Application of Reductive 13C-Methylation of Lysines to Enhance the Sensitivity of Conventional NMR Methods

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
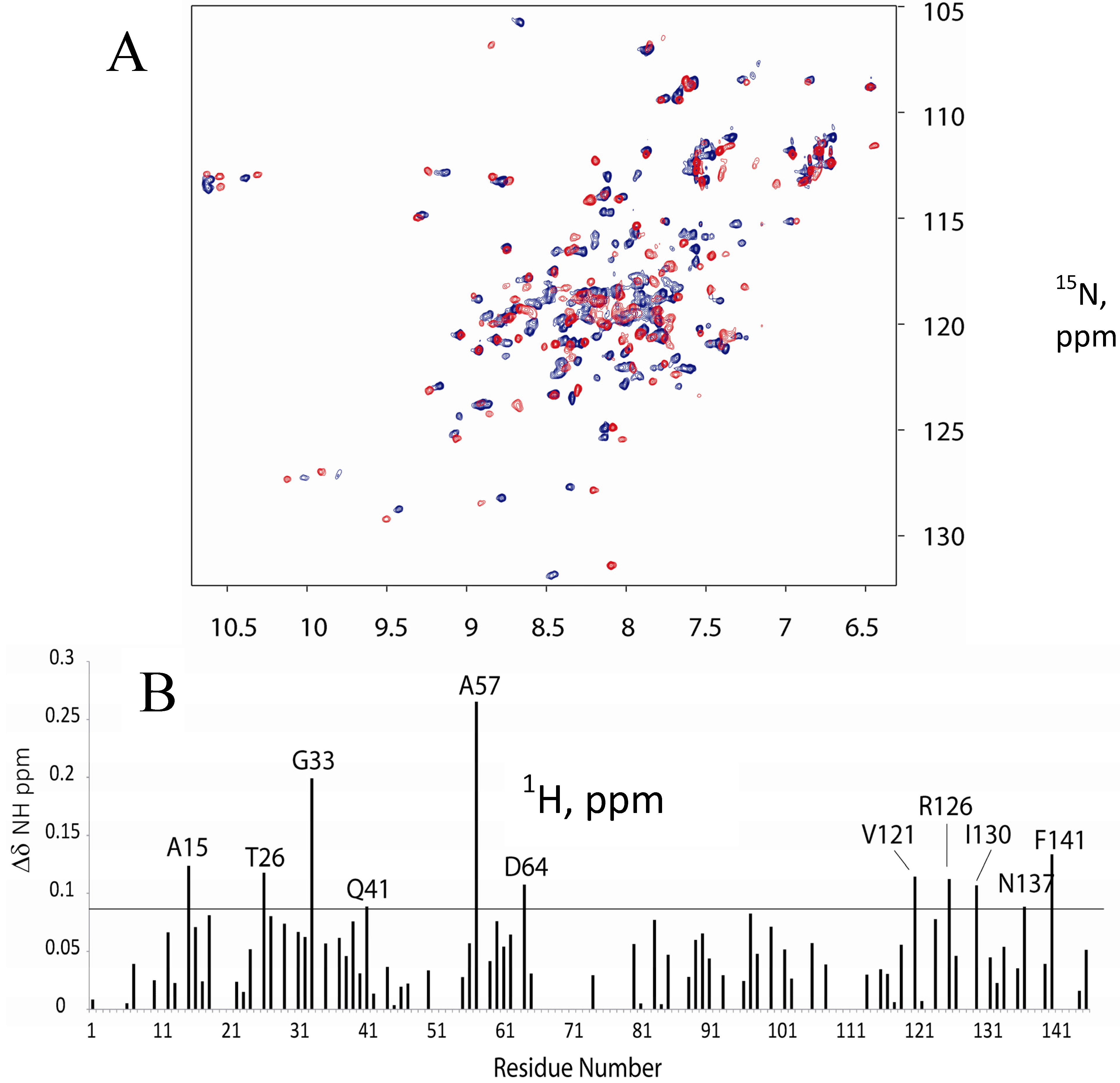
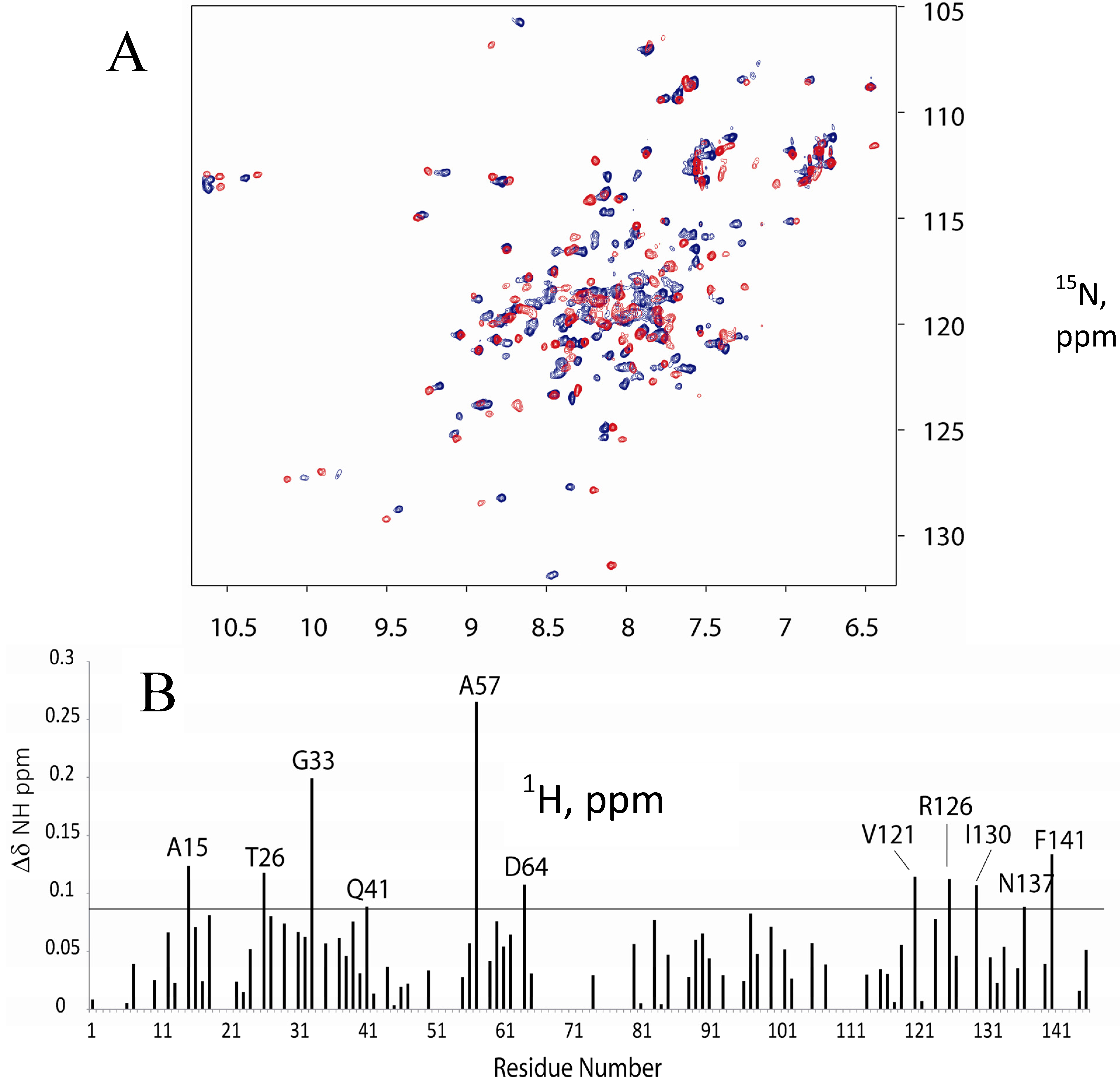
2.1. 15N-HSQC Titration of 15N-CaM with FM-HVR does not Reveal a Continuous Binding Interface
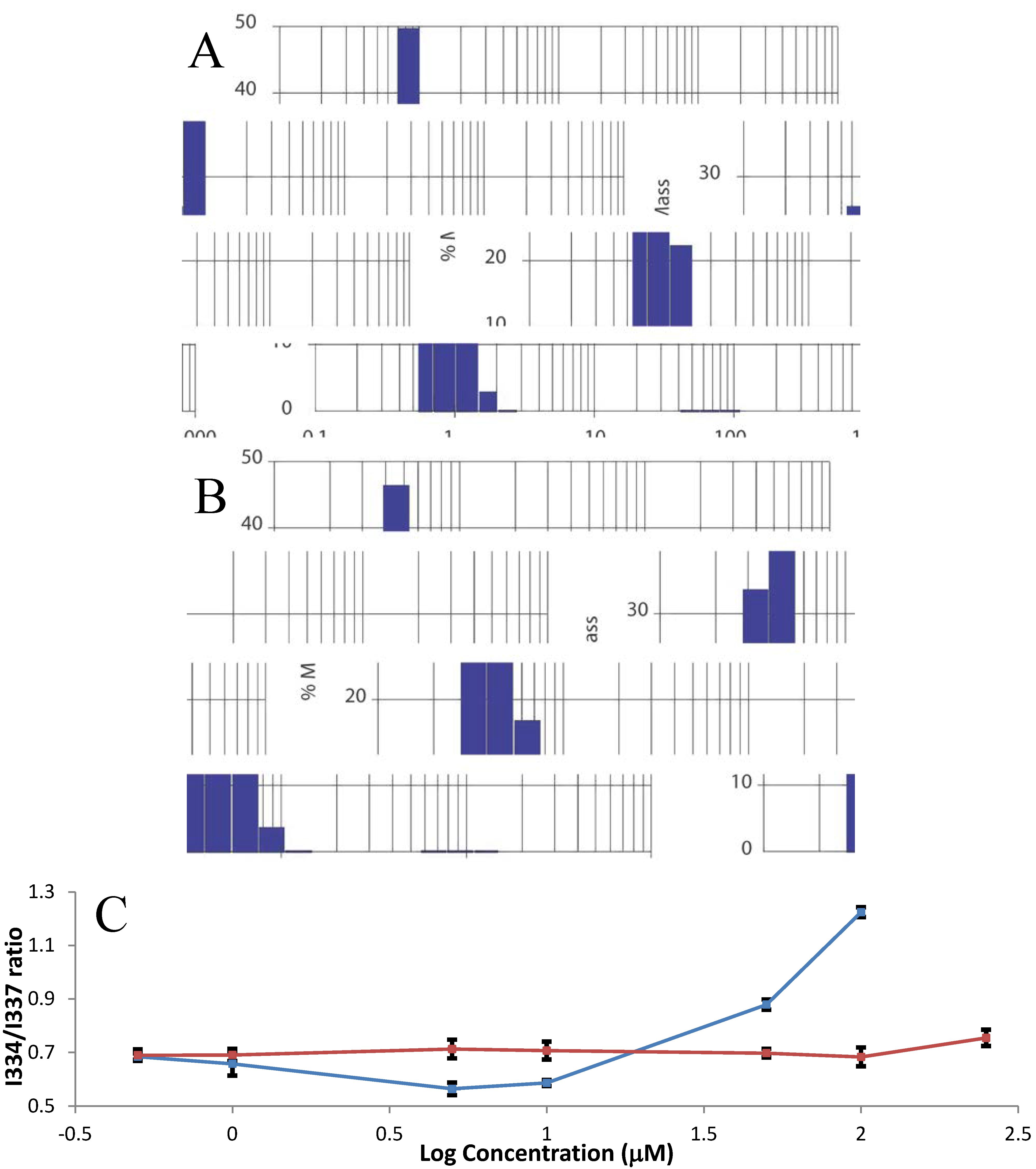
2.2. Farnesylation and Methylation of HVR Cause Aggregation
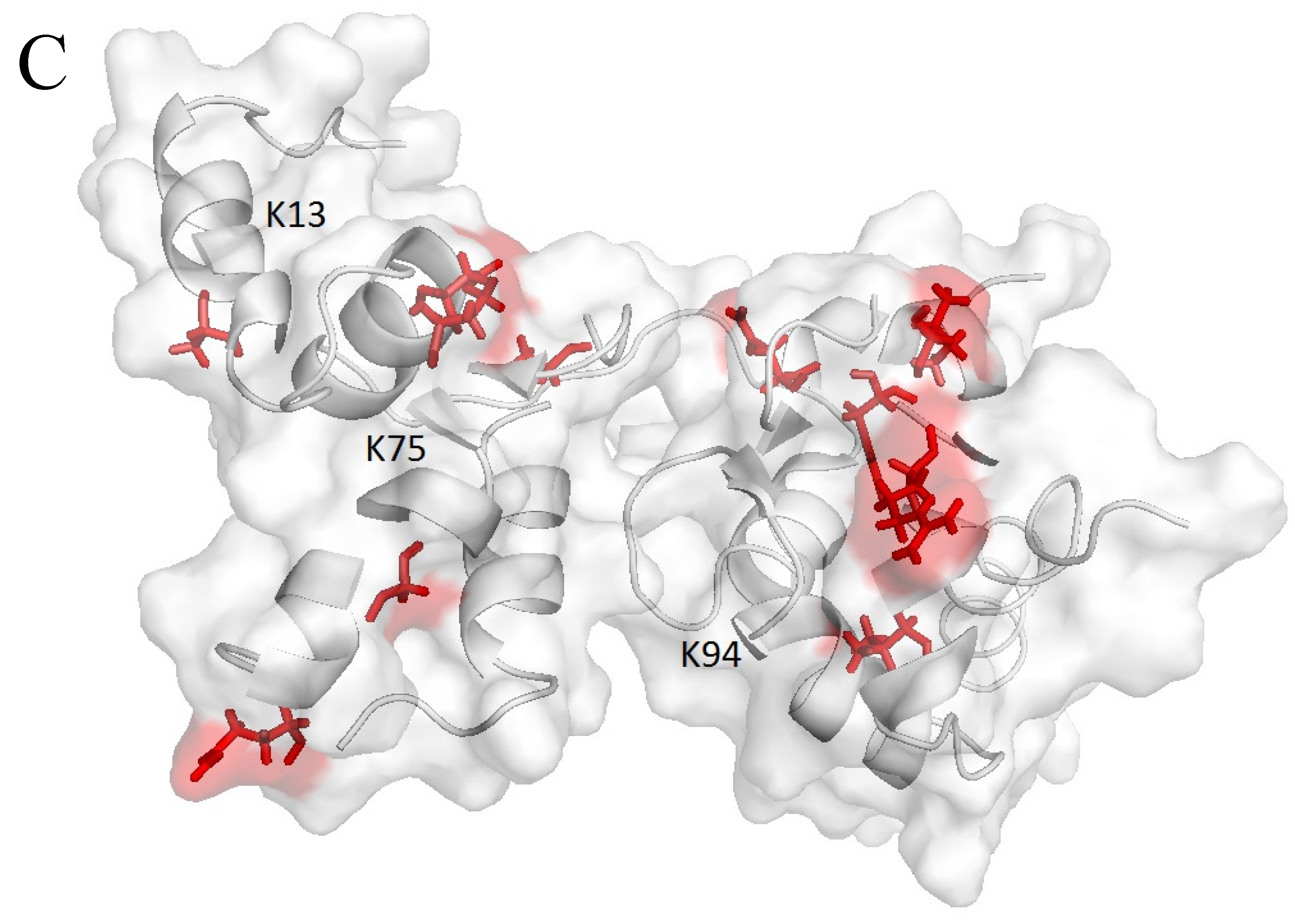
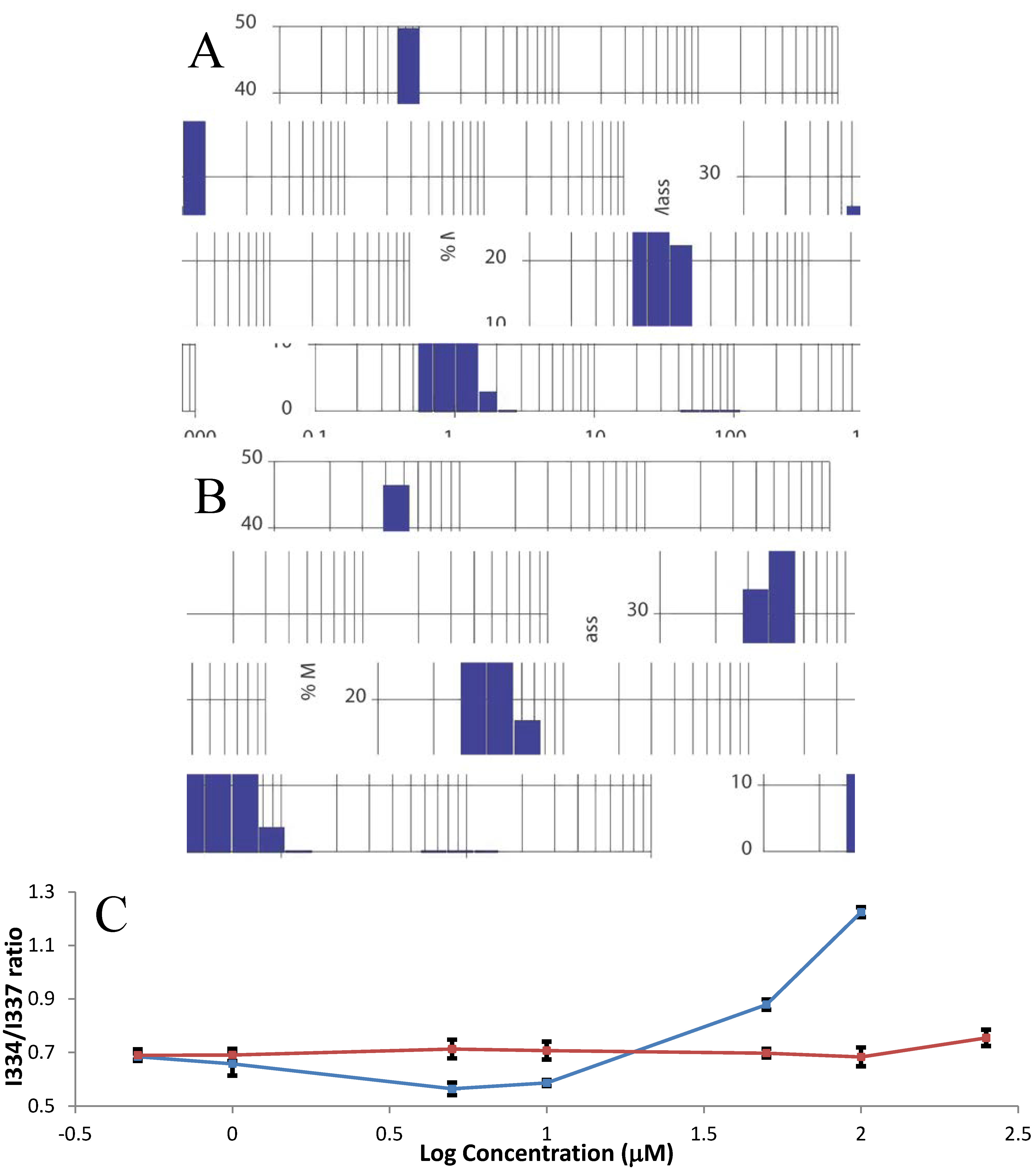
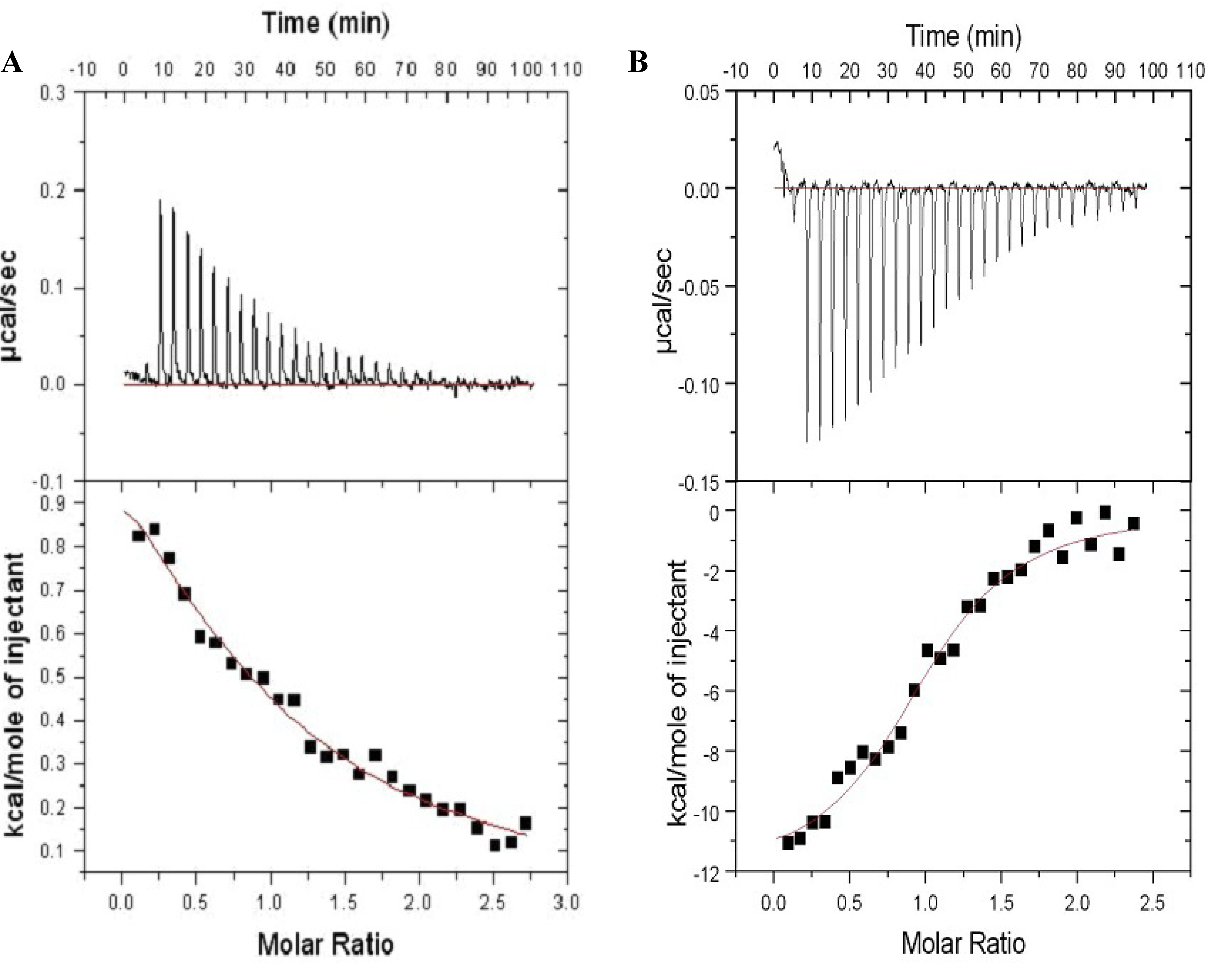
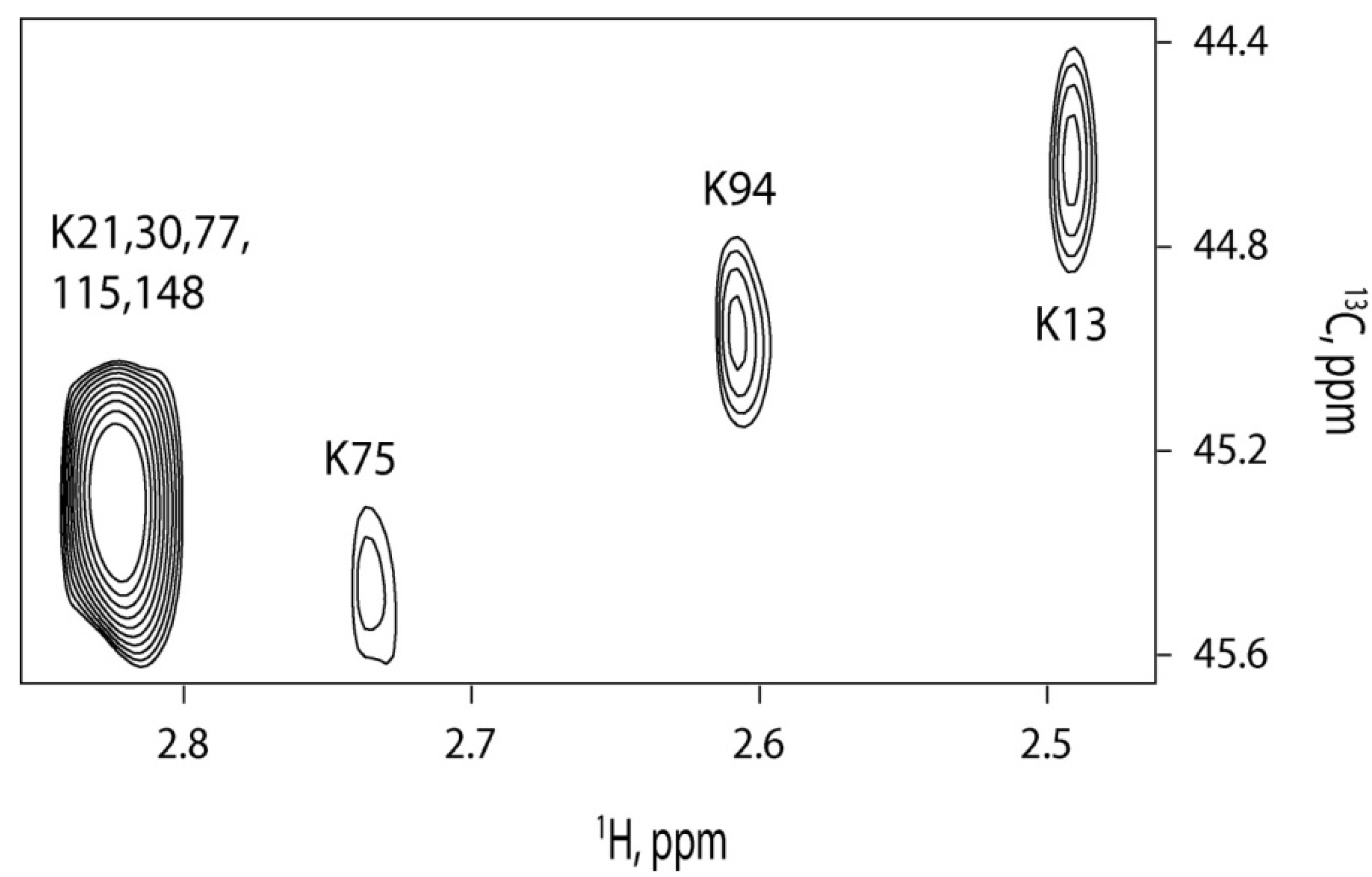
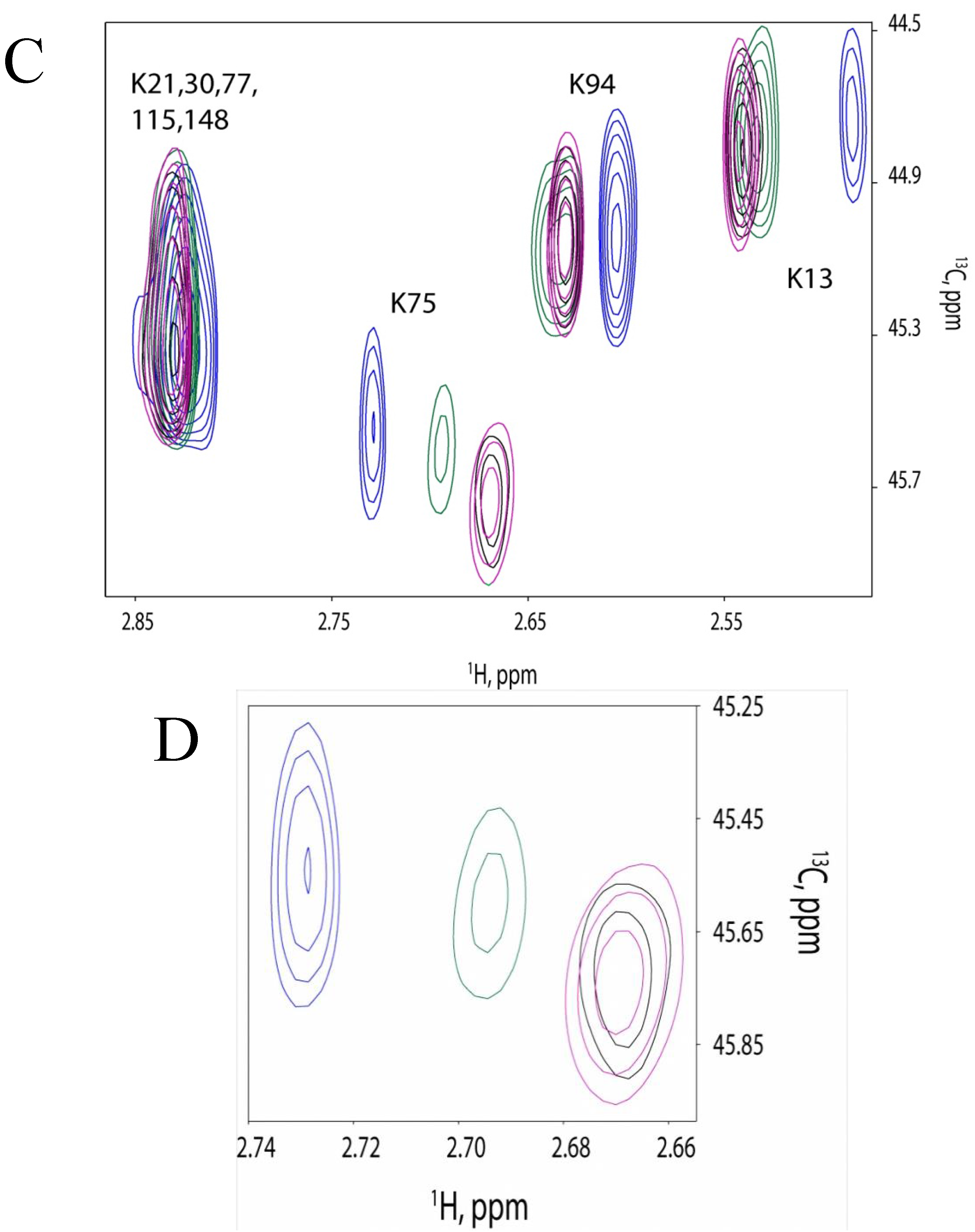
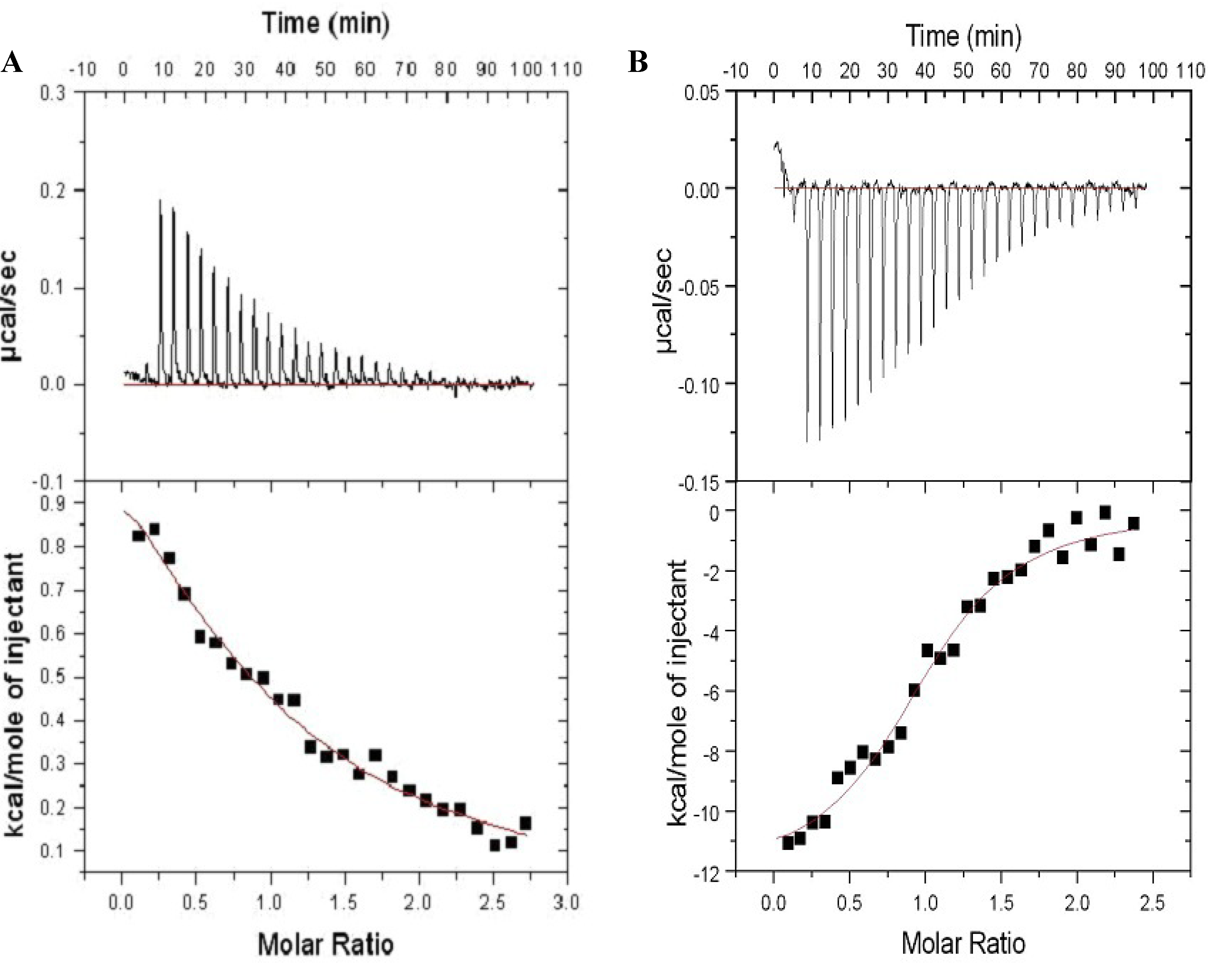
2.3. Farnesylation Increases Affinity of the Hypervariable Region for CaM
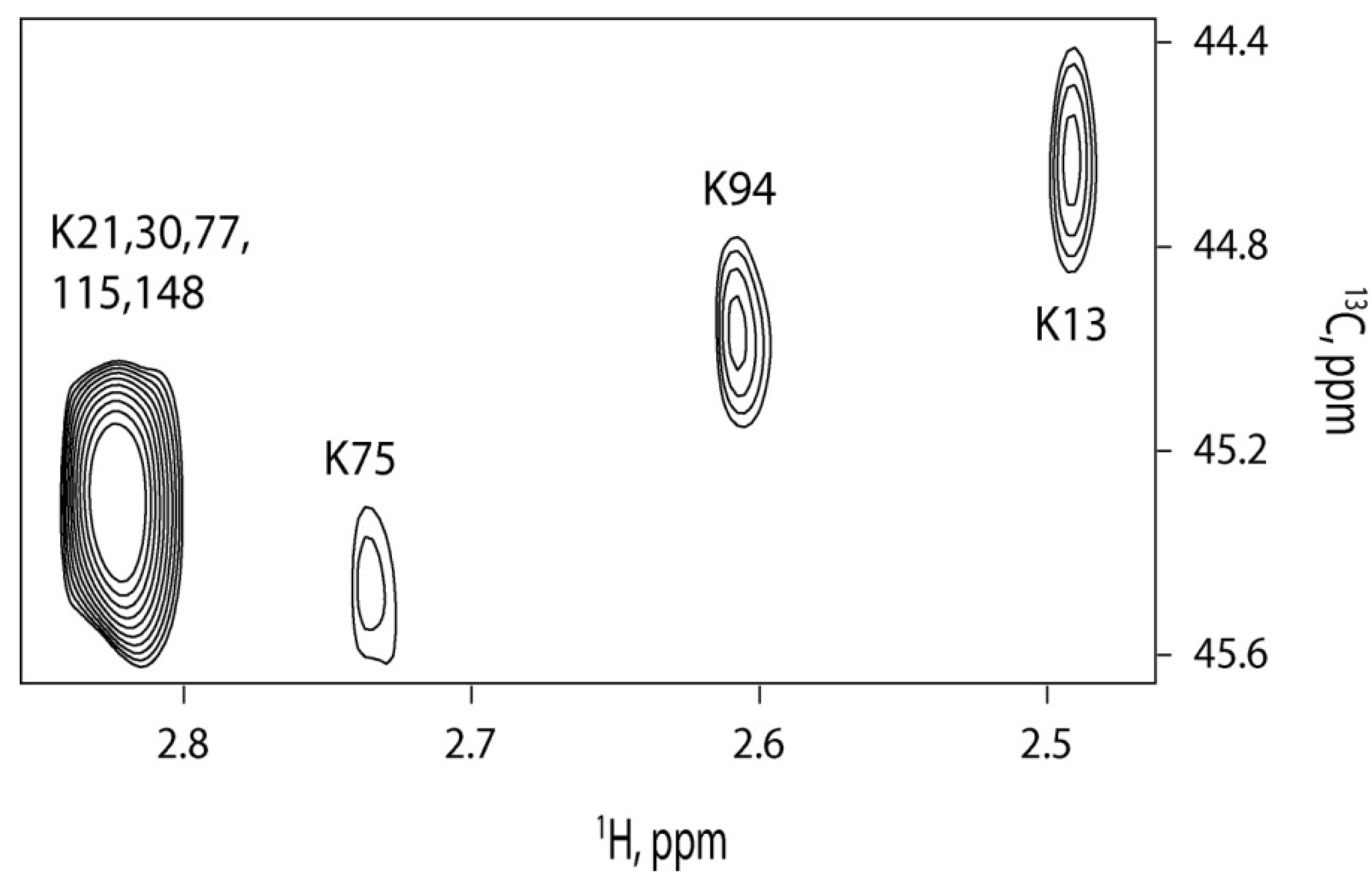
2.4. 13C-HSQC of Reductively Methylated CaM can be Performed at Nanomolar Protein Concentration
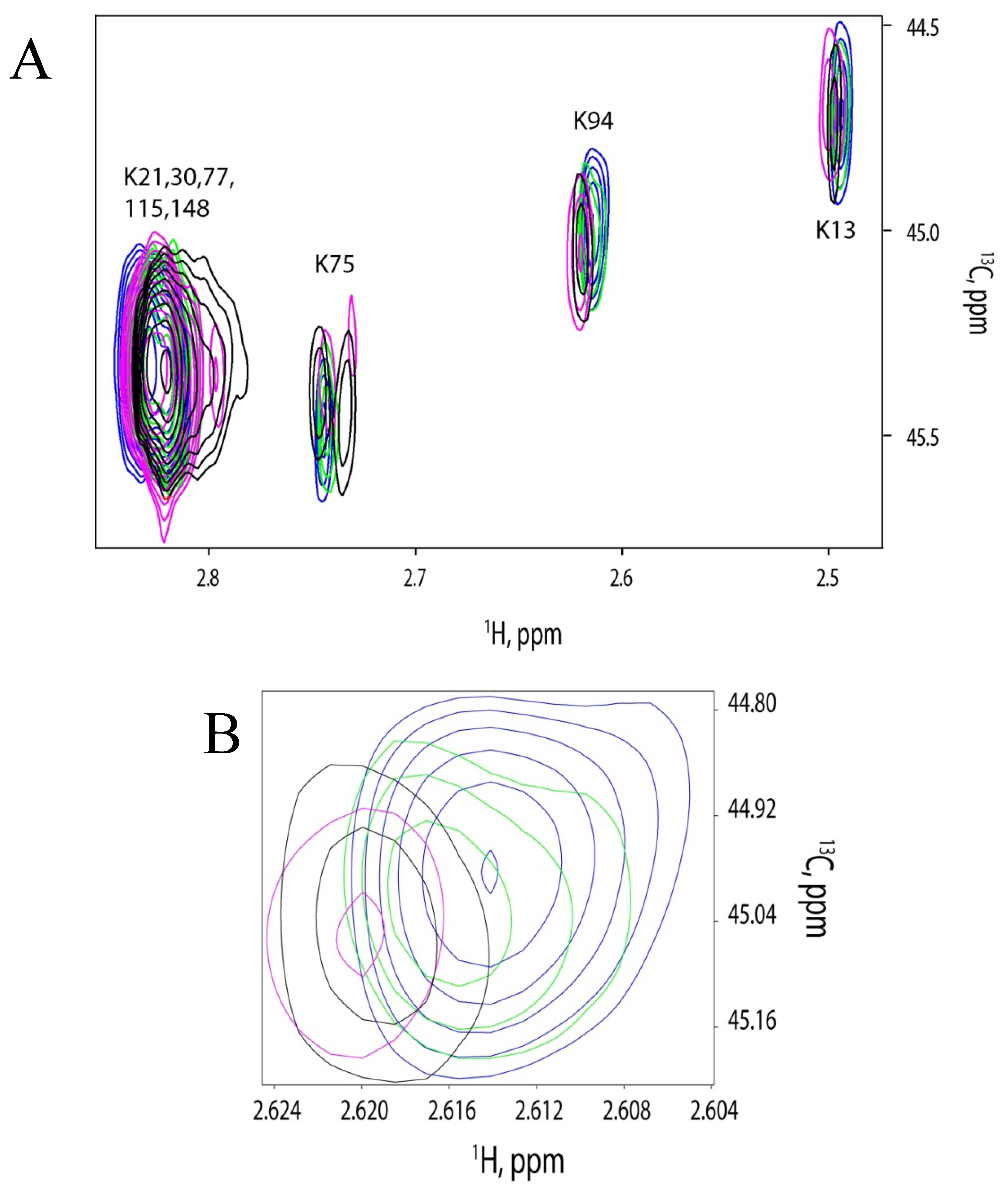

2.5. Discussion
3. Experimental
3.1. Isothermal Titration Calorimetry (ITC)
3.2. Particle Size Analysis
3.3. Fluorescence Experiments
3.4. NMR Experiments

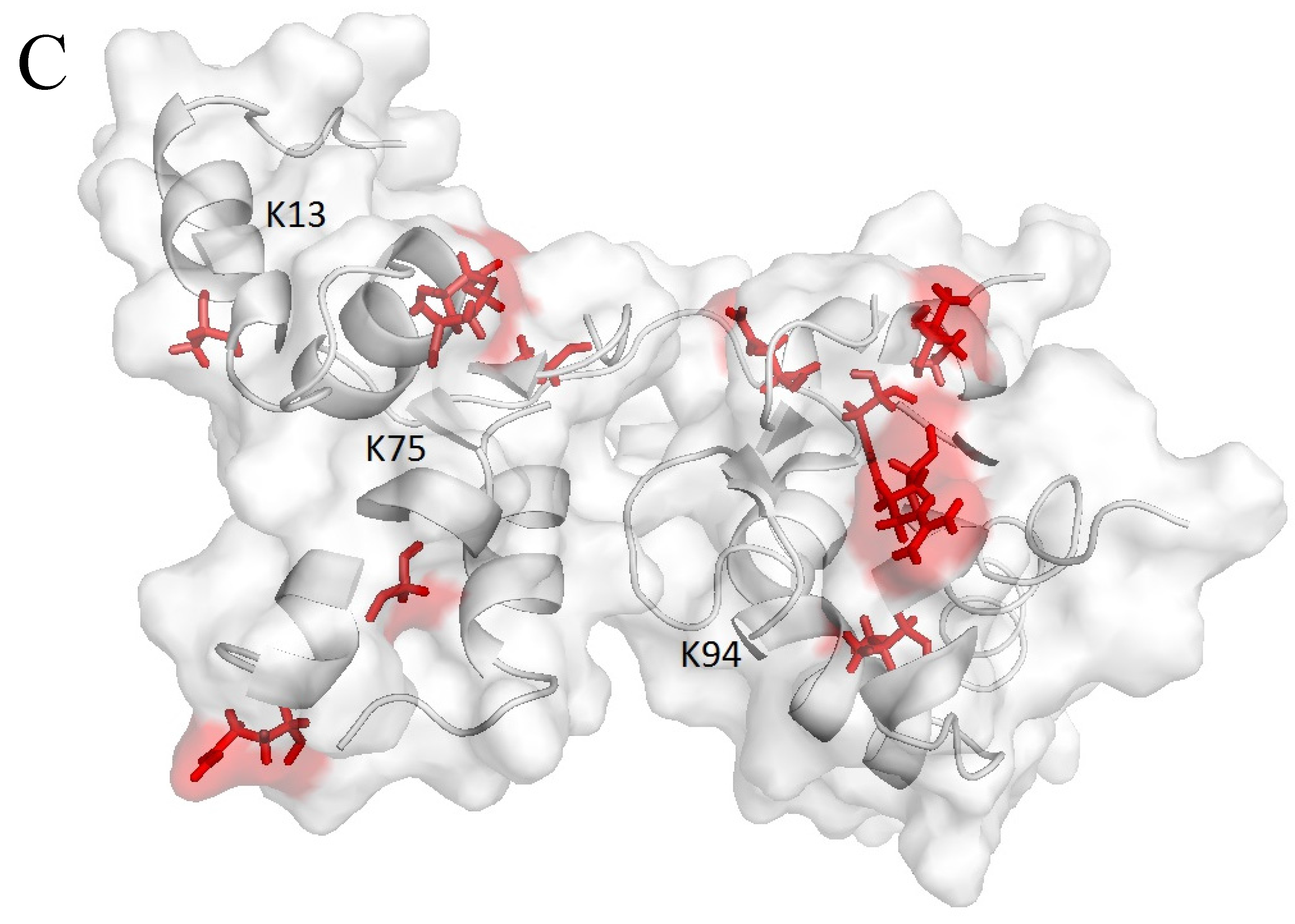
3.5. Reductive Methylation of CaM
3.6. Protein Purification
3.7. Modification of the Hypervariable Region
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Friday, B.B.; Adjei, A.A. K-ras as a target for cancer therapy. Biochim. Biophys. Acta 2005, 1756, 127–144. [Google Scholar]
- Dergham, S.T.; Dugan, M.C.; Kucway, R.; Du, W.; Kamarauskiene, D.S.; Vaitkevicius, V.K.; Crissman, J.D.; Sarkar, F.H. Prevalence and clinical significance of combined K-ras mutation and p53 aberration in pancreatic adenocarcinoma. Int. J. Pancreatol. 1997, 21, 127–143. [Google Scholar]
- Shibata, D.; Almoguera, C.; Forrester, K.; Dunitz, J.; Martin, S.E.; Cosgrove, M.M.; Perucho, M.; Arnheim, N. Detection of c-K-ras mutations in fine needle aspirates from human pancreatic adenocarcinomas. Cancer Res. 1990, 50, 1279–1283. [Google Scholar]
- Tabernero, J.; Salazar, R.; Casado, E.; Martinelli, E.; Gomez, P.; Baselga, J. Targeted therapy in advanced colon cancer: the role of new therapies. Ann. Oncol. 2004, 15 (Suppl 4), 55–62. [Google Scholar]
- Der, C.J.; Krontiris, T.G.; Cooper, G.M. Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes of Harvey and Kirsten sarcoma viruses. Proc. Natl. Acad. Sci. USA 1982, 79, 3637–3640. [Google Scholar] [CrossRef]
- Zhang, M.; Vogel, H.J. Determination of the side chain pKa values of the lysine residues in calmodulin. J. Biol. Chem. 1993, 268, 22420–22428. [Google Scholar]
- Razzaque, M.A.; Komoike, Y.; Nishizawa, T.; Inai, K.; Furutani, M.; Higashinakagawa, T.; Matsuoka, R. Characterization of a novel KRAS mutation identified in Noonan syndrome. Am. J. Med. Genet. 2012, 158A, 524–532. [Google Scholar] [CrossRef]
- Gelb, B.D.; Tartaglia, M. Noonan syndrome and related disorders: dysregulated RAS-mitogen activated protein kinase signal transduction. Hum. Mol. Gen. 2006, 15, R220–R226. [Google Scholar] [CrossRef]
- Plowman, S.J.; Berry, R.L.; Bader, S.A.; Luo, F.; Arends, M.J.; Harrison, D.J.; Hooper, M.L.; Patek, C.E. K-ras 4A and 4B are co-expressed widely in human tissues, and their ratio is altered in sporadic colorectal cancer. J. Exp. Clin. Cancer Res. 2006, 25, 259–267. [Google Scholar]
- Esteban, L.M.; Vicario-Abejon, C.; Fernandez-Salguero, P.; Fernandez-Medarde, A.; Swaminathan, N.; Yienger, K.; Lopez, E.; Malumbres, M.; McKay, R.; Ward, J.M.; et al. Targeted genomic disruption of H-ras and N-ras, individually or in combination, reveals the dispensability of both loci for mouse growth and development. Mol. Cell. Biol. 2001, 21, 1444–1452. [Google Scholar] [CrossRef]
- Johnson, L.; Greenbaum, D.; Cichowski, K.; Mercer, K.; Murphy, E.; Schmitt, E.; Bronson, R.T.; Umanoff, H.; Edelmann, W.; Kucherlapati, R.; et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997, 11, 2468–2481. [Google Scholar] [CrossRef]
- Villalonga, P.; Lopez-Alcala, C.; Bosch, M.; Chiloeches, A.; Rocamora, N.; Gil, J.; Marais, R.; Marshall, C.J.; Bachs, O.; Agell, N. Calmodulin binds to K-Ras, but not to H- or N-Ras, and modulates its downstream signaling. Mol. Cell. Biol. 2001, 21, 7345–7354. [Google Scholar] [CrossRef]
- Abraham, S.J.; Nolet, R.P.; Calvert, R.J.; Anderson, L.M.; Gaponenko, V. The hypervariable region of K-Ras4B is responsible for its specific interactions with calmodulin. Biochemistry 2009, 48, 7575–7583. [Google Scholar] [CrossRef]
- Lopez-Alcala, C.; Alvarez-Moya, B.; Villalonga, P.; Calvo, M.; Bachs, O.; Agell, N. Identification of essential interacting elements in K-Ras/calmodulin binding and its role in K-Ras localization. J. Biol. Chem. 2008, 283, 10621–10631. [Google Scholar]
- Gomes, A.V.; Barnes, J.A.; Vogel, H.J. Spectroscopic characterization of the interaction between calmodulin-dependent protein kinase I and calmodulin. Arch. Biochem. Biophys. 2000, 379, 28–36. [Google Scholar] [CrossRef]
- Chin, D.; Sloan, D.J.; Quiocho, F.A.; Means, A.R. Functional consequences of truncating amino acid side chains located at a calmodulin-peptide interface. J. Biol. Chem. 1997, 272, 5510–5513. [Google Scholar] [CrossRef]
- Kranz, J.K.; Lee, E.K.; Nairn, A.C.; Wand, A.J. A direct test of the reductionist approach to structural studies of calmodulin activity: relevance of peptide models of target proteins. J. Biol. Chem. 2002, 277, 16351–16354. [Google Scholar]
- Liao, J.; Planchon, S.M.; Wolfman, J.C.; Wolfman, A. Growth factor-dependent AKT activation and cell migration requires the function of c-K(B)-Ras versus other cellular ras isoforms. J. Biol. Chem. 2006, 281, 29730–29738. [Google Scholar] [CrossRef]
- Bosch, M.; Gil, J.; Bachs, O.; Agell, N. Calmodulin inhibitor W13 induces sustained activation of ERK2 and expression of p21(cip1). J. Biol. Chem. 1998, 273, 22145–22150. [Google Scholar] [CrossRef]
- Villalonga, P.; Lopez-Alcala, C.; Chiloeches, A.; Gil, J.; Marais, R.; Bachs, O.; Agell, N. Calmodulin prevents activation of Ras by PKC in 3T3 fibroblasts. J. Biol. Chem. 2002, 277, 37929–37935. [Google Scholar]
- Fivaz, M.; Meyer, T. Reversible intracellular translocation of KRas but not HRas in hippocampal neurons regulated by Ca2+/calmodulin. J. Cell. Biol. 2005, 170, 429–441. [Google Scholar] [CrossRef]
- Abraham, S.J.; Hoheisel, S.; Gaponenko, V. Detection of protein-ligand interactions by NMR using reductive methylation of lysine residues. J. Biomol. NMR 2008, 42, 143–148. [Google Scholar] [CrossRef]
- Abraham, S.J.; Kobayashi, T.; Solaro, R.J.; Gaponenko, V. Differences in lysine pKa values may be used to improve NMR signal dispersion in reductively methylated proteins. J. Biomol. NMR 2009, 43, 239–246. [Google Scholar] [CrossRef]
- Ismail, S.A.; Chen, Y.X.; Rusinova, A.; Chandra, A.; Bierbaum, M.; Gremer, L.; Triola, G.; Waldmann, H.; Bastiaens, P.I.; Wittinghofer, A. Arl2-GTP and Arl3-GTP regulate a GDI-like transport system for farnesylated cargo. Nat. Chem. Biol. 2011, 7, 942–949. [Google Scholar] [CrossRef]
- Takai, Y.; Sasaki, T.; Matozaki, T. Small GTP-binding proteins. Physiol. Rev. 2001, 81, 153–208. [Google Scholar]
- Kowluru, A. Small G proteins in islet beta-cell function. Endocr. Rev. 2010, 31, 52–78. [Google Scholar] [CrossRef]
- Kowluru, A. Protein prenylation in glucose-induced insulin secretion from the pancreatic islet beta cell: a perspective. J. Cell. Mol. Med. 2008, 12, 164–173. [Google Scholar] [CrossRef]
- Linder, M.E.; Deschenes, R.J. Palmitoylation: policing protein stability and traffic. Nat. Rev. Mol. Cell Biol. 2007, 8, 74–84. [Google Scholar] [CrossRef]
- Wu, L.J.; Xu, L.R.; Liao, J.M.; Chen, J.; Liang, Y. Both the C-terminal polylysine region and the farnesylation of K-RasB are important for its specific interaction with calmodulin. PLoS One 2011, 6, e21929. [Google Scholar]
- Rayment, I. Reductive alkylation of lysine residues to alter crystallization properties of proteins. Methods Enzymol. 1997, 276, 171–179. [Google Scholar] [CrossRef]
- Schanda, P.; Brutscher, B. Very fast two-dimensional NMR spectroscopy for real-time investigationof dynamic events in proteins on the time scale of seconds. J. Am. Chem. Soc. 2005, 127, 8014–8015. [Google Scholar] [CrossRef]
- Ganzhorn, A.J.; Lepage, P.; Pelton, P.D.; Strasser, F.; Vincendon, P.; Rondeau, J.M. The contribution of lysine-36 to catalysis by human myo-inositol monophosphatase. Biochemistry 1996, 35, 10957–10966. [Google Scholar] [CrossRef]
- Gao, G.; Prasad, R.; Lodwig, S.N.; Unkefer, C.J.; Beard, W.A.; Wilson, S.H.; London, R.E. Determination of lysine pK values using [5–13C]lysine: Application to the lyase domain of DNA Pol beta. J. Am. Chem. Soc. 2006, 128, 8104–8105. [Google Scholar]
- Iwahara, J.; Jung, Y.S.; Clore, G.M. Heteronuclear NMR spectroscopy for lysine NH(3) groups in proteins: unique effect of water exchange on (15)N transverse relaxation. J. Am. Chem Soc. 2007, 129, 2971–2980. [Google Scholar] [CrossRef]
- Larda, S.T.; Bokoch, M.P.; Evanics, F.; Prosser, R.S. Lysine methylation strategies for characterizing protein conformations by NMR. J. Biomol. NMR 2012, 54, 199–209. [Google Scholar] [CrossRef]
- Wilhelm, M.; Zhao, C.L.; Wang, Y.C.; Xu, R.L.; Winnik, M.A.; Mura, J.L.; Riess, G.; Croucher, M.D. Polymer Micelle Formation. 3. Poly(Styrene-Ethylene Oxide) Block Copolymer Micelle Formation in Water — a Fluorescence Probe Study. Macromolecules 1991, 24, 1033–1040. [Google Scholar]
- Means, G.E.; Feeney, R.E. Reductive alkylation of amino groups in proteins. Biochemistry 1968, 7, 2192–2201. [Google Scholar] [CrossRef]
- Waddell, W.J. A simple ultraviolet spectrophotometric method for the determination of protein. J. Lab. Clin. Med. 1956, 48, 311–314. [Google Scholar]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chavan, T.S.; Abraham, S.; Gaponenko, V. Application of Reductive 13C-Methylation of Lysines to Enhance the Sensitivity of Conventional NMR Methods. Molecules 2013, 18, 7103-7119. https://doi.org/10.3390/molecules18067103
Chavan TS, Abraham S, Gaponenko V. Application of Reductive 13C-Methylation of Lysines to Enhance the Sensitivity of Conventional NMR Methods. Molecules. 2013; 18(6):7103-7119. https://doi.org/10.3390/molecules18067103
Chicago/Turabian StyleChavan, Tanmay S., Sherwin Abraham, and Vadim Gaponenko. 2013. "Application of Reductive 13C-Methylation of Lysines to Enhance the Sensitivity of Conventional NMR Methods" Molecules 18, no. 6: 7103-7119. https://doi.org/10.3390/molecules18067103