.70 | 0.00 | 0.00 |
| Hb | +0.39 | +0.44 | 0.00 | 0.00 |
| Fe | +0.50 | +0.40 | +1.25 | +1.04 |
| Por | −0.48 | −0.59 | −0.14 | −0.07 |
| SH | −0.02 | +0.01 | −0.11 | +0.03 |
| Group | Charge | Spin population | ||
|---|---|---|---|---|
| In fragments | In complex | In fragments | In complex | |
| Ha | +0.38 | +0.45 | 0.00 | 0.00 |
| O | −0.77 | −0.83 | 0.00 | +0.01 |
| Hb | +0.39 | +0.38 | 0.00 | 0.00 |
| Fe | +0.70 | +0.70 | +1.18 | +1.28 |
| Por | −0.23 | −0.17 | −0.51 | −0.55 |
| Oxo | −0.40 | −0.48 | +0.90 | +0.81 |
| SH | −0.06 | −0.06 | −0.54 | −0.55 |
| Group | Charge | |
|---|---|---|
| O | −0.77 | −0.75 |
| Ha1 | +0.39 | +0.41 |
| Ha2 | +0.39 | +0.41 |
| O | −0.77 | −0.84 |
| Hb1 | +0.39 | +0.40 |
| Hb2 | +0.39 | +0.37 |
3. Computational Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef]
- Omura, T.; Sato, R. A new cytochrome in liver microsomes. J. Biol. Chem. 1962, 237, PC1375–PC1376. [Google Scholar]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2278. [Google Scholar] [CrossRef]
- Sligar, S.G.; Makris, T.M.; Denisov, I.G. Thirty years of microbial P450 monooxygenase research: peroxo-heme intermediates—The central bus station in heme oxygenase catalysis. Biochem. Biophys. Res. Commun. 2005, 338, 346–354. [Google Scholar] [CrossRef]
- Groves, J.T. High-valent iron in chemical and biological oxidations. J. Inorg. Biochem. 2006, 100, 434–447. [Google Scholar] [CrossRef]
- Schlichting, I.; Berendzen, J.; Chu, K.; Stock, A.M.; Maves, S.A.; Benson, D.E.; Sweet, R.M.; Ringe, D.; Petsko, G.A.; Sligar, S.G. The catalytic pathway of cytochrome P450cam at atomic resolution. Science 2000, 287, 1615–1622. [Google Scholar] [CrossRef]
- Schöneboom, J.C.; Cohen, S.; Lin, H.; Shaik, S.; Thiel, W. Quantum mechanical/molecular mechanical investigation of the mechanism Of C−H hydroxylation of camphor by cytochrome P450cam: Theory supports a two-state rebound mechanism. J. Am. Chem. Soc. 2004, 126, 4017–4034. [Google Scholar] [CrossRef]
- Guallar, V.; Baik, M.-H.; Lippard, S.J.; Friesner, R.A. Peripheral heme substituents control the hydrogen-atom abstraction chemistry in cytochromes P450. Proc. Natl. Acad. Sci. USA 2003, 100, 6998–7002. [Google Scholar] [CrossRef]
- Guallar, V.; Friesner, R.A. Cytochrome P450CAM enzymatic catalysis cycle: A quantum mechanics/molecular mechanics study. J. Am. Chem. Soc. 2004, 126, 8501–8508. [Google Scholar] [CrossRef]
- Altun, A.; Guallar, V.; Friesner, R.A.; Shaik, S.; Thiel, W. The effect of heme environment on the hydrogen abstraction reaction of camphor in P450cam catalysis: A QM/MM study. J. Am. Chem. Soc. 2006, 128, 3924–3925. [Google Scholar]
- Kumar, D.; Altun, A.; Shaik, S.; Thiel, W. Water as biocatalyst in cytochrome P450. Faraday Discuss 2011, 148, 373–383. [Google Scholar] [CrossRef]
- Hirao, H. Energy Decomposition Analysis of the protein environmental effect: The case of cytochrome P450cam compound I. Chem. Lett. 2011, 40, 1179–1181. [Google Scholar] [CrossRef]
- Von Hopffgarten, M.; Frenking, G. Energy decomposition analysis. WIREs Comput. Mol. Sci. 2012, 2, 43–62. [Google Scholar] [CrossRef]
- Su, P.; Li, H. Energy decomposition analysis of covalent bonds and intermolecular interactions. J.Chem. Phys. 2009, 131, 014102. [Google Scholar]
- Misquitta, A.J.; Podeszwa, R.; Jeziorski, B.; Szalewicz, K. Intermolecular potentials based on symmetry-adapted perturbation theory with dispersion energies from time-dependent density-functional calculations. J. Chem. Phys. 2005, 123, 214103. [Google Scholar] [CrossRef]
- Glendening, E.D.; Streitwieser, A. natural energy decomposition analysis: an energy partitioning procedure for molecular interactions with application to weak hydrogen bonding, strong ionic, and moderate donor-acceptor interactions. J. Chem. Phys. 1994, 100, 2900–2909. [Google Scholar] [CrossRef]
- Bagus, P.S.; Hermann, K.; Charles, W.; Bauschlicher, J. A new analysis of charge transfer and polarization for ligand-metal bonding: Model studies of Al4CO and Al4NH3. J. Chem. Phys. 1984, 80, 4378–4386. [Google Scholar] [CrossRef]
- Kitaura, K.; Morokuma, K. A new energy decomposition scheme for molecular interactions within the hartree-fock approximation. Int. J. Quantum Chem. 1976, 10, 325–340. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. A theoretical study of the ethylene-metal bond in complexes between copper(1+), silver(1+), gold(1+), platinum(0) or platinum(2+) and ethylene, based on the Hartree-Fock-Slater transition-state method. Inorg. Chem. 1979, 18, 1558–1565. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. Carbon Monoxide, Carbon Monosulfide, molecular nitrogen, phosphorus trifluoride, and methyl isocyanide as σ donors and π acceptors. A theoretical study by the Hartree-Fock-Slater transition-state method. Inorg. Chem. 1979, 18, 1755–1759. [Google Scholar] [CrossRef]
- Stevens, W.J.; Fink, W.H. Frozen fragment reduced variational space analysis of hydrogen bonding interactions. Application to the water dimer. Chem. Phys. Lett. 1987, 1, 15–22. [Google Scholar] [CrossRef]
- Mo, Y.R.; Gao, J.L.; Peyerimhoff, S.D. Energy decoposition analysis of intermolecular interactions using a block-localized wavefunction approach. J. Chem. Phys. 2000, 112, 5530–5538. [Google Scholar] [CrossRef]
- Hirao, H. Reactive Bond Orbitals: A localized resonance-structure approach to charge transfer. Chem. Phys. Lett. 2007, 443, 141–146. [Google Scholar] [CrossRef]
- Khaliullin, R.Z.; Cobar, E.A.; Lochan, R.C.; Bell, A.T.; Head-Gordon, M. Unravelling the origin of intermolecular interactions using absolutely localized molecular orbitals. J. Phys. Chem. A 2007, 111, 8753–8765. [Google Scholar] [CrossRef]
- Hirao, H. The effects of protein environment and dispersion on the formation of ferric-superoxide species in myo-inositol oxygenase (MIOX): A combined ONIOM(DFT:MM) and energy decomposition analysis. J. Phys. Chem. B 2011, 115, 11278–11285. [Google Scholar] [CrossRef]
- Schöneboom, J.; Thiel, W. The resting state of P450cam: A QM/MM Study. J. Phys. Chem. B 2004, 108, 7468–7478. [Google Scholar] [CrossRef]
- Ortiz de Montellano, P.R. Cytochrome P450: Structure, Mechanism, and Biochemistry., 3rd ed.; Ortiz de Montellano, P.R., Ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2005. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Gordon, M.S.; Schmidt, M.W. Theory and Applications of Computational Chemistry; Dykstra, C.E., Frenking, K.S., Scuseria, G.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Fonseca Guerra, C.; Snijders, J.G.; te Velde, G.; Baerends, E.J. Towards an Order-N DFT method. Theor. Chem. Acc. 1998, 99, 391–403. [Google Scholar]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Thellamurege, N.; Hirao, H. Water Complexes of Cytochrome P450: Insights from Energy Decomposition Analysis. Molecules 2013, 18, 6782-6791. https://doi.org/10.3390/molecules18066782
Thellamurege N, Hirao H. Water Complexes of Cytochrome P450: Insights from Energy Decomposition Analysis. Molecules. 2013; 18(6):6782-6791. https://doi.org/10.3390/molecules18066782
Chicago/Turabian StyleThellamurege, Nandun, and Hajime Hirao. 2013. "Water Complexes of Cytochrome P450: Insights from Energy Decomposition Analysis" Molecules 18, no. 6: 6782-6791. https://doi.org/10.3390/molecules18066782