3. Experimental
Sterols and NaBH
4 were purchased from Merck Co. (Shanghai, China) All chemicals and solvents were analytical grade and solvents were purified by general methods before being used. Melting points were determined on an X4 apparatus and were uncorrected. Infrared spectra were measured with a Thermo Scientific IS-10 Spectrophotometer. The
1H and
13C-NMR spectra were recorded in CDCl
3 on a Bruker AV-300 spectrometer at working frequencies 300 and 75 MHz. Chemical shifts are expressed in parts per million (
δ) values and coupling constants (
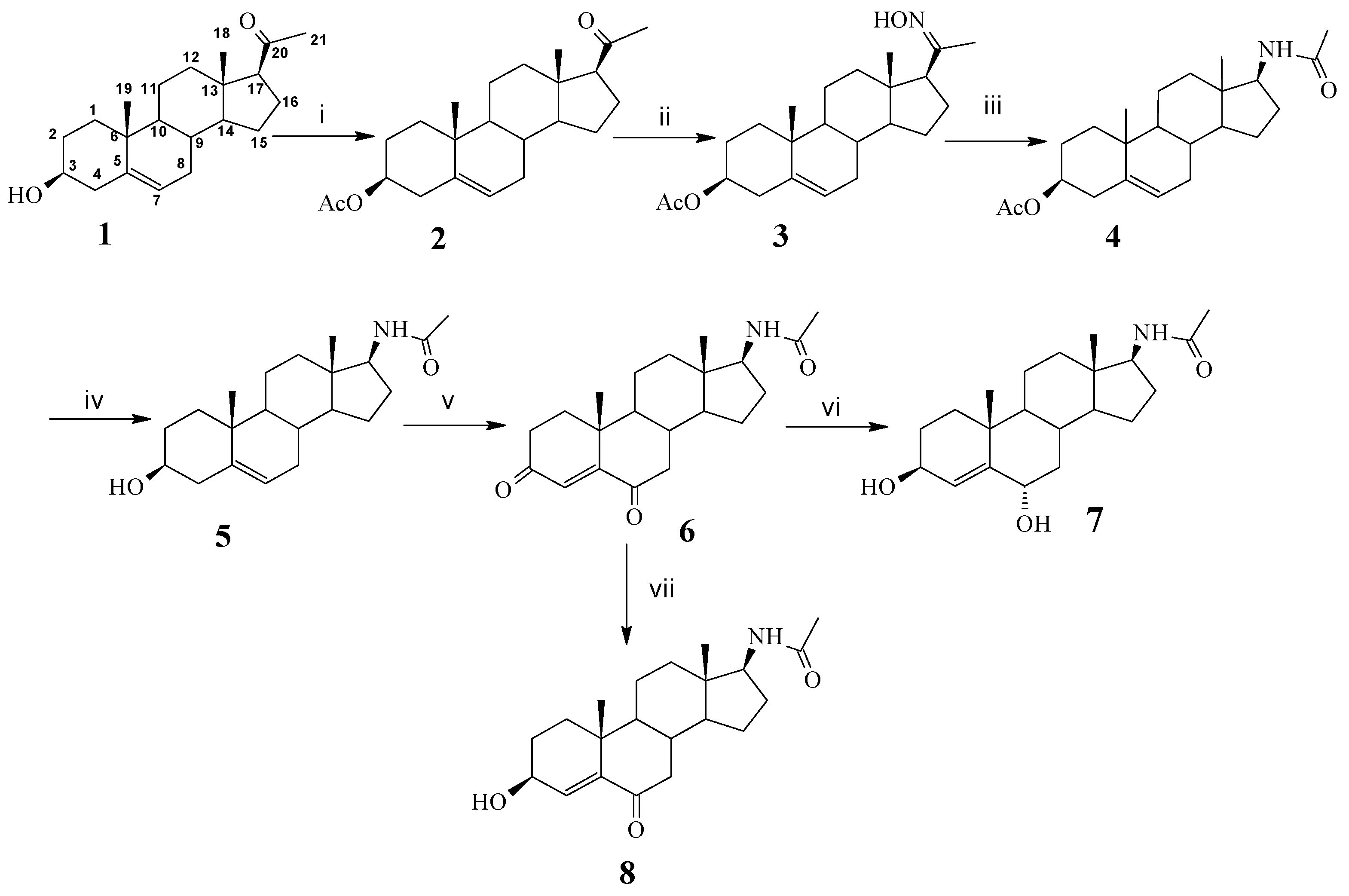
J) in Hertz. LREIMS were recorded on a Thermo-DSQ instrument, while HREIMS were measured on a Agilent 6210 TOFMS instrument. 3-Acetoxy-17-acetamidoandrost-5-ene (
4) and 3,12-dioxydeoxycholic acid (
10) were prepared according to the literature [
6] and [
20], respectively.
Synthesis of 3-hydroxy-17-acetamidoandrost-5-en (5). K2CO3 solution (13%, 20 mL) was added to a solution of compound 4 (438 mg, 3.32 mmol) in CH3OH (200 mL). The reaction mixture was heated under reflux for 1 h. After completion of the reaction as indicated by TLC, the solvent was removed under vacuum. CH2Cl2 (200 mL) was added to dissolve the solid and the resulting solution was washed with cold water and saturated brine. After drying over anhydrous sodium sulfate, the solvent was removed under reduced pressure, and the resulting crude product was purified by chromatography on silica gel using methanol/dichloromethane (30:1) as eluent to give 709 mg (76%) of 5 as a white solid. θ mp 196–197 °C; IR (KBr) ν/cm−1: 3354, 2962, 2937, 1646, 1556, 1433, 1372, 1315; 1H-NMR (CDCl3, 300 MHz) δ: 0.721 (3H, s, 18-CH3), 1.028 (3H, s, 19-CH3), 2.002 (3H, s, N-COCH3), 2.36–2.24 (2H, m, C4-H), 3.58–3.51 (1H, m, C3-H), 3.908 (1H, q, J = 9.0, C17-H), 5.302 (1H, brd, J = 9.9, N-H), 5.367(1H, dd, J = 3.6, 1.8, C6-H); 13C-NMR (CDCl3, 75 MHz) δ: 169.9 (-NHCOCH3), 140.9 (5-C), 121.3 (6-C), 71.7 (3-C), 58.9 (17-C), 52.8 (9-C), 50.1 (13-C), 42.7 (4-C), 42.2 (14-C), 37.2 (10-C), 36.8(1-C), 36.7 (12-C), 32.0 (8-C), 31.6 (7-C), 31.5 (16-C), 28.7 (2-C), 27.7 (15-C), 23.6 (21-C), 20.6 (11-C), 19.4 (19-C), 11.9 (18-C); ESI-MS m/z: 332 (M+1)+.
Synthesis of 17-acetamidoandrost-4-en-3,6-dione (6). Jones’ reagent (0.7 mL, 0.267 mol/L) was added dropwise to the solution of 5 (709 mg) in acetone (30 mL) in 20 min. The mixture was stirred at room temperature for 1 h and then neutralized with 10% K2CO3 solution. The suspension was poured over a silica gel column and eluted with ethyl acetate. The solvent was removed under reduced pressure. The residue was purified by chromatography on silica gel using methanol/dichloromethane (20:1) as elution to give 590 mg (80%) of 6 as white solid. θ mp 198–199 °C; IR (KBr) ν/cm−1: 3321, 2941, 2864, 1691, 1654, 1556, 1442, 1384; 1H-NMR (CDCl3, 300 MHz) δ: 0.728 (3H, s, 18-CH3), 1.150 (3H, s, 19-CH3), 1.976 (3H, s, N-COCH3), 2.17–2.10 (2H, m, C7-βH and C2-αH), 2.495 (1H, dd, J = 13.8, 5.1, C7-αH), 2.662 (1H, dd, J = 15.9, 3.9, C2-βH), 3.937 (1H, q, J = 9.0, C17-H), 5.582 (1H, d, J = 9.0, N-H), 6.149 (1H, s, C4-H); 13C-NMR (CDCl3, 300 MHz) δ: 201.8 (6-C), 199.3 (3-C), 170.1 (-NHCOCH3), 160.7 (5-C), 125.6 (4-C), 58.5 (14-C), 52.7 (17-C), 50.9 (9-C), 46.3 (13-C), 43.0 (10-C), 39.7 (1-C), 36.3 (12-C), 35.5 (7-C), 34.3 (8-C), 33.9 (16-C), 28.2 (2-C), 23.4 (15-C), 23.2 (21-C), 20.5 (11-C), 17.6 (19-C), 11.9 (18-C); ESI-MS m/z: 344 (M+1)+.
Synthesis of 3,6-dihydroxy-17-acetamidoandrost-4-en (7). NaBH4 (42 mg, 1.1 mmol) was added at room temperature to a solution of 6 (124 mg, 0.36 mmol) in CH3OH (50 mL) in one portion. After 30 min, the reaction was stopped. The solution was neutralized with 1 M HCl. After evaporation of the majority of MeOH under reduced pressure, the residue was extracted with ethyl acetate (15 mL × 3). The organic layer was washed with cold water and saturated brines. After drying over anhydrous sodium sulfate, the solvent was removed under reduced pressure and a crude product was obtained. After recrystallization from methanol, 95 mg (78%) of compound 7 was obtained as a white solid. θ mp 198–200 °C; IR (KBr) ν/cm−1: 3331, 2941, 2866, 1654, 1442, 1384, 1217, 1131; 1H-NMR (CDCl3, 300 MHz) δ: 0.703 (3H, s, 18-CH3), 1.008 (3H, s, 19-CH3), 1.988 (3H, s, N-COCH3), 3.57–3.47 (1H, m, C3-H), 3.93–3.80 (1H, m, C6-H), 4.13 (1H, dd, J = 7.0, 1.5, C17-H), 5.36 (1H, d, J = 2.1, C4-H), 5.40 (1H, br s, N-H); 13C-NMR (CDCl3, 75 MHz) δ: 170.0 (-NHCOCH3), 140.9 (5-C), 121.3 (4-C), 71.6 (6-C), 68.8 (3-C), 58.9 (17-C), 52.8 (14-C), 50.1 (9-C), 42.7 (13-C), 42.2 (7-C), 37.2 (10-C), 36.8 (1-C), 36.5 (12-C), 32.1 (8-C), 31.6 (2-C), 31.5 (16-C), 28.7 (15-C), 23.6 (21-C), 20.6 (11-C), 19.4 (19-C), 12.0 (18-C); ESI-MS m/z: 348 (M+1)+.
Synthesis of 3-hydroxy-17-acetamidoandrost-4-en-6-one (8). NaBH4 (42 mg, 1.1 mmol) was added to a solution of 6 (124 mg, 0.36 mmol) and NiCl2·6H2O (87 mg, 0.37 mmol) in CH3OH (15 mL) in the interval of 5 min at room temperature. After 5 min, the reaction was stopped. The solution was neutralized with 1 M HCl. After evaporation of the majority of MeOH under reduced pressure, water of 15 mL was added. Then the residue was extracted with ethyl acetate (15 × 3 mL). The resulting solution was washed with cold water and saturated brine. After drying over anhydrous sodium sulfate, the solvent was removed under reduced pressure, and the resulting crude product was purified by flash chromatography on silica gel using methanol/dichloromethane (20:1) as the eluent. Compound 8 was obtained as white solid (75 mg, 60%), θ mp 187–188 °C; IR(KBr) ν/cm–1: 3330, 2942, 2865, 1675, 1442, 1385, 1218; 1H-NMR (CDCl3, 300 MHz) δ: 0.747 (3H, s, 18-CH3), 1.196 (3H, s, 19-CH3), 2.004 (3H, s, N-COCH3), 2.22–2.14 (2H, m, C16-H), 2.418 (1H, dd, J = 12.9, 4.5, C7-αH), 3.929 (1H, q, J = 9.0, C17-H), 4.338 (1H, ddd, J = 11.4, 4.2, 1.2, C3-βH), 5.330 (1H, br d, J = 7.5, N-H), 6.196 (1H, d, J = 1.2, C4-H); 13C-NMR (CDCl3, 75 MHz) δ: 199.3 (6-C), 170.0 (-NHCO), 169.9 (5-C), 119.9 (4-C), 68.5 (3-C), 58.6 (17-C), 53.7 (9-C), 51.7 (14-C), 42.9 (13-C), 40.9 (7-C), 39.0 (10-C), 36.5 (8-C), 36.3 (1-C), 34.3 (12-C), 33.7 (2-C), 28.6 (16-C), 23.6 (15-C), 23.5 (21-C), 20.6 (11-C), 18.3 (19-C), 12.1 (18-C); ESI-MS m/z: 346 (M+1)+.
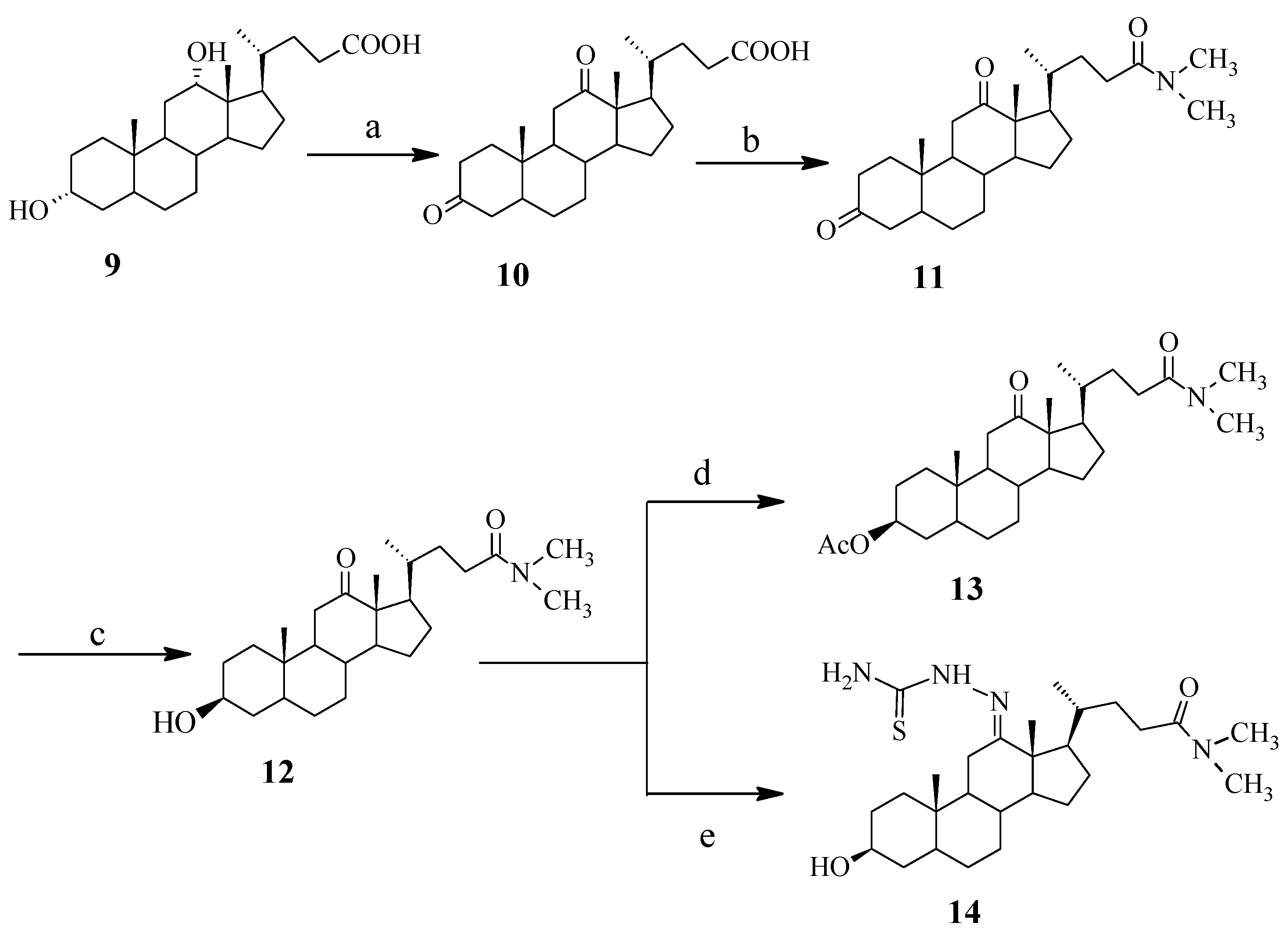
Synthesis of N,N-dimethyl-3,12-dioxo-7-deoxycholic amide (11). Oxalyl chloride (200 µL, 2.23 mmol) was added to a solution of 10 (535 mg, 1.38 mmol) and pyridine (150 µL, 1.80 mmol) in toluene (25 mL) at ice bath temperature. The mixture was stirred for 2 h. After completion of the reaction as indicated by TLC, a solution of dimethyl amine (540 µL) was added dropwise. The solution was stirred for 10 min at 0 °C and 10 mL ice water was added to the solution. The solution was extracted with dichloromethane (30 × 3 mL). The organic solution was washed with cold water and saturated brine. After drying over anhydrous sodium sulfate, the solvent was removed under reduced pressure, and the resulting crude product was purified by chromatography on silica gel using petroleum/ethyl acetate (1:2) as the eluent. Compound 11 was obtained as a white solid (480 mg, 71%), θ mp 271–272 °C; IR(KBr) ν/cm–1: 2929, 2859, 1695, 1634; 1H-NMR (CDCl3, 300 MHz) δ: 0.797 (3H, d, J = 5.7, 21-CH3), 1.000 (3H, s, 18-CH3), 1.050 (3H, s, 19-CH3), 2.13–2.04 (2H, m), 2.37–2.18 (3H, m), 2.58–2.48 (2H, m, C11-H), 2.872 (3H, s, NCH3), 2.955 (3H, s, NCH3); 13C-NMR (CDCl3, 75 MHz) δ: 214.2 (12-C), 212.1 (3-C), 173.4 (24-C), 58.5 (14-C), 57.5 (17-C), 46.5 (9-C), 44.2 (13-C), 43.6 (5-C), 42.1 (4-C), 38.3 (8-C), 37.3 (11-C), 36.8 (1-C), 36.7 (2-C), 35.6 (10-C), 35.5 (N-C), 35.4 (N-C), 35.3 (20-C), 30.6 (7-C), 30.2 (23-C), 27.4 (22-C), 26.5 (15-C), 25.4 (6-C), 24.3 (16-C), 22.1 (21-C), 18.7 (19-C), 11.7 (18-C); ESI-MS m/z: 416 (M+1)+.
Synthesis of N,N-dimethyl-3-hydroxy-12-oxo-7-deoxycholic amide (12). Compound 12 was prepared according to the procedure of 7. Yield 30%, θ mp 279–280 °C; IR(KBr) ν/cm–1: 3465, 2925, 2859, 1707, 1621; 1H-NMR (CDCl3, 300 MHz) δ: 0.843 (3H, d, J = 6.3, 21-CH3), 0.986 (3H, s, 19-CH3), 0.995 (3H, s, 18-CH3), 2.26–2.17 (3H, m), 2.49-2.32 (2H, m, C11-H), 2.912 (3H, s, NCH3), 2.994 (3H, s, NCH3), 3.63-3.53 (1H, m, C3-H); 13C-NMR (CDCl3, 75 MHz) δ: 215.2 (12-C), 173.7 (24-C), 71.2 (3-C), 58.7 (14-C), 57.5 (17-C), 46.4 (13-C), 44.1 (5-C), 41.5 (9-C), 38.1 (8-C), 37.3 (11-C), 36.3 (1-C), 35.6 (4-C), 35.4 (10-C), 35.3 (N-C), 35.3 (N-C), 35.2 (20-C), 30.6 (2-C), 30.3 (23-C), 30.2 (22-C), 27.5 (6-C), 27.1 (15-C), 26.1 (16-C), 24.3 (7-C), 22.8 (21-C), 18.8 (18-C), 11.7 (19-C); ESI-MS m/z: 418 (M+1)+.
Synthesis of N,N-dimethyl-3-acetoxy-12-oxo-7-deoxycholic amide (13). Compound 13 was prepared by the esterification of 12 using acetic anhydride in pyridine. Yield: 78%. θ mp 153–154 °C; IR(KBr) ν/cm–1: 3440, 2953, 1730, 1700, 1656, 1465, 1447; 1H-NMR (CDCl3, 300 MHz)δ: 0.885 (3H, d, J = 6.3, 21-CH3), 1.035 (6H, br s, 18-CH3 and 19-CH3), 2.025 (3H, s, CH3CO-), 2.951 (3H, s, NCH3), 3.025 (3H, s, NCH3), 4.659–4.763 (1H, m, C3-H); 13C-NMR (CDCl3, 75 MHz) δ: 214.9 (12-C), 173.6 (24-C), 170.6 (CH3CO-), 73.7 (3-C), 58.7 (14-C), 57.6 (17-C), 46.5 (13-C), 44.1 (5-C), 41.4 (9-C), 38.1 (8-C). 37.3 (11-C), 35.7 (1-C), 35.4 (4-C), 35.3 (N-C), 34.9 (N-C), 32.1 (20-C), 30.7 (2-C), 30.3 (23-C), 27.5 (10-C), 26.9 (22-C), 26.4 (6-C), 26.4 (15-C), 24.4 (16-C), 22.7 (7-C), 21.4 (21-C), 18.8 (18-C), 11.7 (19-C); ESI-MS m/z: 460 (M+1)+.
Synthesis of N,N-dimethyl-3-hydroxy-12-thiosemicarbazone-7-deoxycholic amide (14). The compound 12 (100 mg, 0.24 mmol) was dissolved in 15 mL of CH3CH2OH and the solution was adjusted to pH ≈ 3–5 by adding a few drops of glacial acetic acid. After the solution was heated to 80 °C, and thiosemicarbazide (32 mg, 0.36 mmol) was added. The mixture was stirred for 10 h at 80 °C until no starting material (the progress of the reaction was monitored by TLC). Then the reaction was terminated and the majority of solvent was evaporated under reduced pressure. Water (10 mL) was added to the mixture which was then extracted with dichloromethane. The organic layer was washed with water and saturated brine. After drying over anhydrous sodium sulfate, the solvent was removed under reduced pressure, and the resulting crude product was purified by flash chromatography on silica gel using dichloromethane/methanol (40:1) as the eluent to give 56 mg of 14 as a white solid (yield: 49%), θ mp 294–295 °C; IR (KBr) ν/cm–1: 3427, 2921, 1634, 1503, 1401; 1H-NMR (CDCl3, 300 MHz): 0.843 (3H, d, J = 6.3, 21-CH3), 0.986 (3H, s, 18-CH3), 0.995 (3H, s, 19-CH3), 2.912 (3H, s, N-CH3), 2.994 (3H, s, N-CH3), 3.577 (1H, m, C3-H), 6.576 (1H, br s, -NH2), 7.234 (1H, br s, -NH2), 9.284 (1H, s, -NH-); 13C-NMR (CDCl3, 75 MHz): 178.8 (C=S), 173.6 (24-C), 162.6 (12-C), 71.1 (3-C), 58.7 (14-C), 57.5 (17-C), 46.4 (13-C), 44.1 (5-C), 41.6 (9-C), 38.1 (8-C), 37.4 (4-C), 36.3 (11-C), 35.7 (10-C), 35.6 (1-C), 35.4 (N-C), 35.3 (20-C), 35.1 (N-C), 30.7 (2-C), 30.3 (22-C), 30.2 (23-C), 27.5 (6-C), 27.1 (15-C), 26.1 (16-C), 24.4 (7-C), 22.8 (21-C), 18.8 (18-C), 11.7 (19-C); ESI-MS m/z: 491(M+1)+.
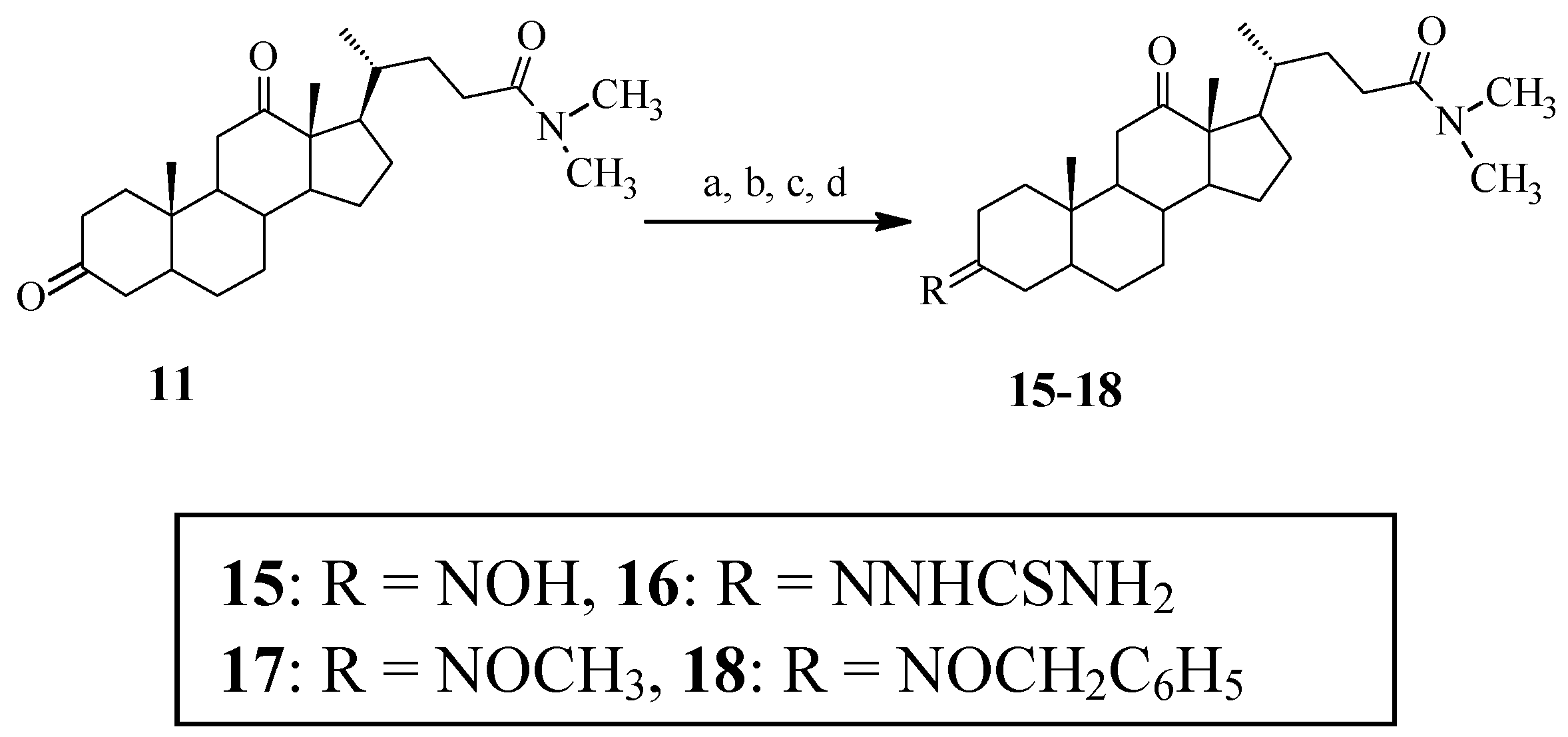
Synthesis of N,N-dimethyl-3-hydroximino-12-oxo-7-deoxycholic amide (15). Compound 11 (91 mg, 0.22 mmol) was dissolved in 95% CH3CH2OH (15 mL). After the mixture was heated to 70 °C, CH3COONa·3H2O (30 mg, 0.22 mmol) and NH2OH·HCl (18 mg, 0.26 mmol) were added. The mixture was stirred for 3 h at 70 °C. Then the reaction was terminated and the majority of solvent was evaporated under reduced pressure. Distilled water was added into the reaction mixture, and the product was extracted with dichloromethane. The combined extracts were washed with water and saturated brine, dried with anhydrous sodium sulfate, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel using dichloromethane/methanol (40:1) as the eluent. Compound 15 was obtained as a white solid (56 mg, 60%). θmp 232–233 °C; IR (KBr) ν/cm–1: 3440, 1691, 1601, 1437, 1392; 1H-NMR (CDCl3, 300 MHz): 0.853 (3H, d, J = 6.3, 21-CH3), 1.025 (3H, s, 18-CH3), 1.035 (3H, s, 19-CH3), 2.534 (2H, m, C11-H), 2.923 (3H, s, N-CH3), 2.999 (3H, s, N-CH3), 3.070 (1H,dd, J = 14.4, 3.9, C2-βH); 13C-NMR (CDCl3, 75 MHz): 214.9 (12-C), 173.7 (24-C), 160.2 (3-C), 58.6 (14-C), 57.6 (17-C), 46.4 (9-C), 44.2 (5-C), 44.0 (13-C), 43.3 (10-C), 41.9 (8-C), 38.3 (11-C), 37.4 (N-C), 36.9 (N-C), 36.1 (20-C), 35.6 (7-C), 35.5 (4-C), 30.6 (23-C), 30.3 (6-C), 27.4 (22-C), 26.6 (2-C), 26.5 (1-C), 25.6 (15-C), 24.8 (16-C), 24.3 (21-C), 22.6 (19-C), 18.8 (18-C); ESI-MS m/z: 431(M+1)+.
Synthesis of N,N-dimethyl-3-thiosemicarbazone-12-oxo-7-deoxycholic amide (16). Compound 16 was prepared similarly to 14, but from the compound 11. Yield: 69%, θmp 282–283 °C. IR (KBr) ν/cm–1: 3432, 2929, 2868, 1703, 1638, 1499; 1H-NMR (CDCl3, 300 MHz): 0.856 (3H, d, J = 5.7, 21-CH3), 1.034 (3H, s, 18-CH3), 1.060 (3H, s, 19-CH3), 2.539 (2H, m, C11-H), 2.926 (3H, s, N-CH3), 3.002 (3H, s, N-CH3), 6.487 (1H, br s, N-H), 7.213 (H, br s, N-H), 8.873 (1H, s, -NH-); 13C-NMR (CDCl3, 75 MHz,): 214.5 (12-C), 178.7 (S=C), 173.6 (24-C), 156.8 (3-C), 58.5 (14-C), 57.5 (17-C), 46.5 (9-C), 44.4 (5-C), 44.1 (13-C), 43.7 (10-C), 42.3 (8-C), 38.3 (11-C), 37.3 (N-C), 36.1 (N-C), 36.0 (20-C), 35.6 (4-C), 35.4 (7-C), 30.6 (23-C), 30.3 (6-C), 29.9 (2-C), 27.4 (22-C), 26.7 (1-C), 25.6 (15-C), 24.3 (16-C), 22.5 (21-C), 18.8 (19-C), 11.8 (18-C); ESI-MS m/z: 489 (M+1)+.
Compounds 17 and 18 were prepared similarly according to the procedure of 15, but CH3ONH2·HCl and PhCH2ONH2·HCl were used as reagents instead of NH2OH·HCl.
N,N-dimethyl-3-O-methyloximino-12-oxo-7-deoxycholic amide (17). Yield: 70%, θmp 188–189 °C. IR (KBr) ν/cm–1: 3436, 2855, 1703, 1654, 1446, 1041; 1H-NMR (CDCl3, 300 MHz): 0.875 (3H, d, J = 6.3, 21-CH3), 1.043 (3H, s, 18-CH3), 1.054 (3H, s, 19-CH3), 2.55–2.40 (2H, m, C11-H), 2.940 (3H, s, N-CH3), 3.014 (3H, s, N-CH3), 3.804 (3H, s, O-CH3); 13C-NMR (CDCl3, 75MHz): 214.7 (12-C), 173.5 (24-C), 160.0 (3-C), 61.0 (-OCH3), 58.6 (14-C), 57.6 (17-C), 46.5 (9-C), 44.2 (13-C), 44.0 (10-C), 43.5 (5-C), 42.0 (8-C), 38.3 (11-C), 37.3 (N-C), 36.1 (N-C), 36.0 (20-C), 35.6 (4-C), 35.4 (7-C), 30.6 (23-C), 30.2 (6-C), 27.4 (22-C), 26.7 (2-C), 26.6 (1-C), 25.6 (15-C), 24.3 (16-C), 22.6 (21-C), 18.8 (19-C), 11.7 (18-C); ESI-MS m/z: 445 (M+1)+.
N,N-dimethyl-3-O-benzyloximino-12-oxo-7-deoxycholic amide (18). Yield: 72%, θmp 148–149 °C. IR (KBr) ν/cm–1: 2970, 2868, 1707, 1625, 1450, 1397; 1H-NMR (CDCl3, 300 MHz): 0.882 (3H, d, J = 6.3, 21-CH3), 1.048 (3H, s, 18-CH3), 1.056 (3H, s, 19-CH3), 2.35–2.45 (1H, m, C11-βH), 2.553 (1H, t, J = 12.6, C11-αH), 2.947 (3H, s, N-CH3), 3.019 (3H, s, N-CH3), 5.052 (2H, s, O-CH2-Ph), 7.36–7.29 (5H, m, -C6H5); 13C-NMR (CDCl3, 75 MHz): 214.7 (12-C), 173.5 (24-C), 160.5 (3-C), [138.2, 128.3, 128.3, 127.9, 127.8, 127.6 (-C6H5)], 75.2 (O-C), 58.6 (14-C), 57.5 (17-C), 46.5 (9-C), 44.3 (13-C), 44.0 (5-C), 43.5 (10-C), 41.9 (8-C), 38.3 (11-C), 37.3 (N-C), 36.1 (N-C), 36.0 (20-C), 35.6 (2-C), 35.4 (7-C), 30.6 (23-C), 30.3 (6-C), 27.4 (1-C), 26.6 (22-C), 25.8 (4-C), 25.6 (15-C), 24.3 (16-C), 22.6 (21-C), 18.8 (19-C), 11.8 (18-C); ESI-MS m/z: 521 (M+1)+.






{kind=link}
{kind=link}
{kind=link}
{kind=link}