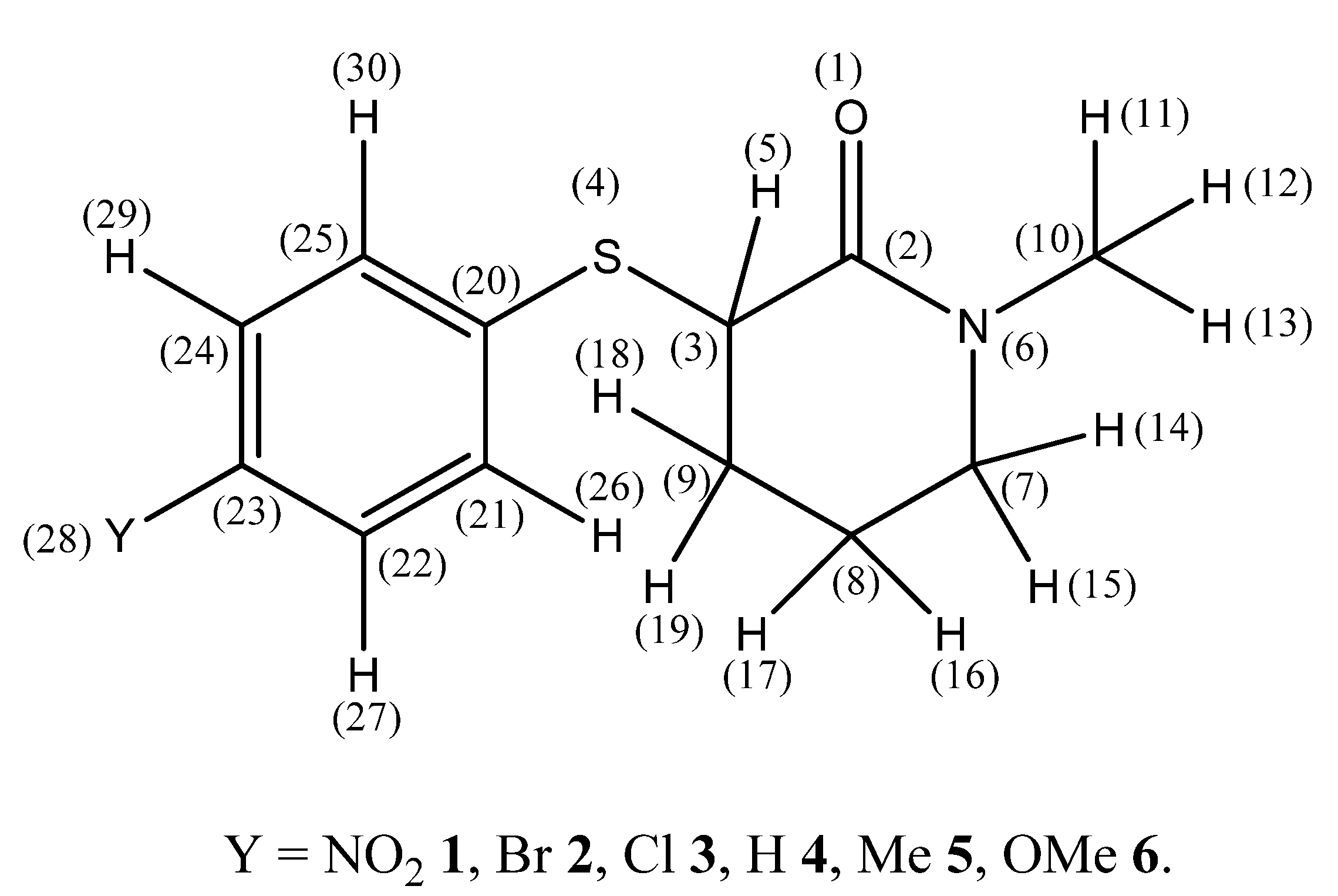
Spectroscopic and Theoretical Studies of Some 3-(4′-Substituted phenylsulfanyl)-1-methyl-2-piperidones


Abstract
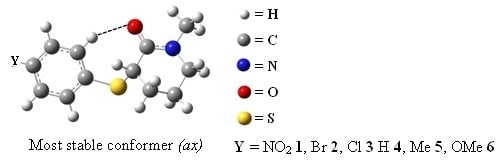
:
1. Introduction
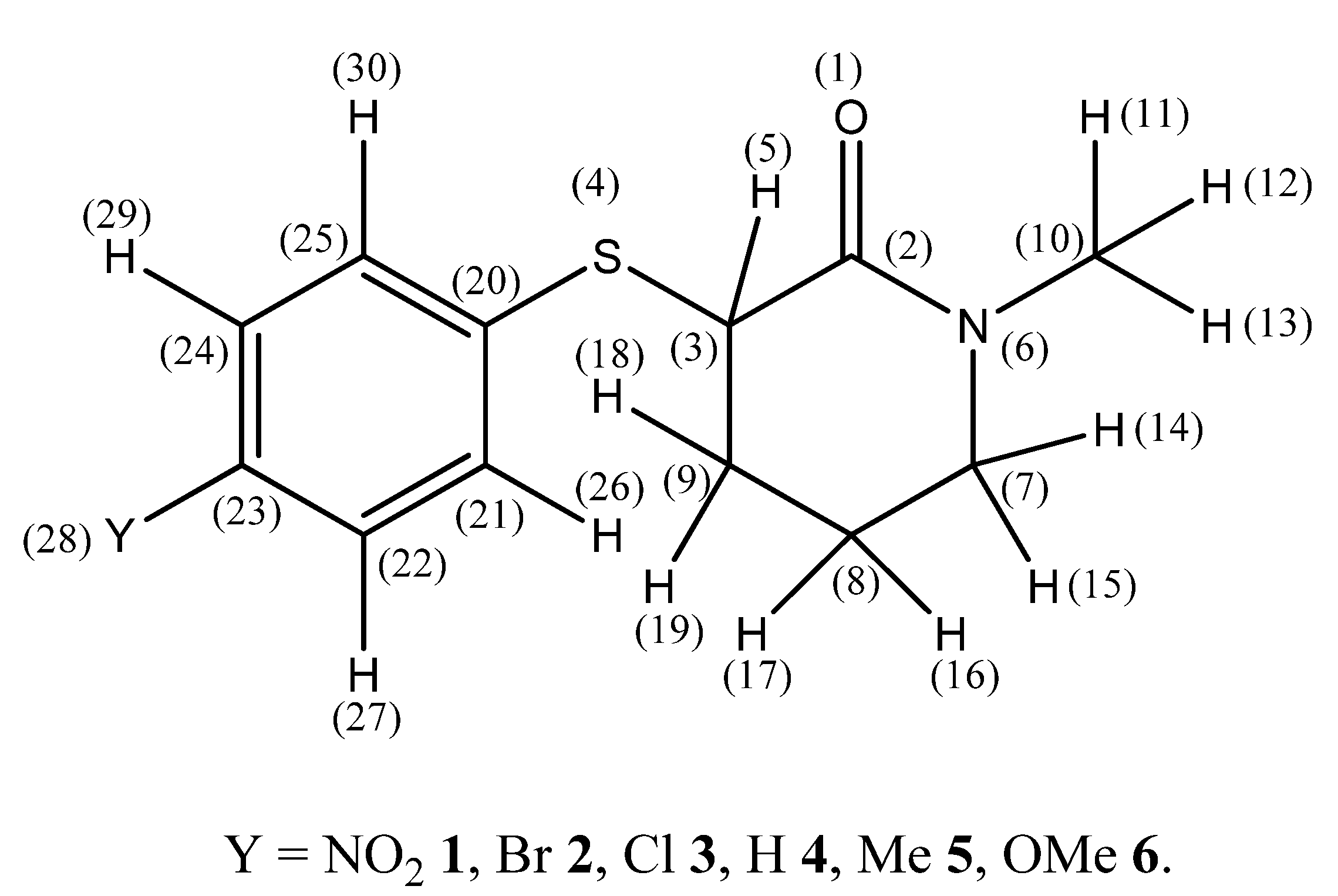
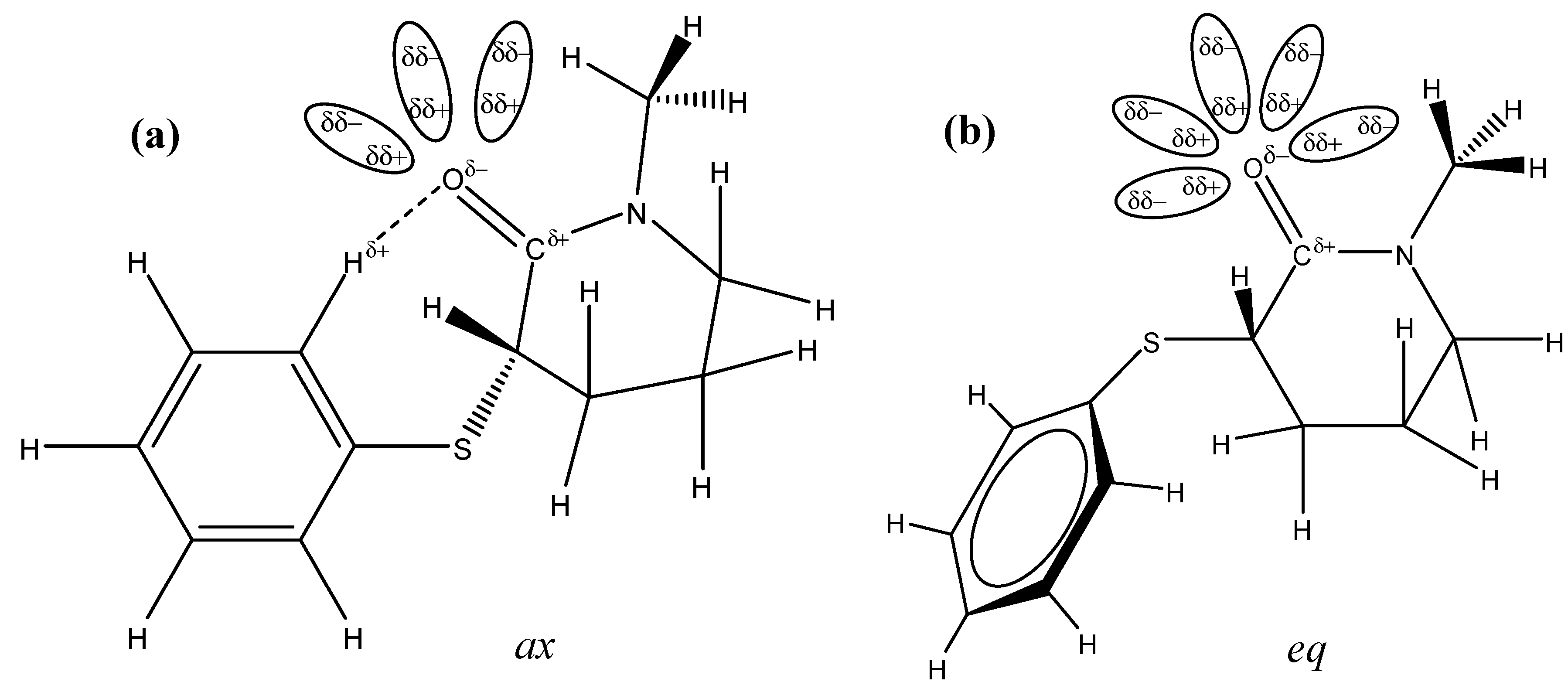
2. Results and Discussion



{kind=link}
{kind=link}
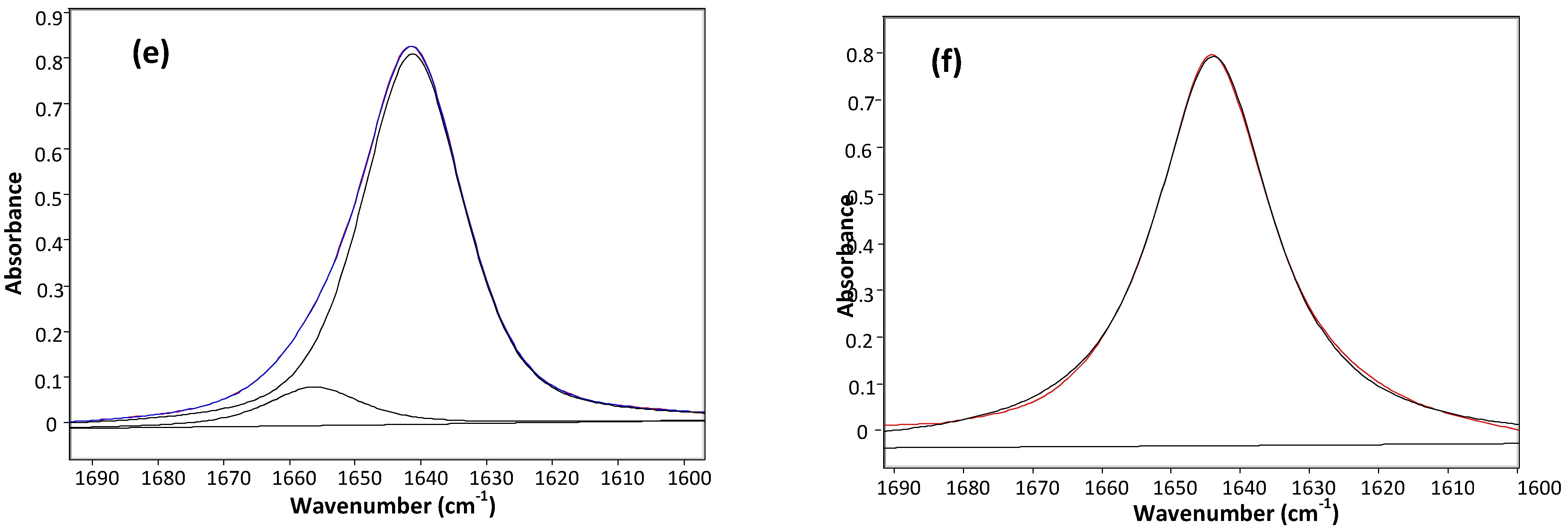{kind=link}
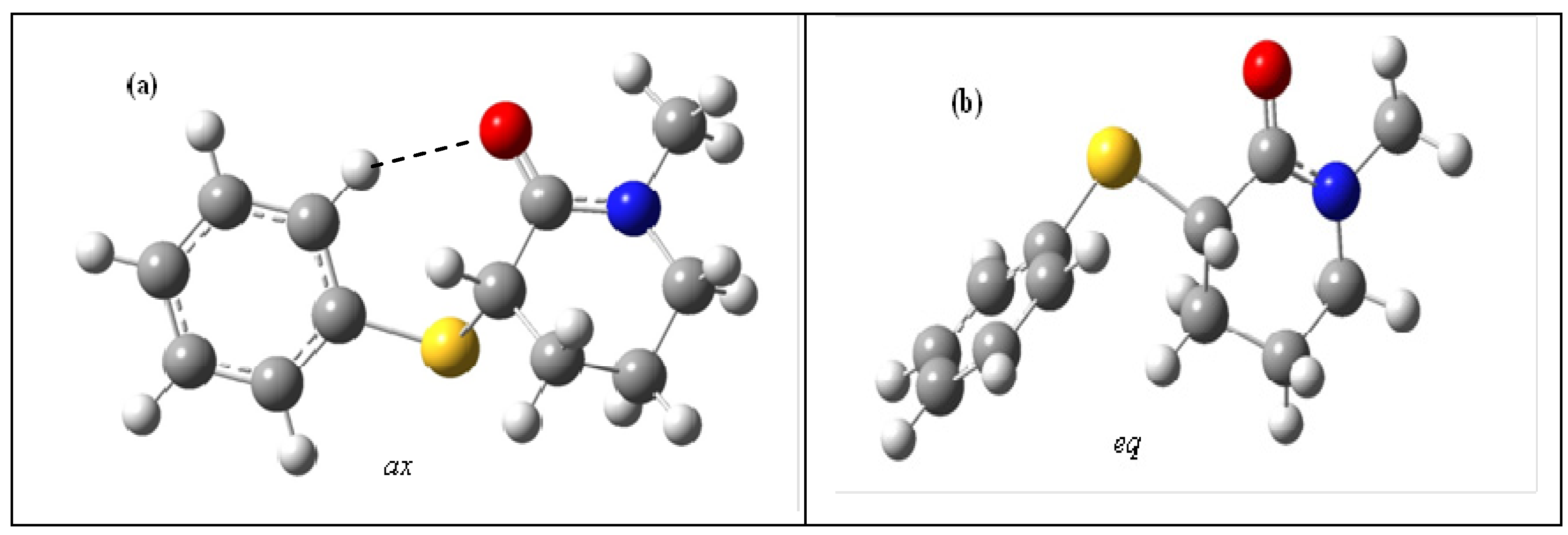{kind=link}
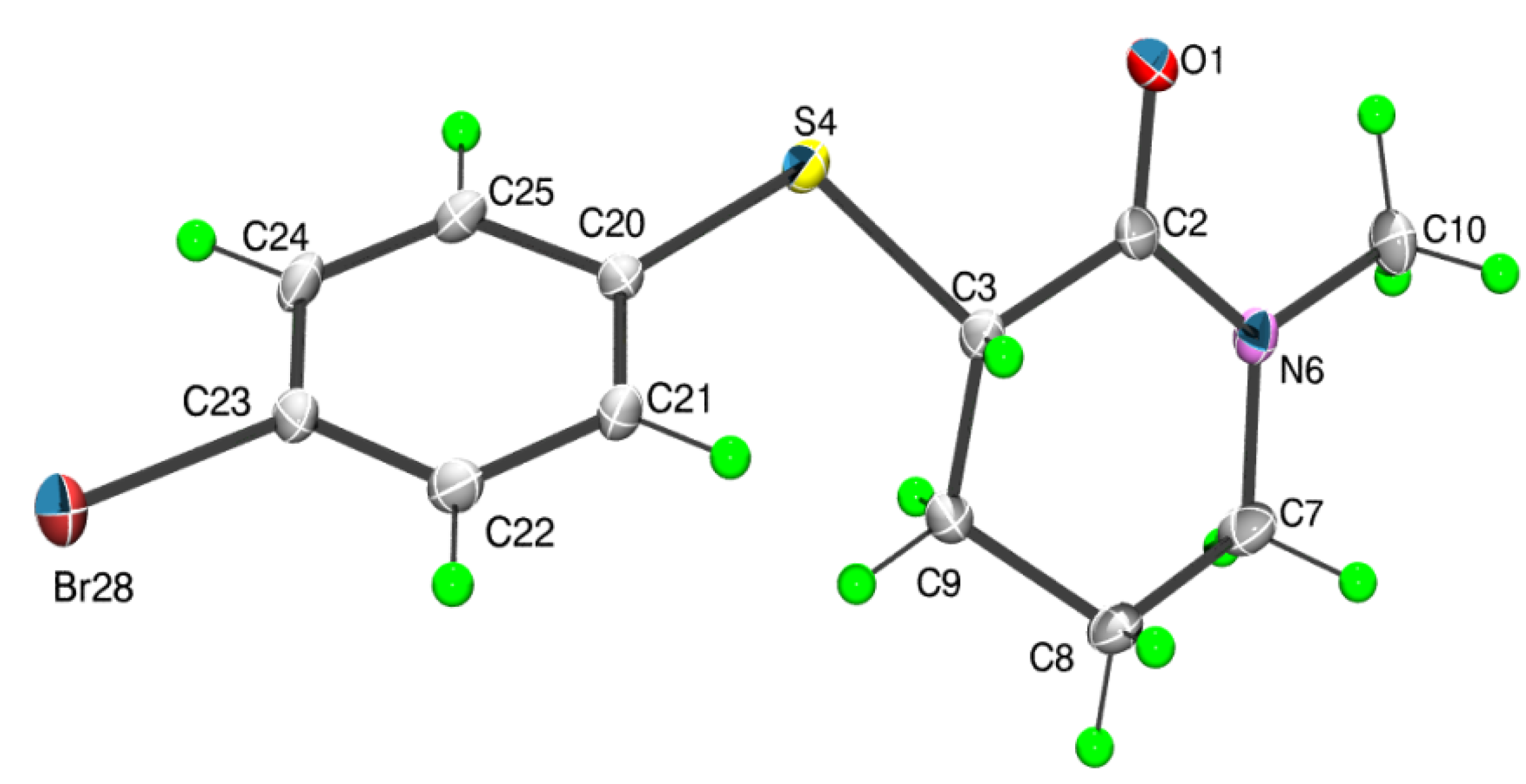{kind=link}
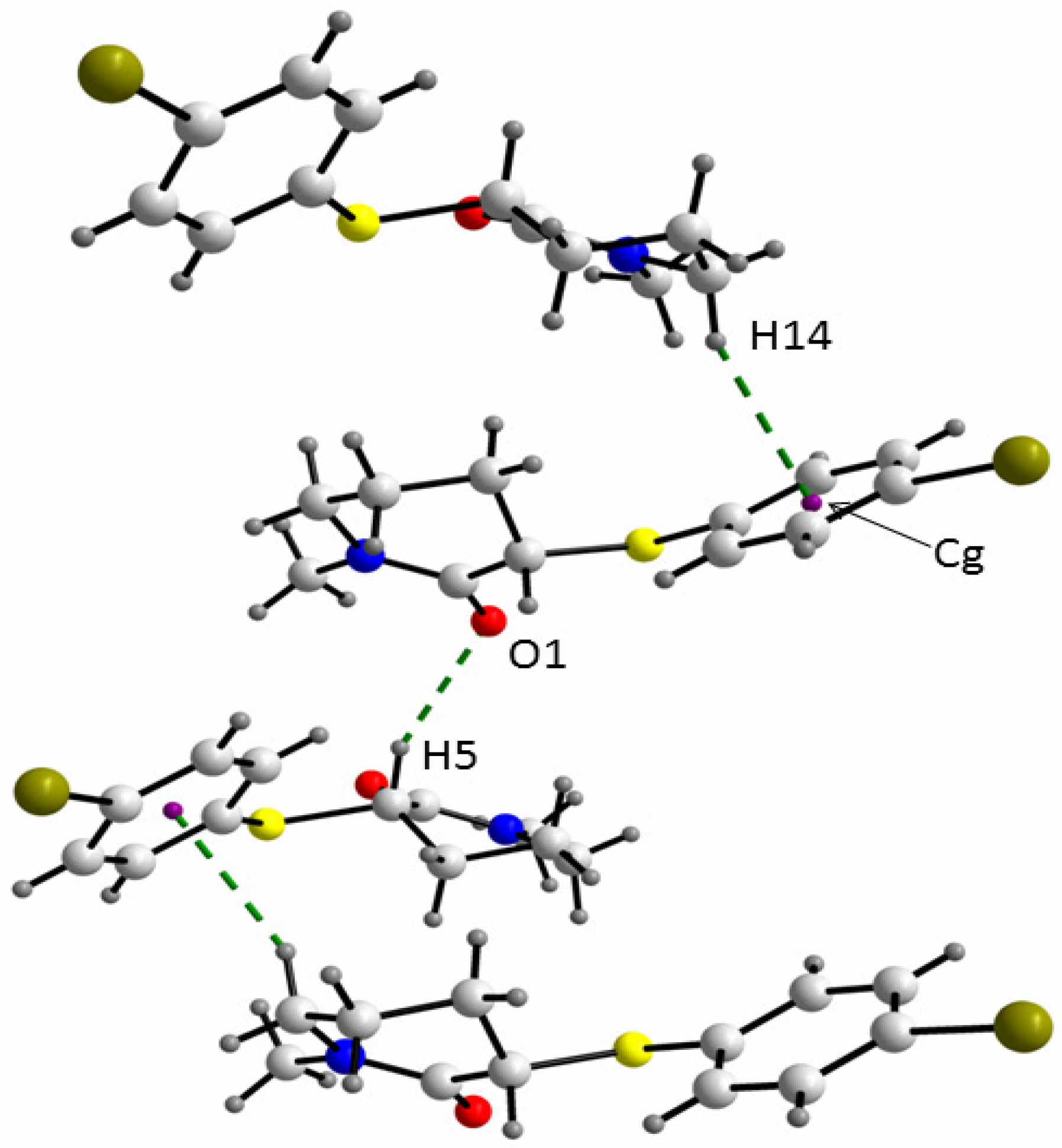{kind=link}
{kind=link}
{kind=link}
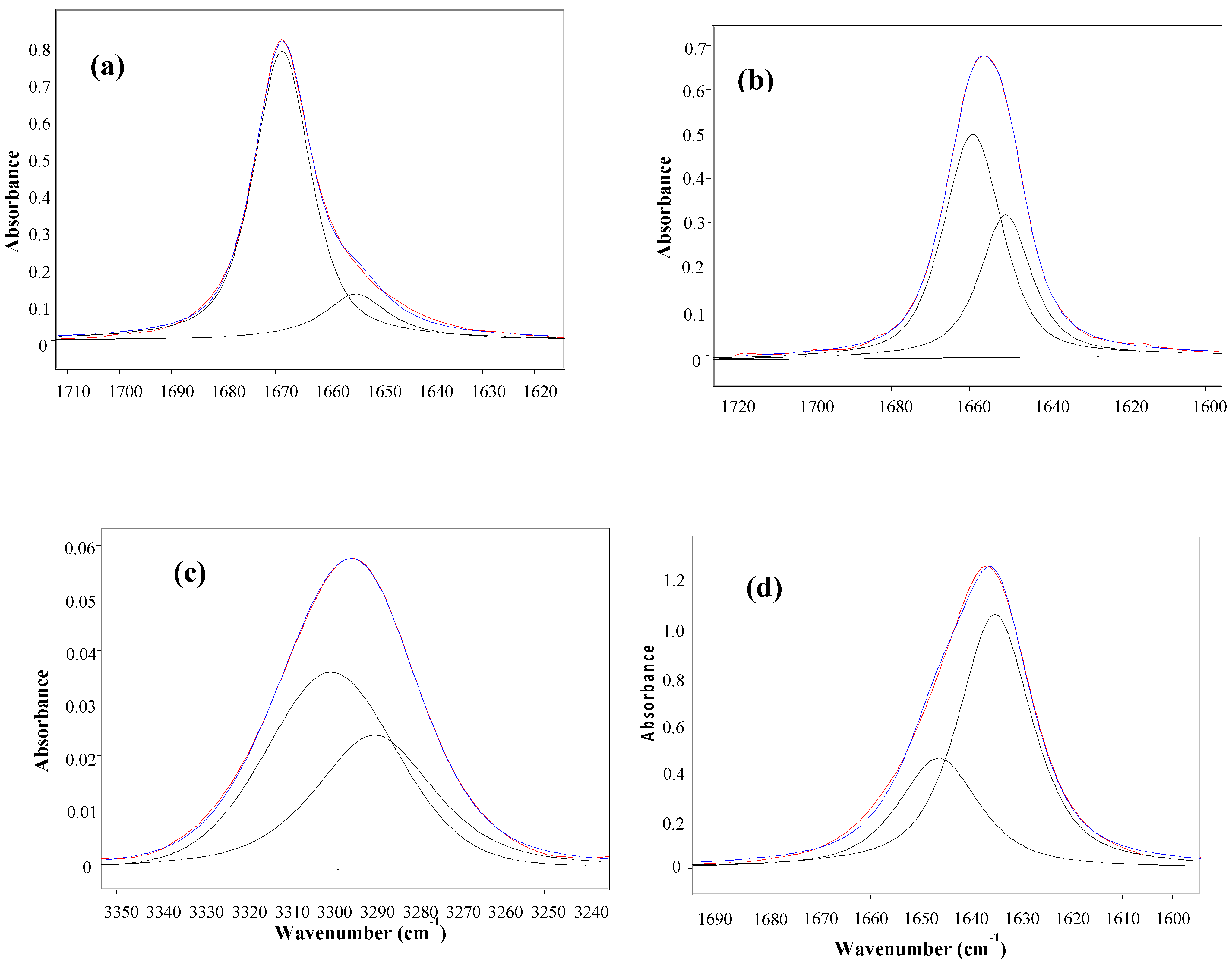
| Compound | Y | n-C6H14 | CCl4 | CHCl3 | CH2Cl2 | CH3CN | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ν | Pa | ν | Pa | νb | P | ν | P | ν | P | ν | P | |||
| 1 | NO2 | -c | - | 1660 | 61 | 3300 | 60 | 1648 | 43 | 1656 | 9 | - | - | |
| - | - | 1652 | 39 | 3289 | 40 | 1637 | 57 | 1641 | 91 | 1645 | 100 | |||
| 2 | Br | 1668 | 84 | 1658 | 68 | 3299 | 63 | 1645 | 33 | 1652 | 12 | - | - | |
| 1656 | 16 | 1650 | 32 | 3291 | 37 | 1635 | 67 | 1641 | 88 | 1644 | 100 | |||
| 3 | Cl | 1668 | 79 | 1660 | 45 | 3306 | 49 | 1647 | 30 | 1656 | 13 | - | - | |
| 1656 | 21 | 1652 | 55 | 3292 | 51 | 1635 | 70 | 1641 | 87 | 1644 | 100 | |||
| 4 | H | 1669 | 83 | 1659 | 62 | 3300 | 59 | 1647 | 32 | 1656 | 9 | - | - | |
| 1654 | 17 | 1651 | 38 | 3290 | 41 | 1635 | 68 | 1641 | 91 | 1644 | 100 | |||
| 5 | Me | 1668 | 72 | 1657 | 57 | 3300 | 57 | 1645 | 16 | 1652 | 12 | - | - | |
| 1659 | 28 | 1651 | 43 | 3286 | 43 | 1634 | 84 | 1640 | 88 | 1643 | 100 | |||
| 6 | OMe | 1668 | 78 | 1660 | 44 | 3301 | 41 | 1647 | 25 | 1652 | 15 | - | - | |
| 1657 | 22 | 1651 | 56 | 3288 | 59 | 1635 | 75 | 1639 | 85 | 1643 | 100 | |||
| Conf. a | E b | P c | μ | νCO | Torsion angles/º d | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| α | β | γ | δ | θ | θ’ | ω | ω' | ||||||
| gas | ax | 0 | 96.8 | 3.62 | 1713.4 | 82.0 | −101.3 | 48.2 | 2.13 | 167.9 | −154.3 | 23.6 | −50.8 |
| eq | 8.45 | 3.2 | 5.39 | 1720.1 | 30.4 | −176.5 | 73.2 | −3.90 | −170.8 | 157.1 | −23.7 | 50.8 | |
| C7H16 | ax | 0 | 84.4 | 4.08 | 1696.7 | 82.0 | −101.9 | 48.4 | 2.06 | 168.2 | −154.5 | 23.3 | −50.7 |
| eq | 4.20 | 15.6 | 6.21 | 1699.0 | 31.6 | −177.4 | 72.9 | −3.74 | −172.2 | 158.2 | −22.8 | 50.3 | |
| CCl4 | ax | 0 | 80.0 | 4.16 | 1694.2 | 81.9 | −102.1 | 49.1 | 2.06 | 168.1 | −154.5 | 23.4 | −50.7 |
| eq | 3.43 | 20.0 | 6.37 | 1694.3 | 31.1 | −176.2 | 73.3 | −3.75 | −171.7 | 157.7 | −23.1 | 50.6 | |
| CHCl3 | ax | 0.17 | 48.3 | 4.61 | 1676.8 | 81.9 | −102.4 | 47.7 | 2.11 | 168.3 | −154.5 | 23.2 | −50.7 |
| eq | 0 | 51.7 | 7.20 | 1673.0 | 33.4 | −178.1 | 72.1 | −3.28 | −174.9 | 159.9 | −20.6 | 49.7 | |
| CH2Cl2 | ax | 2.01 | 30.8 | 4.89 | 1668.9 | 80.6 | −104.3 | 48.5 | 2.57 | 169.3 | −155.7 | 23.1 | −50.1 |
| eq | 0 | 69.2 | 7.62 | 1663.8 | 32.7 | −178.4 | 71.4 | −3.20 | −174.3 | 159.1 | −20.8 | 50.9 | |
| CH3CN | ax | 3.93 | 16.9 | 5.18 | 1659.5 | 80.0 | −105.6 | 48.9 | 2.60 | 169.8 | −156.2 | 22.6 | −49.8 |
| eq | 0 | 83.1 | 8.10 | 1652.3 | 32.1 | −178.4 | 70.7 | −3.11 | −173.2 | 158.5 | −21.7 | 50.2 | |
| X-ray e | - | - | - | - | - | 31.0 (3) | −176.2 (2) | −155.5(2) | −4.1 (4) | −177.6(3) | 157.2(3) | −14.3(4) | 53.1 (3) |
| Conf. | O(1) | C(2) | S(4) | N(6) | H(26) | H(30) | H(5) | H(11) | H(16) | H(17) |
|---|---|---|---|---|---|---|---|---|---|---|
| ax | −0.557 | 0.525 | −0.321 | −0.187 | 0.131 | 0.102 | 0.048 | 0.088 | 0.028 | 0.065 |
| eq | −0.537 | 0.605 | −0.320 | −0.311 | 0.103 | 0.061 | 0.027 | 0.058 | 0.030 | 0.045 |
| Conf. a | O[1]...S[4] b | Δl c | O[1]...H[11] d | Δl | O[1]...H[26] d | Δl | O[1]...H[5] d | Δl |
|---|---|---|---|---|---|---|---|---|
| ax | 3.37 | +0.05 | 2.28 | −0.44 | 2.35 | −0.37 | 2.52 | −0.20 |
| eq | 2.89 | −0.43 | 2.27 | −0.45 | 4.75 | +2.03 | 2.83 | +0.11 |

| Orbitals | ax | eq |
|---|---|---|
| LPN6→π*C2=O1 | 65.3 | 50.6 |
| LPO1→σ*C2-N6 | 25.4 | 25.5 |
| LPO1→σ*C2-C3 | 18.9 | 20.3 |
| LPO1→σ*C21-H26 | 3.0 | - |
| LPO1→σ*C10-H11 | 1.0 | 1.2 |
| LPO1→ σ*S4-C20 | - a | 0.9 |
| LPS4→ π*C2=O1 | 2.0 | - |
| LPS4→ π *C20-C21 | 8.3 | 2.7 |
| σC3-S4→π*C2=O1 | 4.2 | 0.7 |
| σC3-S4→σ*C2-N6 | - | 3.4 |
| πC2=O1→σ*C3-S4 | 1.7 | 0.6 |
| π*C2=O1→σ*C3-S4 | 5.5 | 0.8 |
| ∑ Eb | 135.3 | 106.7 |


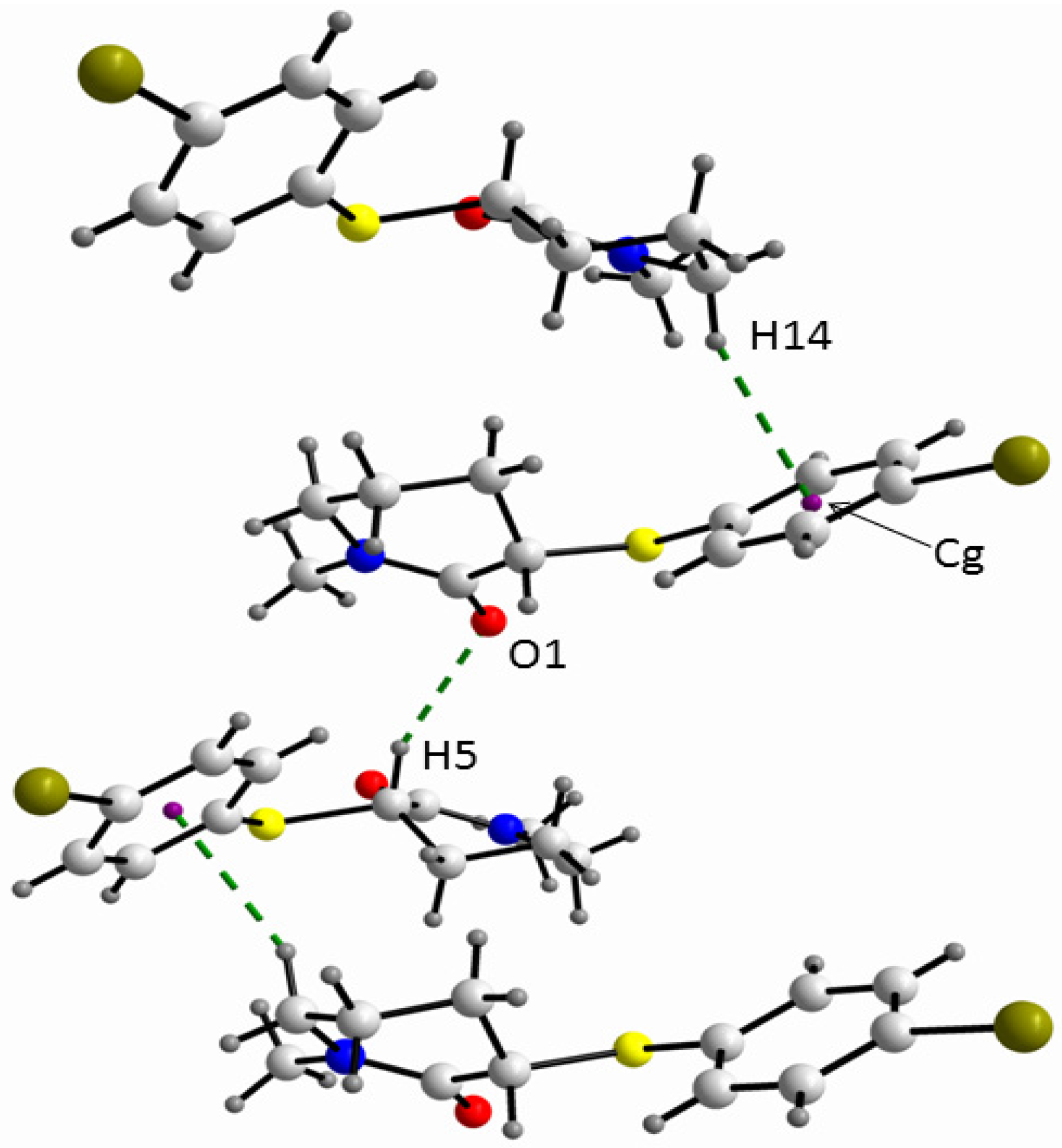
3. Experimental
3.1. Materials
3.2. IR Measurements
| Compd. | Y | Mp (°C) | 1H- and 13C-NMR a |
|---|---|---|---|
| 1 | NO2 | 77–78 | 1H-NMR: 8.14–8.12 (m, 2H), 7.64–7.63 (m, 2H), 4.06 (t, 1H, J = 5.5 Hz), 3.41–3.32 (m, 2H), 2.99 (s, 3H), 2.30–2.27 (m, 1H), 2.09–2.03 (m, 2H), 1.92–1.88 (m, 1H). 13C-NMR: 167.83, 146.07, 145.77, 128.67, 123.86, 49.82, 46.90, 35.36, 28.68, 20.76. |
| 2 | Br | 64–64 | 1H-NMR: 7.44–7.40 (m, 4H), 3.82 (t, 1H, J = 5.7Hz), 3.33–3.26 (m. 2H), 2.96 (s, 3H), 2.11–2.04 (m, 2H), 1.99–1.94 (m, 1H), 1.82–1.77 (m, 1H). 13C-NMR: 168.36, 134.27, 134.07, 132.19, 121.82, 50.06, 49.02, 35.47, 28.83, 20.59. |
| 3 | Cl | b | 1H-NMR: 7.53–7.51 (m, 2H), 7.30–7.29 (m, 2H), 3.84 (t, 1H, J = 5.7Hz), 3.36–3.28 (m, 2H), 2.99 (s, 3H), 2.17–2.08 (m, 2H), 2.02–1.96 (m, 1H), 1.83–1.80 (m, 1H) 13C-NMR: 168.19, 133.77, 133.64, 133.33, 129.06, 49.86, 48.96, 35.27, 28.62, 20.38. |
| 4 | H | 38–39 | 1H-NMR: 7.56–7.54 (m, 2H), 7.32–7.28 (m, 2H), 7.26–7.24 (m, 1H), 3.88 (t, 1H, J = 5.0 Hz), 3.30–3.24 (m, 2H), 2.97 (s, 3H), 2.12–2.07 (m, 2H), 1.98–1.94 (m, 1H), 1.77–1.74 (m, 1H). 13C-NMR: 168.47, 134.96, 132.61, 129.12, 127.61, 50.08, 49.03, 35.44, 28.75, 20.46. |
| 5 | CH3 | 44–45 | 1H-NMR: 7.47–7.46 (m, 2H), 7.15–7.13 (m, 2H), 3.83 (t, 1H, J = 5,5Hz), 3.34–3.25 (m, 2H), 2.98 (s, 3H), 2.35 (s, 3H), 2.13–2.05 (m, 2H), 1.96-1.92 (m, 1H), 1.80–1.75 (m, 1H) 13C-NMR: 168.37, 137.77, 133.25, 130.79, 129.71, 49.89, 49.21, 35.23, 28.35, 21.12, 20.12. |
| 6 | OCH3 | b | 1H-NMR: δ (ppm): 7.51–7.49 (m, 2H), 6.85–6.83 (m, 2H), 3.79 (s, 3H), 3,71 (t, 1H, J = 6,0Hz), 3.25–3,23 (m, 2H), 2.94 (s, 3H), 2.10–2,01 (m, 2H), 1.94–1,89 (m, 1H), 1.75–1,71 (m, 1H). 13C-NMR: 168.36, 159.85, 135.93, 124.61, 114.50, 55.31, 49.89, 49.80, 35.20, 28.28, 20.12 |
| Compd | Y | Molecular formula | Analysis (%) | |||
|---|---|---|---|---|---|---|
| C | H | N | ||||
| 1 | NO2 | C12H14N2O3S | Calc.Found | 54.1254.34 | 5.305.23 | 10.5210.46 |
| 2 | Br | C12H14BrNOS | Calc. Found | 48.0147.91 | 4.704.77 | 4.674.79 |
| 3 | Cl | C12H14ClNOS | Calc. Found | 56.3556.26 | 5.525.22 | 5.485.27 |
| 4 | H | C12H15NOS | Calc. Found | 65.1265.12 | 6.836.56 | 6.336.46 |
| 5 | CH3 | C13H17NOS | Calc. Found | 66.3466.23 | 7.287.20 | 5.956.01 |
| 6 | OCH3 | C13H17NO2S | Calc. Found | 62.1262.13 | 6.826.81 | 5.575.55 |
3.3. X-ray Measurements
3.3.1. Data Collection and Processing
3.4. Theoretical Calculations
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References and Notes
- Vinhato, E.; Olivato, P.R.; Zukerman-Schpector, J.; Dal Colle, M. Conformational Analysis of some N,N-diethyl-2-[(4′-substituted) phenylthio] acetamides. Spectrochim. Acta Part A 2013, submitted. [Google Scholar]
- Warshel, A.; Levitt, M.; Lifson, S. Consistent Force Field for Calculation of Vibrational Spectra and Conformations of Some Amides and Lactam Rings. J. Mol. Spectrosc. 1970, 33, 84–99. [Google Scholar] [CrossRef]
- Treschanke, L.; Rademacher, P. Electronic structure and conformational properties of the amide linkage: Part 1. Geometric and electronic structure of lactams as determined by MNDO calculations. J. Mol. Struct. (Theochem) 1985, 122, 35–45. [Google Scholar] [CrossRef]
- Kuze, N.; Funahashi, H.; Ogawa, M.; Tariji, H.; Ohta, Y.; Usami, T.; Sakaizumi, T.; Ohashi, O. Microwave Spectrum and Molecular Conformation of delta-Valerolactam. J. Mol. Spectrosc. 1999, 198, 381–386. [Google Scholar] [CrossRef]
- Boudreault, N.; Ball, R.G.; Bayly, C.; Bernstein, M.A.; Leblanc, Y. Conformational analysis of δ-lactams. Tetrahedron 1994, 50, 7947–7968. [Google Scholar] [CrossRef]
- Romers, C.; Rutten, E.W.M.; van Driel, C.A.A.; Sanders, W.W. a-chloro-δ-valerolactam. Acta Cryst. 1967, 22, 893–899. [Google Scholar] [CrossRef]
- Zukerman-Schpector, J.; Olivato, P.R.; Cerqueira, C.R., Jr.; Vinhato, E.; Tiekink, E.R.T. 1-Methyl-3-phenylsulfonyl-2-piperidone. Acta Cryst. 2008, E64, o835–o836. [Google Scholar]
- Olivato, P.R.; Santos, J.M.M.; Cerqueira, C.R., Jr.; Vinhato, E.; Zukerman-Schpector, J.; Ng, S.W.; Tiekink, E.R.T.; Dal Colle, M. Conformational preferences for some 3-(4´-substituted phenylsulfonyl)-1-methyl-2-piperidones through spectroscopic and theoretical studies. J. Mol. Struct. 2012, 1028, 97–106. [Google Scholar]
- Lide, D.R. (Ed.) CRC Handbook of Chemistry and Physics; CRC Press: Boca Raton, FL, USA, 2005.
- Colthup, N.B.; Daly, L.H.; Wiberley, S.E. Introduction to Infrared and Raman Spectroscopy, 3rd ed.; Academic Press: New York, NY, USA, 1990; pp. 27–33. [Google Scholar]
- Bellamy, L.J. Advances in Infrared Group Frequencies; Chapman and Hall: London, UK, 1975; pp. 141–142. [Google Scholar]
- Gaset, A.; Lafaille, A.; Verdier, A.; Lattes, A. Infrared spectra of α-aminoketones: conformational study and evidence of an enol form. Bull. Soc. Chim. Fr. 1968, 4108–4112. [Google Scholar]
- Jones, D.; Modelli, A.; Olivato, P.R.; Dal Colle, M.; De Palo, M.; Distefano, G. Ab initio and electron spectroscopy study of carbonyl derivatives. J. Chem. Soc. Perkin Trans. 2 1994, 7, 1651–1656. [Google Scholar]
- Mujika, J.I.; Matxain, J.M.; Eriksson, L.A.; Lopez, X. Resonance structures of the amide bond: The advantages of planarity. Chem. Eur. J. 2006, 12, 7215–7224. [Google Scholar] [CrossRef]
- MP2/6-31G+(d,p) calculations for 4 in the gas phase are presented in Table S1 (Supplementary information).
- Single point PCM calculations at the MP2/6-31G+(d,p) level for 4 in solvents of increasing relative permissivity are presented in Table S2.
- Mulliken charges are presented in Table S3.
- Katritzky, A.R.; Topsom, R. Infrared intensities: A guide to intramolecular interactions in conjugated systems. Chem. Rev. 1977, 77, 639–658. [Google Scholar] [CrossRef]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhlod, F. NBO Version 3.1 (Implemented in Gaussian 03 package of Programs). Available online: http://archive.osc.edu/supercomputing/software/apps/gaussian03.shtml (accessed on 25 June 2013).
- Occupancies for donor and acceptor orbital for the ax and eq conformers of 4 are presented in TableS4.
- Levin, C.C.; Hoffmann, R.; Hehre, W.J.; Hudec, J. Orbital Interaction in Amino-ketones. J. Chem. Soc. Perkin 2 1973, 210–220. [Google Scholar]
- Eisenstein, O.; Ahn, N.T.; Devaquet, J.A.; Cantacuzene, J.; Salem, L. Lone pairs in organic molecules: Energetic and orientational non-equivalence: Stereochemical consequences. Tetrahedron 1974, 30, 1717–1723. [Google Scholar] [CrossRef]
- Zoretic, P.A.; Soja, P. Sulfenylation and selenenylation of lactams. J. Org. Chem. 1976, 41, 3587–3589. [Google Scholar] [CrossRef]
- Hashmat, A.M.; McDermott, M. Oxidation of thiols to disulfides with molecular bromine on hydrated silica gel support. Tetrahedron Lett. 2002, 43, 6271–6273. [Google Scholar] [CrossRef]
- Brummond, K.M.; Gesenberg, K.D. Alpha-chlorination of ketones using p-toluenesulfonyl chloride. Tetrahedron Lett. 1999, 40, 2231–2234. [Google Scholar] [CrossRef]
- Grams/32 curve-fitting program, version 4.04 Level II; Galactic Industries Corporation: Salem, NH, USA, 1991–1998.
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.J. Completion and Refinement of Crystal Structures with SIR92. J. Appl. Crystallogr. 1993, 26, 343–350. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Farrugia, L.J. ORTEP-3 for Windows - A version of ORTEP-III with a Graphical User Interface (GUI) J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON, A Multipurpose Crystallographic Tool. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- ChemAxon, Marvin 5.1.10. Available online: http://www.chemaxon.com (accessed on 25 June 2013).
- Brandenburg, K.; Putz, H. Diamond—Crystal and Molecular Structure Visualization; Crystal Impact: Bonn, Germany, 2012. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03; Revision E.01; Gaussian Inc.: Wallingford, CT., USA, 2004. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. 1988, A 38, 3098–3100. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar]
- Hehre, W.J.; Radom, L.; Schleyer, P.V.R.; Pople, J.A. Ab Initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J. Chem. Phys. 2002, 117, 43–54. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comp. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–6 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Olivato, P.R.; Santos, J.M.M.; Contieri, B.; Cerqueira, C.R., Jr.; Rodrigues, D.N.S.; Vinhato, E.; Zukerman-Schpector, J.; Colle, M.D. Spectroscopic and Theoretical Studies of Some 3-(4′-Substituted phenylsulfanyl)-1-methyl-2-piperidones. Molecules 2013, 18, 7492-7509. https://doi.org/10.3390/molecules18077492
Olivato PR, Santos JMM, Contieri B, Cerqueira CR Jr., Rodrigues DNS, Vinhato E, Zukerman-Schpector J, Colle MD. Spectroscopic and Theoretical Studies of Some 3-(4′-Substituted phenylsulfanyl)-1-methyl-2-piperidones. Molecules. 2013; 18(7):7492-7509. https://doi.org/10.3390/molecules18077492
Chicago/Turabian StyleOlivato, Paulo R., Jean M. M. Santos, Bruna Contieri, Carlos R. Cerqueira, Jr., Daniel N. S. Rodrigues, Elisângela Vinhato, Julio Zukerman-Schpector, and Maurizio Dal Colle. 2013. "Spectroscopic and Theoretical Studies of Some 3-(4′-Substituted phenylsulfanyl)-1-methyl-2-piperidones" Molecules 18, no. 7: 7492-7509. https://doi.org/10.3390/molecules18077492