Oligonucleotide Tagging for Copper-Free Click Conjugation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Results and Discussion
2.1. 5′-Labeling and Conjugation of Oligonucleotides
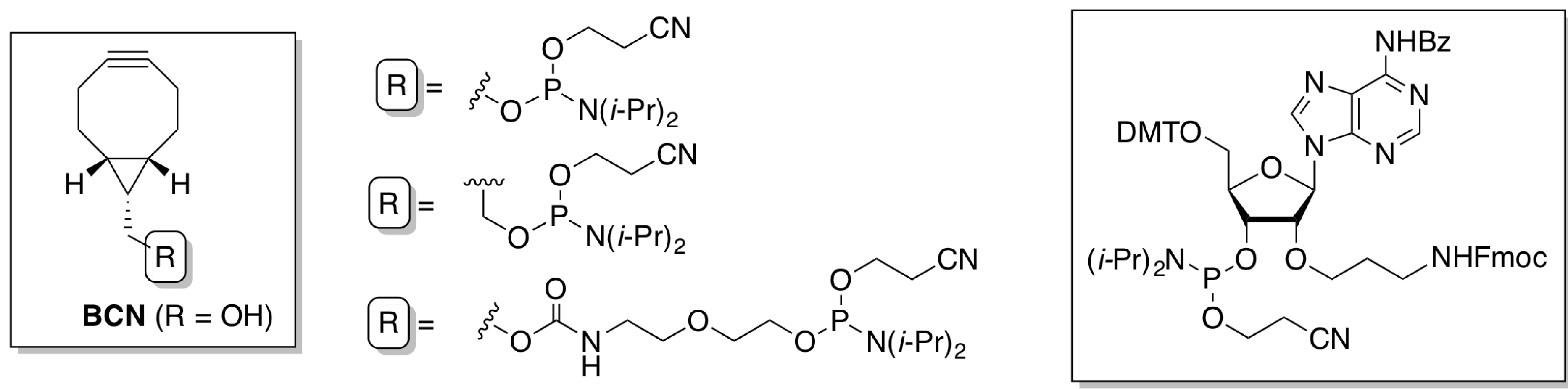
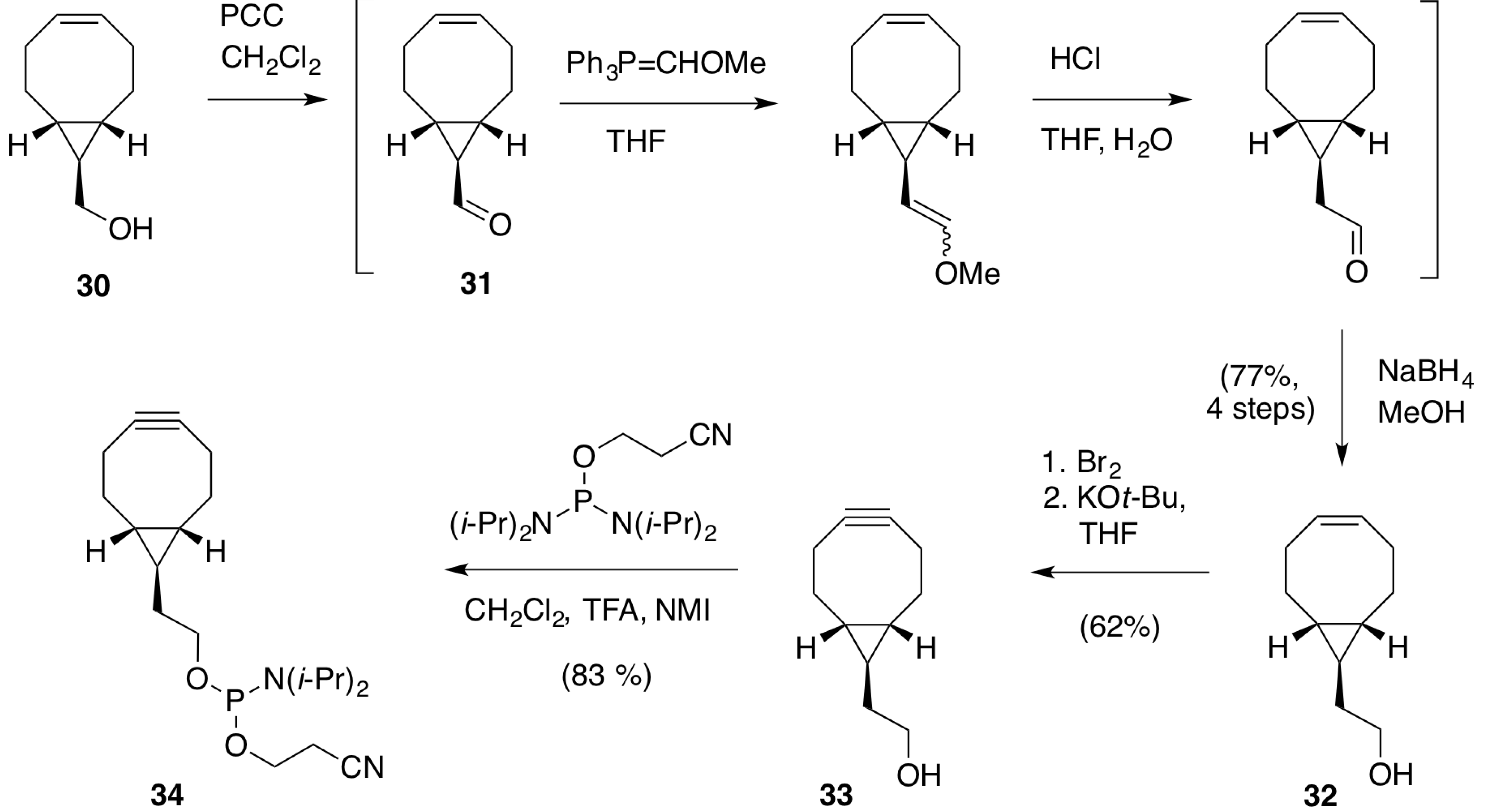
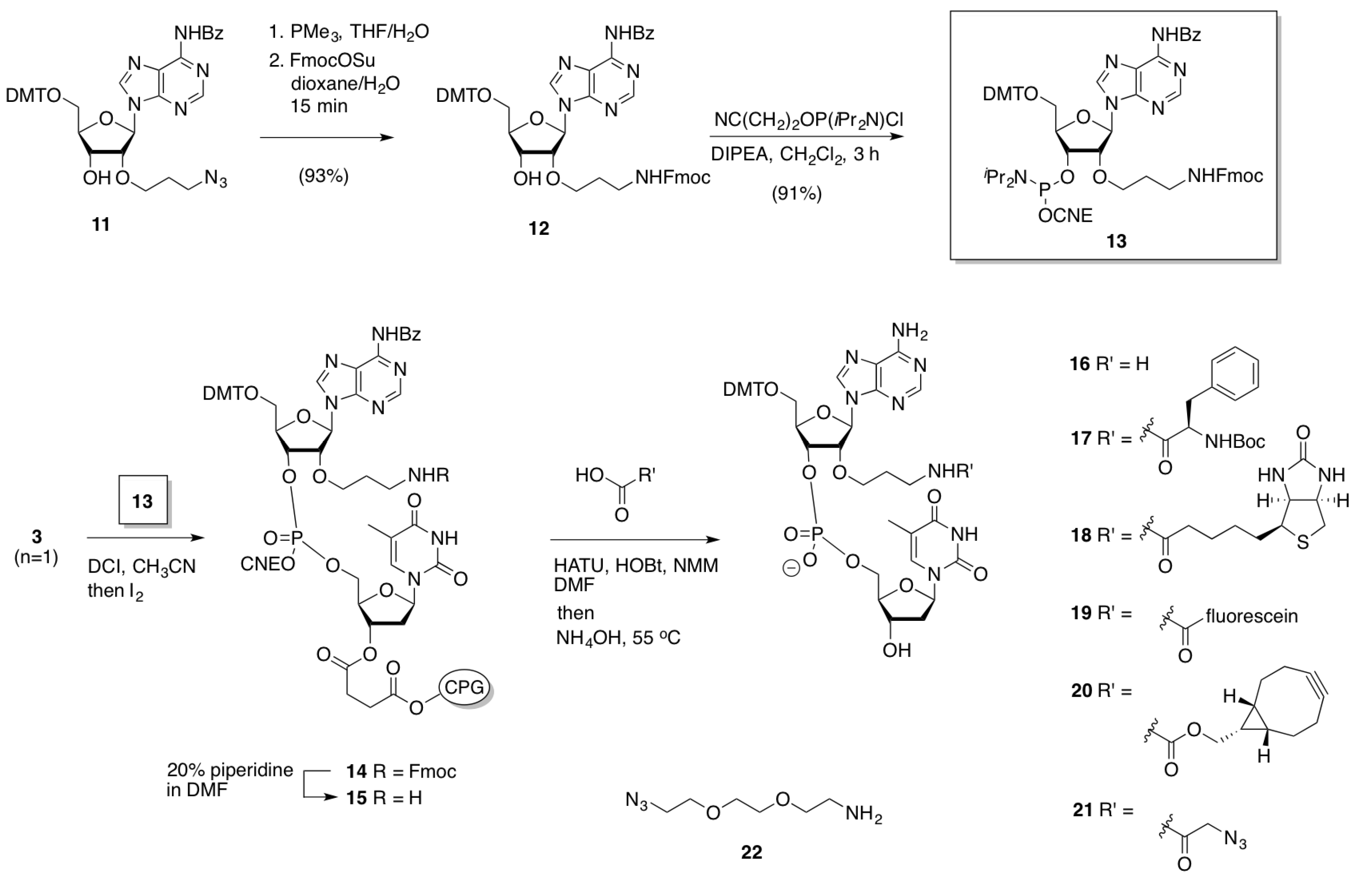
2.1.1. Preparation of BCN-Phosphoramidites

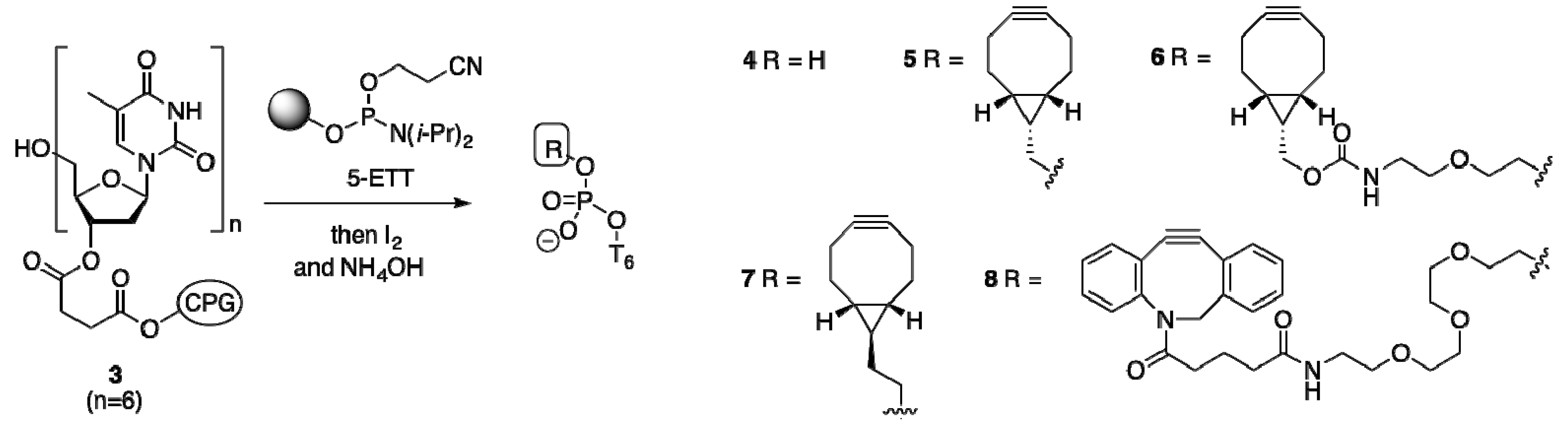
2.1.2. Activation and Incorporation of BCN-Phosphoramidites


2.1.3. Comparison of BCN-Containing Oligonucleotides to Dibenzofused Cyclooctynes

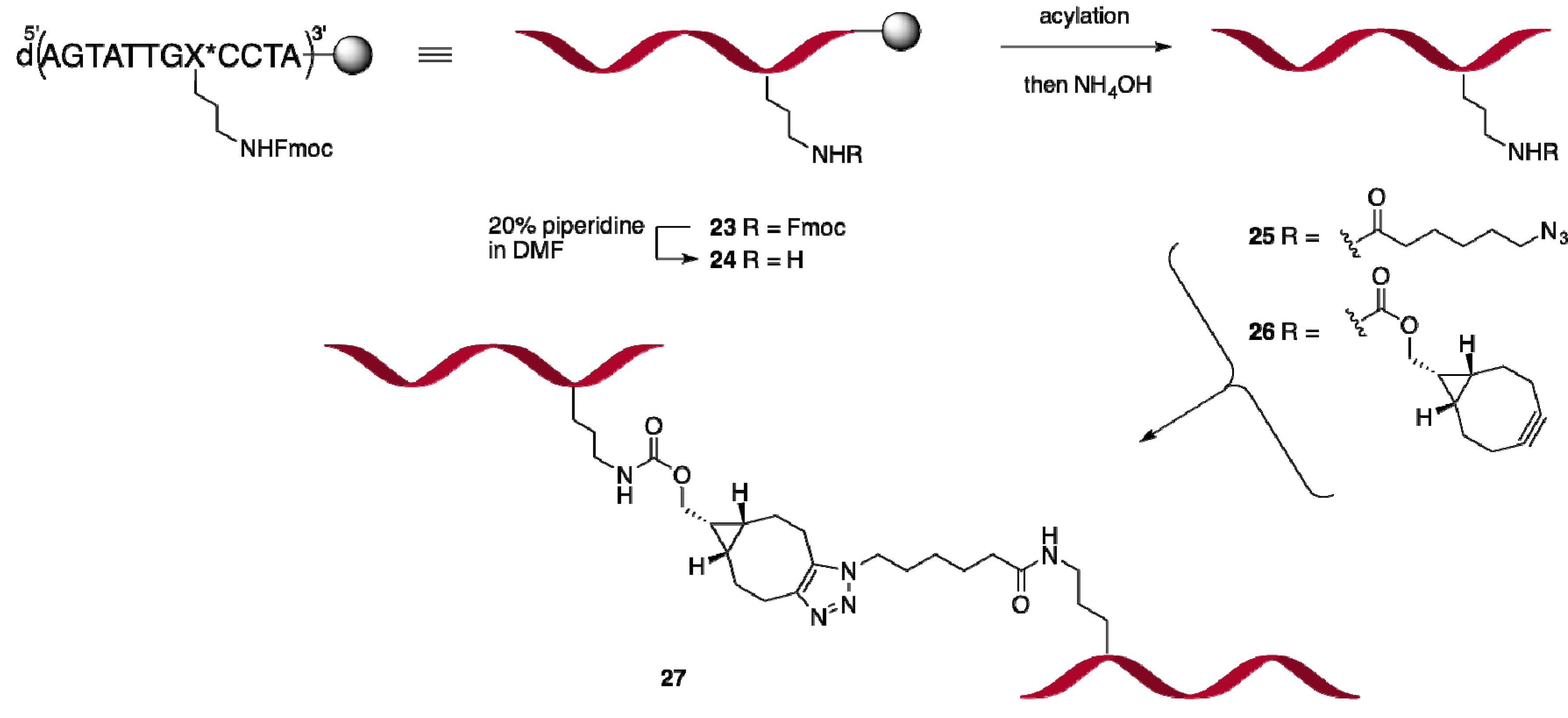
2.2. 3′-Fmoc-Aminopropyl Adenosine for Internal Labeling and Conjugation of Oligonucleotides
2.2.1. Preparation, Incorporation and Model Studies


2.2.2. Oligonucleotide Dimerization

2.2.3. Oligonucleotide-Polythiophene Hybrid

3. Experimental
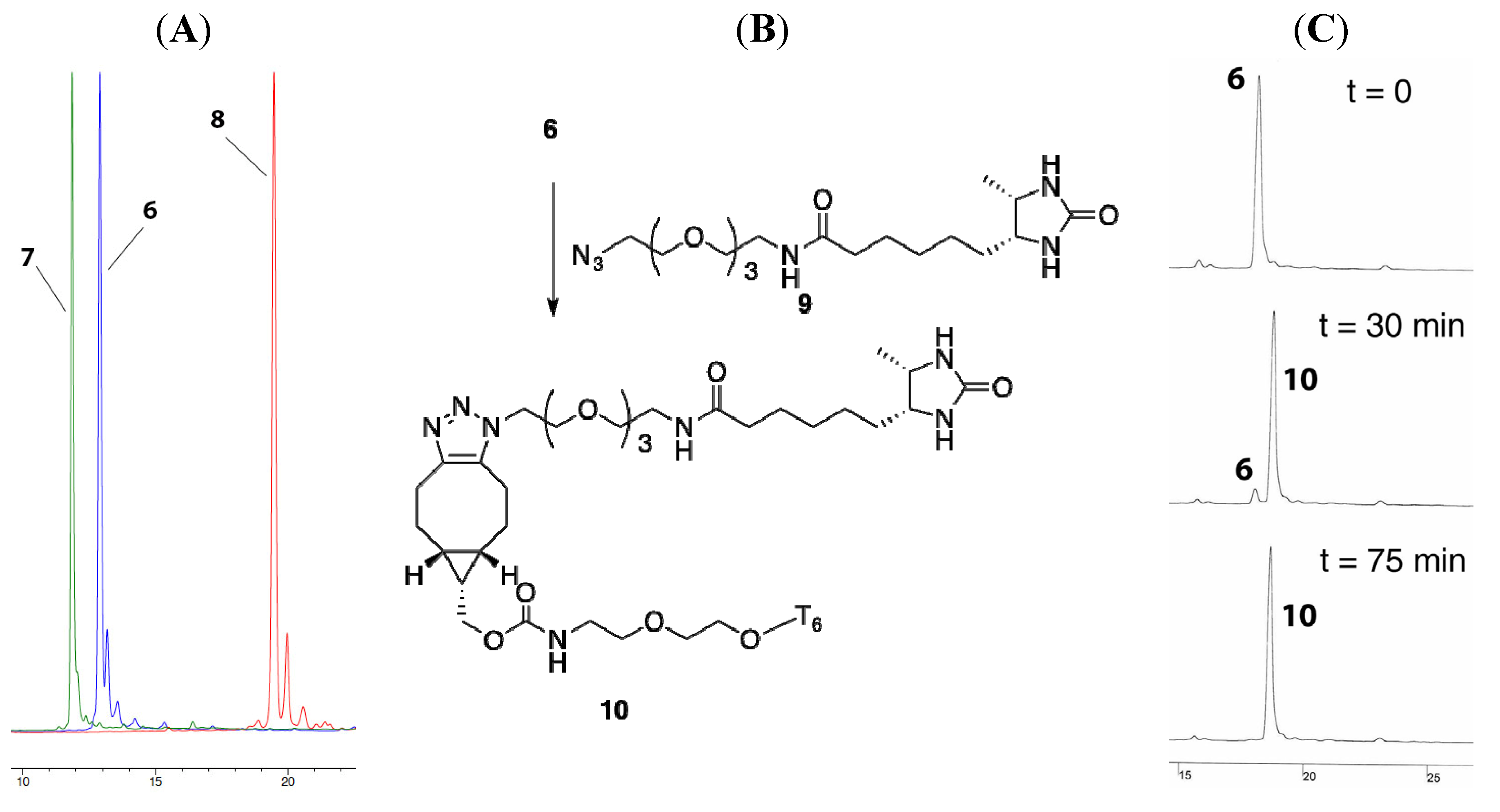
HPLC-analysis of 6, 7 and 8
SPAAC reaction of 6 and 9
Synthesis of dinucleotides 16–21
General procedure for the synthesis of 17–21
SPAAC reactions and analysis of 20 with 21 and 22
Synthesis and conjugation of dodecamer ONs 25 and 26
Dimerization of 25 and 26 to give 27
Preparation of 28
Conjugation of 26 to polymer 28
Preparation of 29
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Pollack, J.R. DNA microarray technology. Introduction. Methods Mol. Biol. 2009, 556, 1–6. [Google Scholar] [CrossRef]
- Dagle, J.M.; Weeks, D.L. Oligonucleotide-based strategies to reduce gene expression. Differentiation 2011, 69, 75–82. [Google Scholar] [CrossRef]
- El-Sagheer, A.H.; Brown, T. Click nucleic acid ligation: Applications in biology and nanotechnology. Acc. Chem. Res. 2012, 45, 1258–1267. [Google Scholar] [CrossRef]
- Sacca, B.; Niemeyer, C.M. Functionalization of DNA nanostructures with proteins. Chem. Soc. Rev. 2011, 40, 5910–5921. [Google Scholar] [CrossRef]
- Niemeyer, C.M. Semisynthetic DNA-Protein Conjugates for Biosensing and Nanofabrication. Angew. Chem. Int. Ed. 2010, 49, 1200–1216. [Google Scholar] [CrossRef]
- Weisbrod, S.H.; Marx, A. Novel strategies for the site-specific covalent labelling of nucleic acids. Chem. Commun. 2001, 5675–5685. [Google Scholar]
- Lönnberg, H. Solid-phase synthesis of oligonucleotide conjugates useful for delivery and targeting of potential nucleic acid therapies. Bioconj. Chem. 2009, 20, 1065–1094. [Google Scholar] [CrossRef]
- Singh, Y.; Murat, P.; Defrancq, E. Recent developments in oligonucleotide conjugation. Chem. Soc. Rev. 2010, 39, 2054–2070. [Google Scholar] [CrossRef]
- Lu, K.; Duan, Q.-P.; Ma, L.; Zhao, D.-X. Chemical strategies for the synthesis of peptide-oligonucleotide conjugates. Bioconj. Chem. 2010, 21, 187–202. [Google Scholar] [CrossRef]
- Singh, Y.; Spinelli, N.; Defrancq, E. Chemical strategies for oligonucleotide-conjugates synthesis. Curr. Org. Chem. 2008, 12, 263–290. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, H. Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Kanan, M.W.; Rozenman, M.M.; Sakurai, K.; Snyder, T.M.; Liu, D.R. Reaction discovery enabled by DNA-templated synthesis and in vitro selection. Nature 2004, 431, 545–549. [Google Scholar] [CrossRef]
- Seela, F.; Pujari, S.S. Azide-Alkyne “Click” Conjugation of 8-Aza-7-deazaadenine-DNA: Synthesis, Duplex Stability and Fluorogenic Dye Labelling. Bioconj. Chem. 2010, 21, 1629–1641. [Google Scholar] [CrossRef]
- Yamada, T.; Peng, C.G.; Matsuda, S.; Addepalli, H.; Jayaprakash, K.N.; Alam, M.R.; Mills, K.; Maier, M.A.; Charisse, K.; Sekine, M.; et al. Versatile site-specific conjugation of small molecules to siRNA using click chemistry. J. Org. Chem. 2001, 76, 1198–1211. [Google Scholar]
- Soriano del Amo, D.; Wang, W.; Jiang, H.; Besanceney, C.; Yan, A.C.; Levy, M.; Liu, Y.; Marlow, F.L.; Wu, P. Biocompatible copper(I) catalysts for in vivo imaging of glycans. J. Am. Chem. Soc. 2010, 132, 16893–16899. [Google Scholar] [CrossRef]
- Kennedy, D.C.; McKay, C.S.; Legault, M.C.B.; Danielson, D.C.; Blake, J.A.; Pegoraro, A.F.; Stolow, A.; Mester, Z.; Pezacki, J.P. Cellular consequences of copper complexes used to catalyze bioorthogonal click reactions. J. Am. Chem. Soc. 2011, 133, 17993–18001. [Google Scholar] [CrossRef]
- Eltepu, L.; Jayaraman, M.; Rajeev, K.G.; Manoharan, M. An immobilized and reusable Cu(I) catalyst for metal ion-free conjugation of ligands to fully deprotected oligonucleotides through click reaction. Chem. Commun. 2013, 49, 184–186. [Google Scholar] [CrossRef]
- Jayaprakash, K.N.; Geng Peng, C.; Butler, D.; Varghese, J.P.; Maier, M.A.; Rajeev, K.G.; Manoharan, M. Non-nucleoside building blocks for copper-assisted and copper-free click chemistry for the efficient synthesis of RNA conjugates. Org. Lett. 2010, 12, 5410–5413. [Google Scholar] [CrossRef]
- Van Delft, P.; Meeuwenoord, N.J.; Hoogendoorn, S.; Dinkelaar, J.; Overkleeft, H.S.; van der Marel, G.A.; Filippov, D.V. Synthesis of oligoribonucleic acid conjugates using a cyclooctyne phosphoramidite. Org. Lett. 2010, 12, 5486–5489. [Google Scholar] [CrossRef]
- Marks, I.S.; Sung Kang, J.; Jones, B.T.; Landmark, K.J.; Cleland, A.J.; Taton, T.A. Strain-promoted “click” chemistry for terminal labeling of DNA. Bioconj. Chem. 2011, 22, 1259–1263. [Google Scholar] [CrossRef]
- Shelbourne, M.; Chen, X.; Brown, T.; Al-Sagheer, A.H. Fast copper-free click DNA ligation by the ring-strain promoted alkyne-azide cycloaddition reaction. Chem. Commun. 2011, 47, 6257–6259. [Google Scholar] [CrossRef]
- Santoshkumar, L.K.; Kang, J.S.; Mullen, D.G.; Hast, M.A.; Beese, L.S.; Distefano, M.D.; Taton, T.A. Covalent protein-oligonucleotide conjugates by copper-free click reaction. Bioorg. Med. Chem. 2012, 20, 4532–4539. [Google Scholar] [CrossRef]
- Ning, X.; Guo, J.; Wolfert, M.A.; Boons, G.-J. Visualizing metabolically labeled glycoconjugates of living cells by copper-free and fast Huisgen cycloadditions. Angew. Chem. Int. Ed. 2008, 47, 2253–2255. [Google Scholar] [CrossRef]
- Richardson, J.A.; Gerowska, M.; Shelbourne, M.; French, D.; Brown, T. Six-colour HyBeacon probes for multiplex genetic analysis. ChemBioChem 2010, 11, 2530–2533. [Google Scholar] [CrossRef]
- Shelbourne, M.; Brown, T. El-Sagheer, A.H.; Brown, T. Fast and efficient DNA crosslinking and multiple orthogonal labelling by copper-free click chemistry. Chem. Commun. 2012, 48, 11184–11186. [Google Scholar] [CrossRef]
- Lietard, J.; Meyer, A.; Vasseur, J.J.; Morvan, F. An efficient reagent for 5'-azido oligonucleotide synthesis. Tetrahedr. Lett. 2007, 48, 8795–8798. [Google Scholar] [CrossRef]
- Miller, G.P.; Kool, E.T. Versatile 5′-functionalization of oligonucleotides on solid support: Amines, azides, thiols, and thioethers via phosphorus chemistry. J. Org. Chem. 2004, 69, 2404–2410. [Google Scholar] [CrossRef]
- Kumar, R.; El-Sagheer, A.H.; Tumpane, J.; Lincoln, P.; Wilhelmsson, L.M.; Brown, T. Template-directed oligonucleotide strand ligation, covalent intramolecular DNA circularization and catenation using click chemistry. J. Am. Chem. Soc. 2007, 129, 6859–6864. [Google Scholar] [CrossRef]
- Lartia, R.; Murat, P.; Dumy, P.; DeFrancq, E. Versatile introduction of azido moiety into oligonucleotides through diazo transfer reaction. Org. Lett. 2011, 13, 5672–5675. [Google Scholar] [CrossRef]
- Wada, T.; Mochizuki, A.; Higashiya, S.; Tsuruoka, H.; Kawahara, S.; Ishikawa, M.; Sekine, M. Synthesis and properties of 2-azidodeoxyadenosine and its incorporation into oligodeoxynucleotides. Tetrahedr. Lett. 2001, 42, 9215–9219. [Google Scholar] [CrossRef]
- Kiviniemi, A.; Virta, R.; Lönnberg, H. Utilization of intrachain 4′-C-azidomethylthymidine for preparation of oligodeoxyribonucleotide conjugates by click chemistry in solution and on a solid support. Bioconj. Chem. 2008, 19, 1726–1734. [Google Scholar] [CrossRef]
- Aigner, M.; Hartl, M.; Fauster, K.; Steger, J.; Bister, K.; Micura, R. Chemical synthesis of site-specifically 2′-azido-modified RNA and potential applications for bioconjugation and RNA interference. ChemBioChem 2011, 12, 47–51. [Google Scholar]
- Fauster, K.; Hartl, M.; Santner, T.; Aigner, M.; Kreutz, C.; Bister, K.; Ennifar, E.; Micura, R. 2′-Azido RNA, a versatile tool for chemical biology: Synthesis, X-ray structure, siRNA applications, click labeling. ACS Chem. Biol. 2012, 7, 581–589. [Google Scholar] [CrossRef]
- Dommerholt, J.; Schmidt, S.; Temming, R.; Hendriks, L.J.A.; Rutjes, F.P.J.T.; van Hest, J.C.M.; Lefeber, D.J.; Friedl, P.; van Delft, F.L. Readily accessible Bicyclononynes for bioorthogonal labeling and three-dimensional imaging of living cells. Angew. Chem. Int. Ed. 2010, 49, 9422–9425. [Google Scholar] [CrossRef]
- Debets, M.F.; van Berkel, S.S.; Dommerholt, J.; Dirks, A.J.; Rutjes, F.P.J.T.; van Delft, F.L. Bioconjugation with strained alkenes and alkynes. Acc. Chem. Res. 2011, 44, 805–815. [Google Scholar] [CrossRef]
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry, 5th ed.; Wiley: Hoboken, NJ, USA, 2001; pp. 222–225. [Google Scholar]
- Jawalekar, A.M.; Meeuwenoord, N.; Cremers, J.G.O.; Overkleeft, H.S.; van der Marel, G.A.; Rutjes, F.P.J.T.; van Delft, F.L. Conjugation of nucleosides and oligonucleotides by [3+2] cycloaddition. J. Org. Chem. 2008, 73, 287–290. [Google Scholar] [CrossRef]
- Borrmann, A.; Milles, J.; Plass, T.; Dommerholt, J.; Verkade, J.M.M.; Wießler, M.; Schultz, C.J.; van Hest, C.M.; van Delft, F.L.; Lemke, E.A. Genetic Encoding of a bicyclo[6.1.0]nonyne-charged amino acid enables fast cellular protein imaging by metal-free ligation. ChemBioChem 2012, 13, 2094–2099. [Google Scholar] [CrossRef]
- Chen, W.; Wang, D.; Dai, C.; Hamelberg, D.; Wang, B. Clicking 1,2,4,5-tetrazine and cyclooctynes with tunable reaction rates. Chem. Commun. 2012, 48, 1736–1738. [Google Scholar] [CrossRef]
- Lang, K.; Davis, L.; Wallace, S.; Mahesh, M.; Cox, D.J.; Blackmann, M.L.; Fox, J.M.; Chin, J.W. Genetic encoding of bicyclononynes and trans-cyclooctenes for site-specific protein labeling in vitro and in live mammalian cells via rapid fluorogenic Diels-Alder reactions. J. Am. Chem. Soc. 2012, 134, 10317–10320. [Google Scholar] [CrossRef]
- Plass, T.; Milles, S.; Koehler, C.; Schultz, C.; Lemke, E.A. Genetically encoded copper-free click chemistry. Angew. Chem. Int. Ed. 2012, 51, 4166–4170. [Google Scholar] [CrossRef]
- Rossin, R.; Renart Verkerk, P.; van den Bosch, S.; Vulders, R.; Verel, I.; Lub, J.; Robillard, M. In vivo chemistry for pretargeted tumor imaging in live mice. Angew. Chem. Int. Ed. 2012, 49, 3375–3378. [Google Scholar]
- Sample Availability: Samples of the compounds 1, 2, 6, 7, 10, 30, 33, 34 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jawalekar, A.M.; Malik, S.; Verkade, J.M.M.; Gibson, B.; Barta, N.S.; Hodges, J.C.; Rowan, A.; Van Delft, F.L. Oligonucleotide Tagging for Copper-Free Click Conjugation. Molecules 2013, 18, 7346-7363. https://doi.org/10.3390/molecules18077346
Jawalekar AM, Malik S, Verkade JMM, Gibson B, Barta NS, Hodges JC, Rowan A, Van Delft FL. Oligonucleotide Tagging for Copper-Free Click Conjugation. Molecules. 2013; 18(7):7346-7363. https://doi.org/10.3390/molecules18077346
Chicago/Turabian StyleJawalekar, Anup M., Sudip Malik, Jorge M. M. Verkade, Brian Gibson, Nancy S. Barta, John C. Hodges, Alan Rowan, and Floris L. Van Delft. 2013. "Oligonucleotide Tagging for Copper-Free Click Conjugation" Molecules 18, no. 7: 7346-7363. https://doi.org/10.3390/molecules18077346