Elaboration of Stable and Antibody Functionalized Positively Charged Colloids by Polyelectrolyte Complexation between Chitosan and Hyaluronic Acid

Abstract
:1. Introduction
2. Results and Discussions
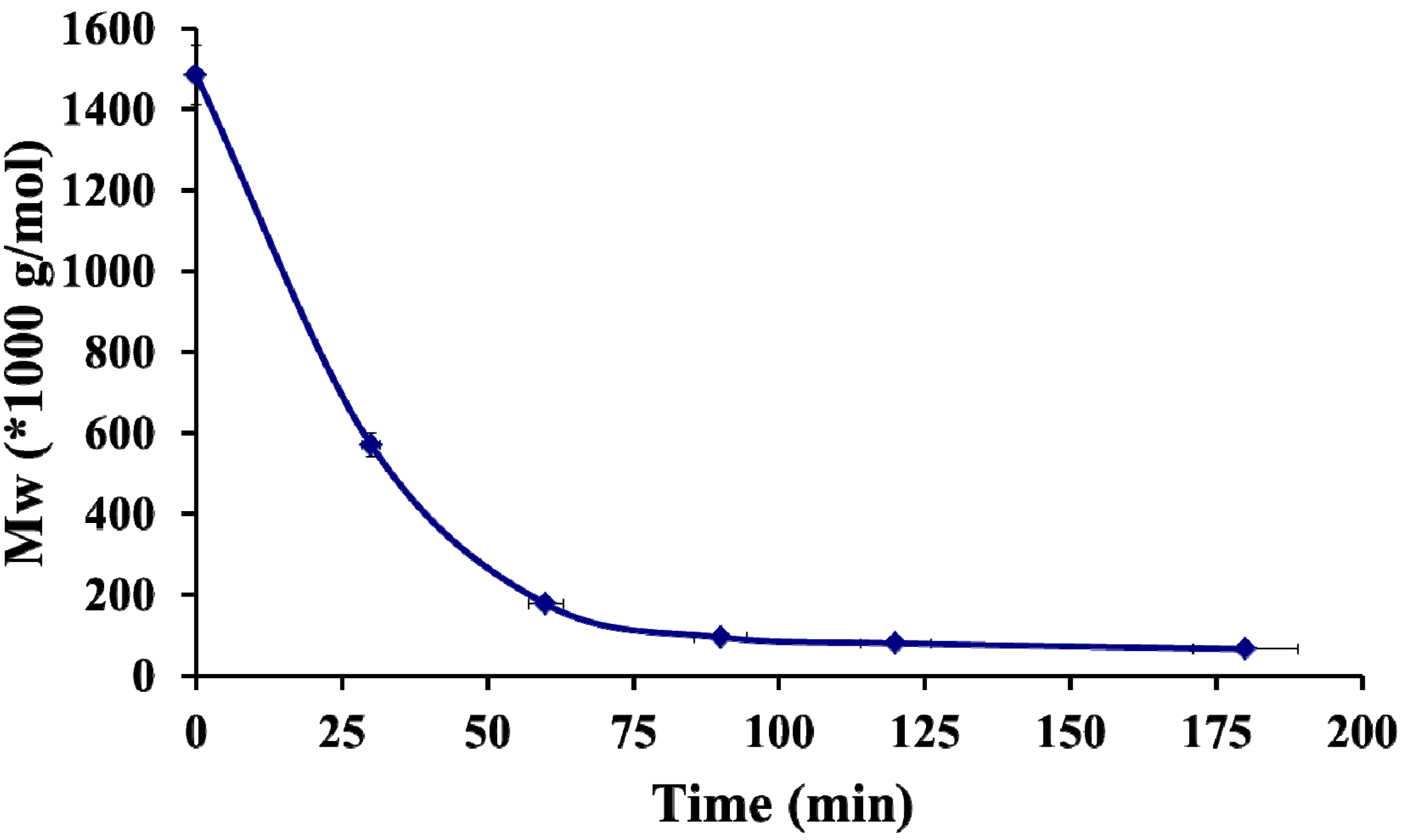
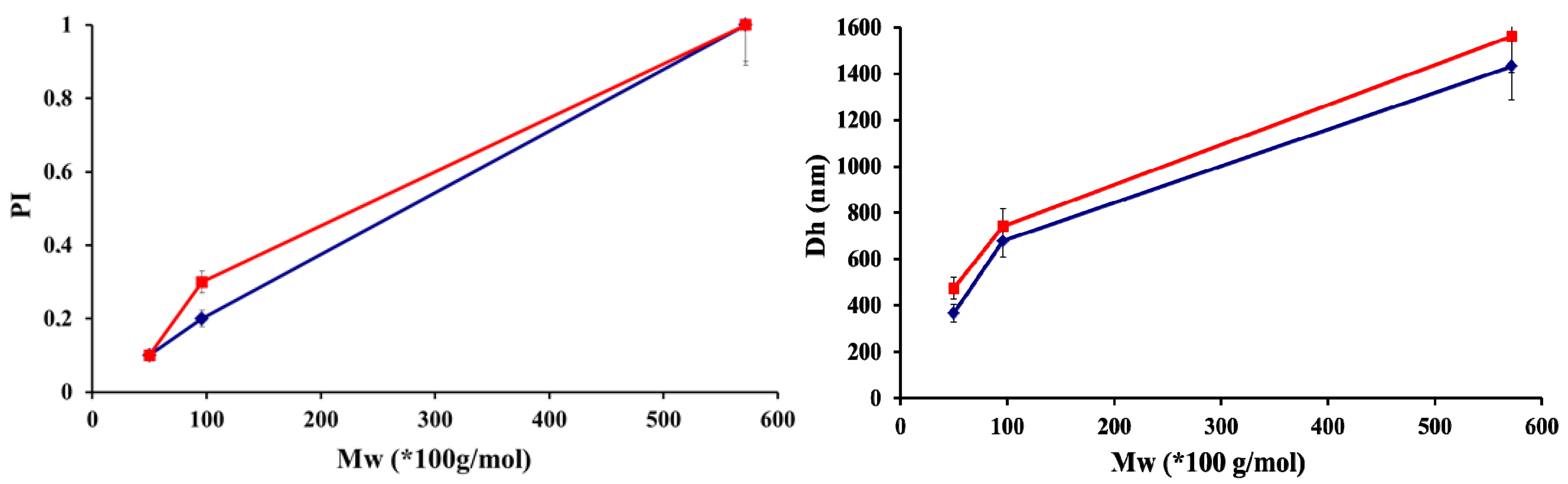
2.1. Physico-Chemical Properties of Chitosan and Hayluronic Acid

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
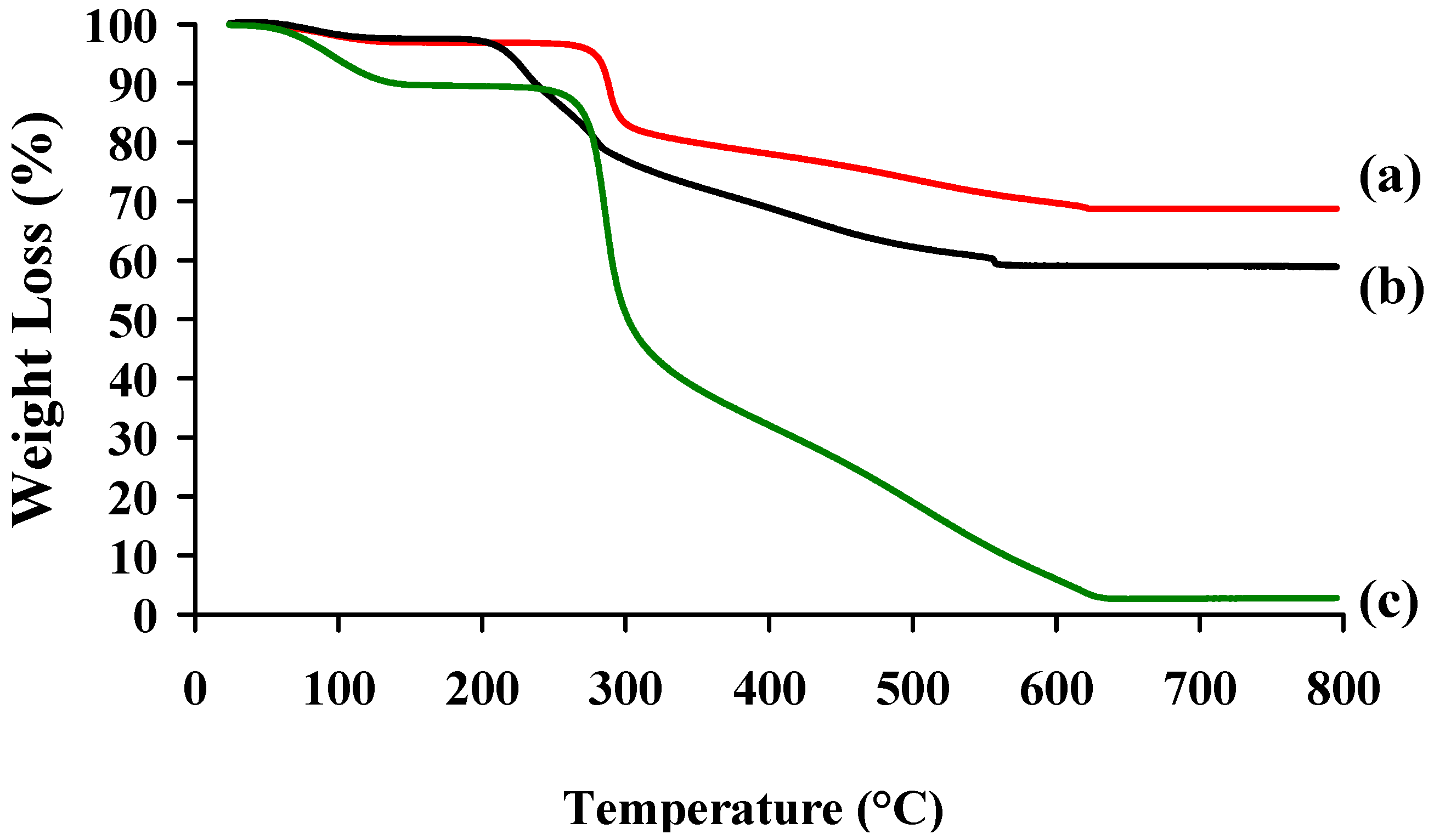{kind=link}
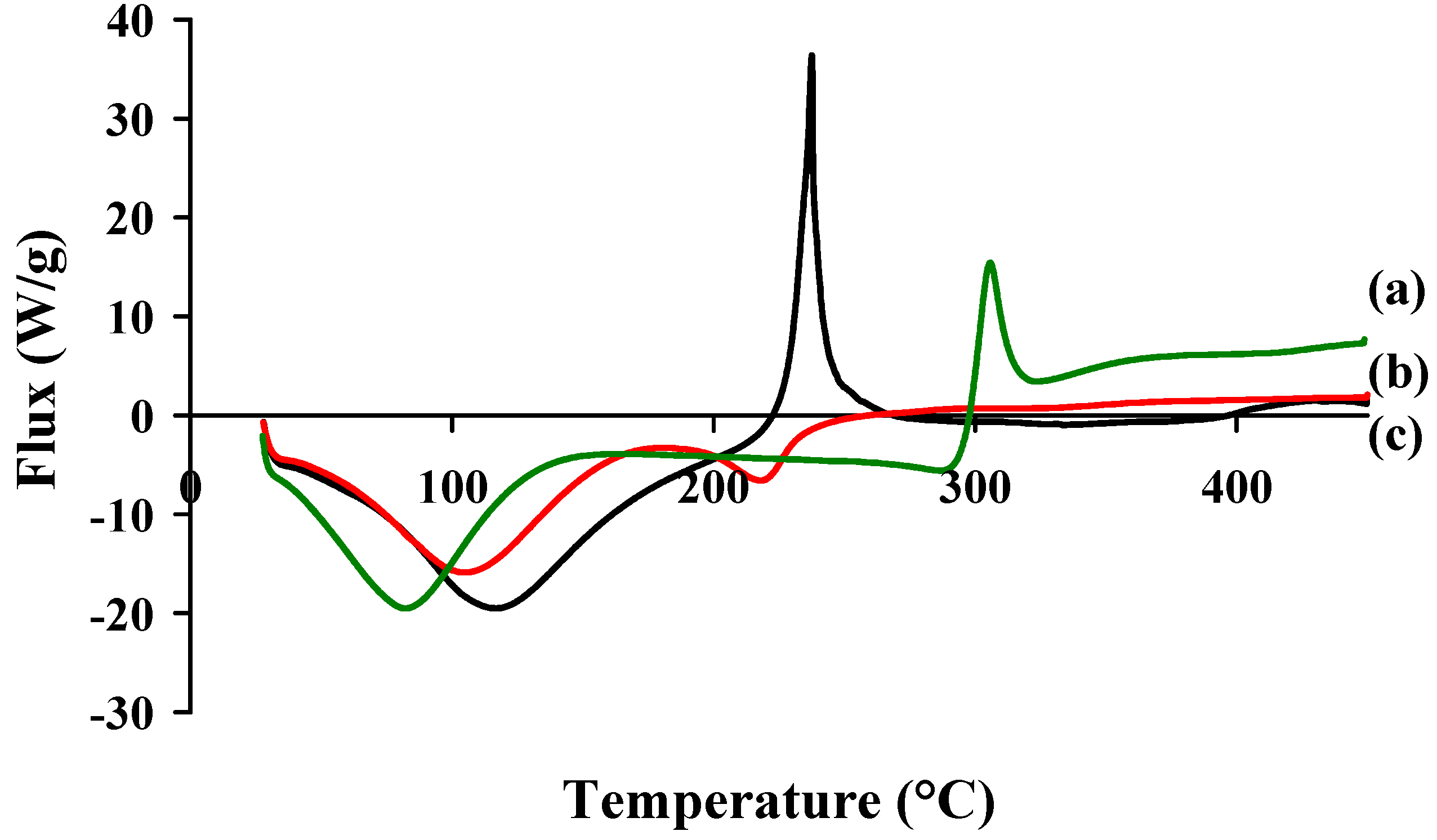{kind=link}
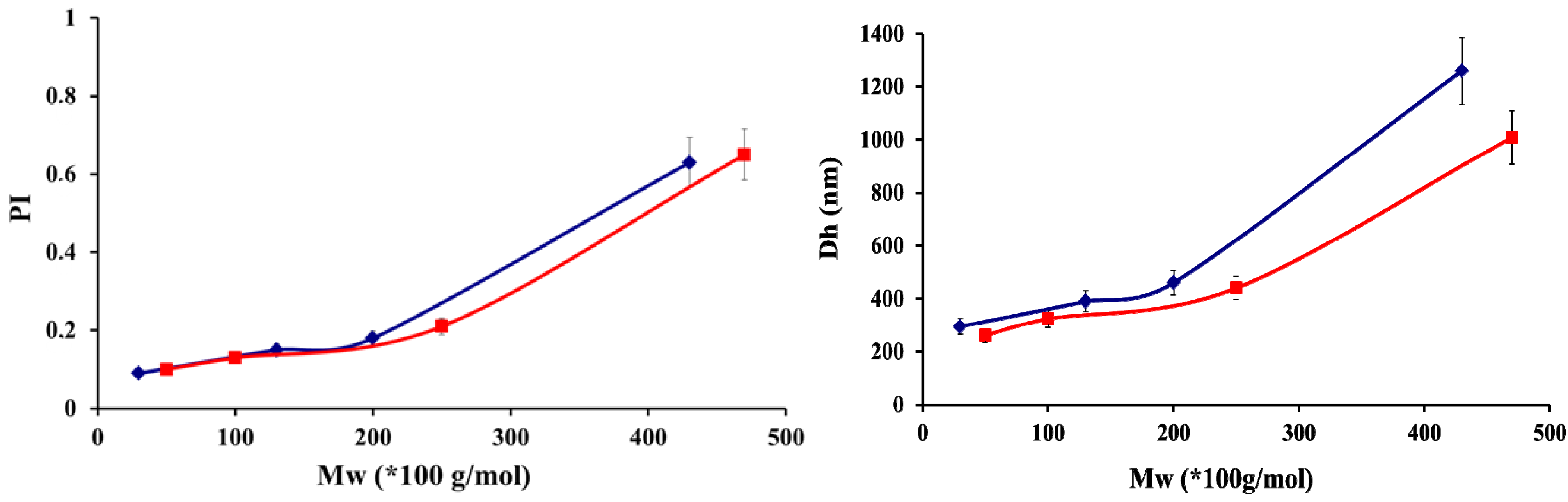
| Chitosan | ||||||||
|---|---|---|---|---|---|---|---|---|
| Low Molar Masses | Medium Molar Masses | High Molar Masses | ||||||
| Mw (g/mol) | DA (%) | PI | Mw (g/mol) | DA (%) | PI | Mw (g/mol) | DA (%) | PI |
| 50 × 103 | 5 | 1.56 | 100 × 103 | 5 | 1.67 | 470 × 103 | 5 | 1.72 |
| 30 × 103 | 48 | 1.61 | 130 × 103 | 48 | 1.47 | 430 × 103 | 48 | 1.52 |
| 250 × 103 | 5 | 1.43 | ||||||
| 200 × 103 | 48 | 1.54 | ||||||
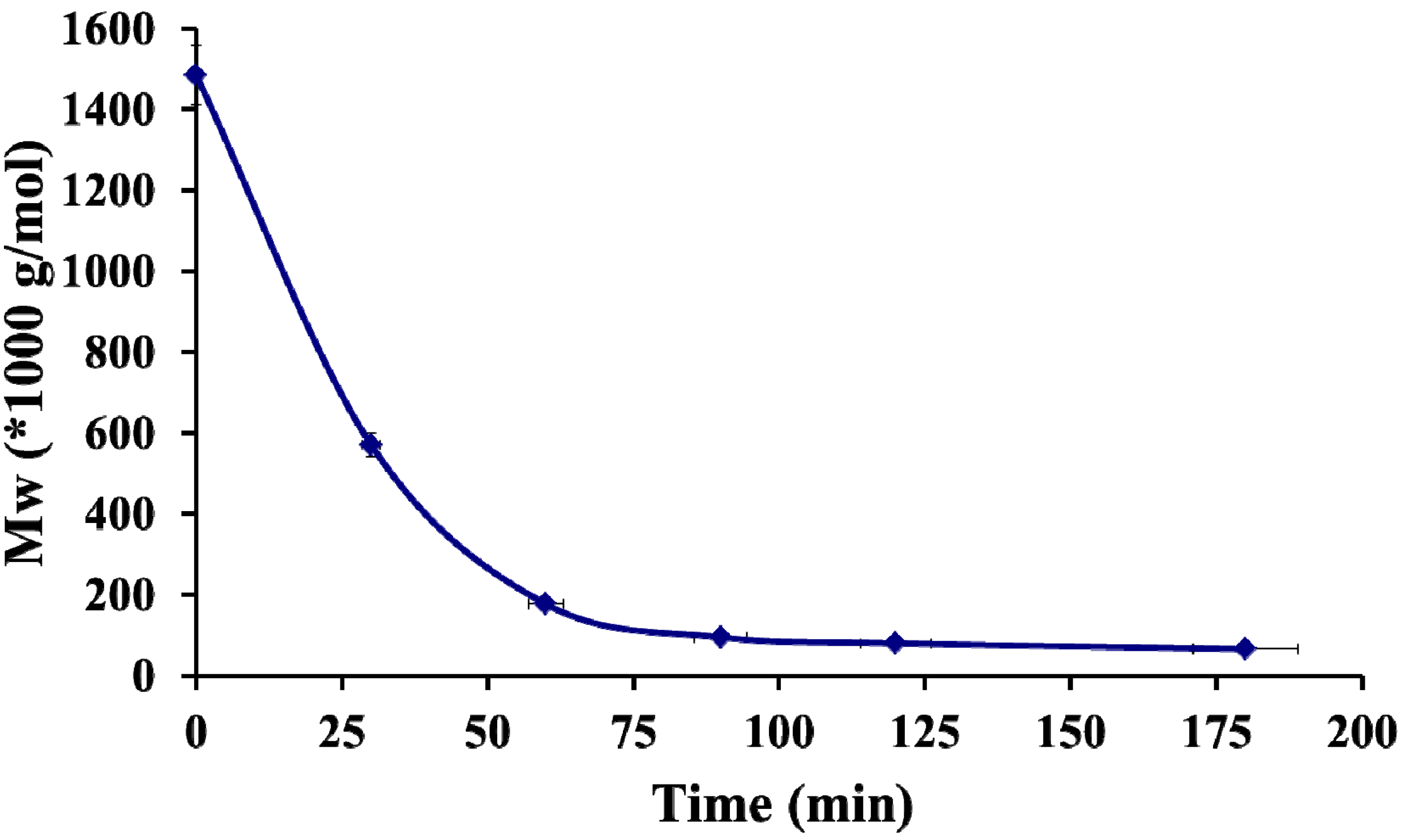
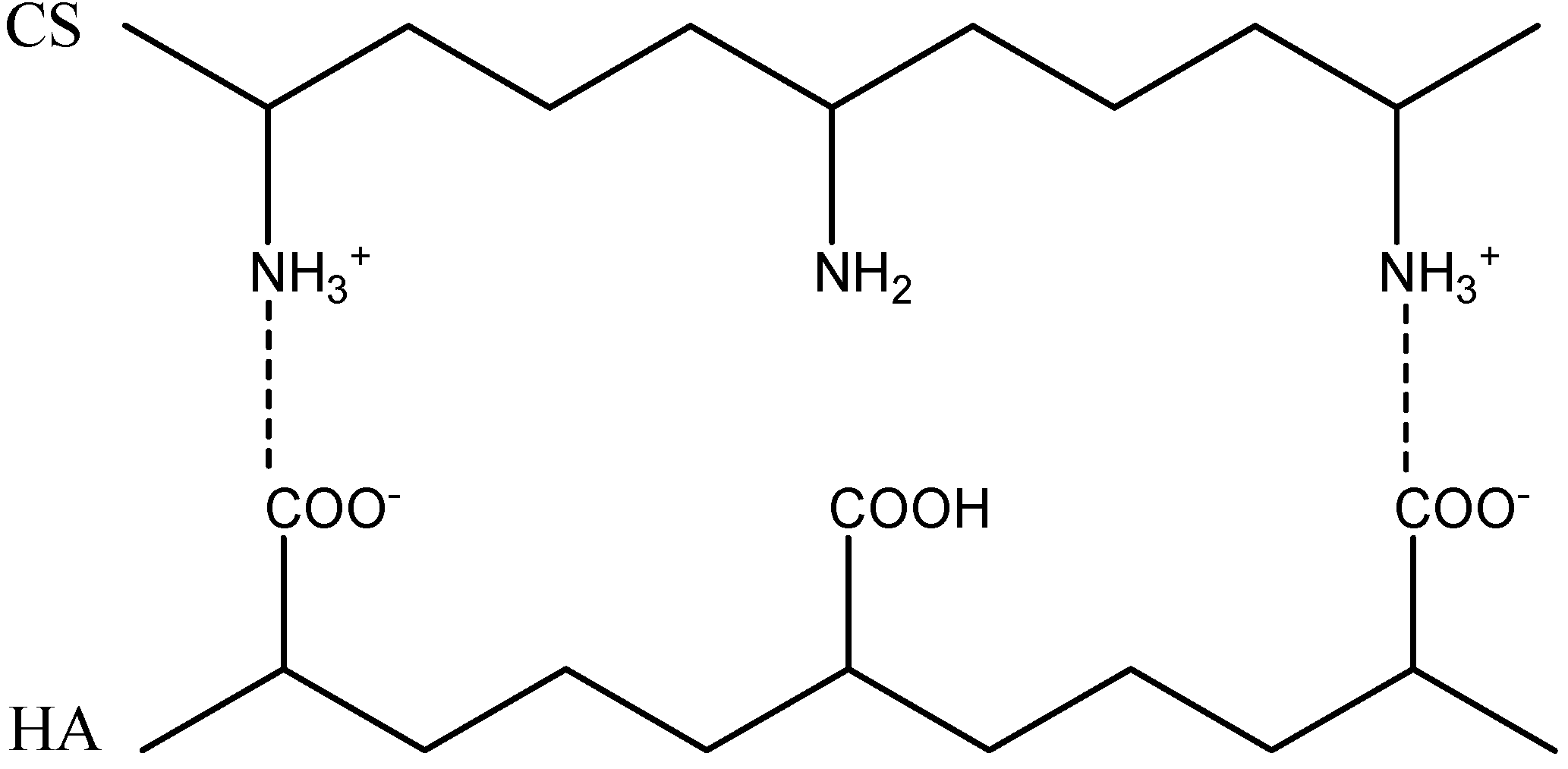
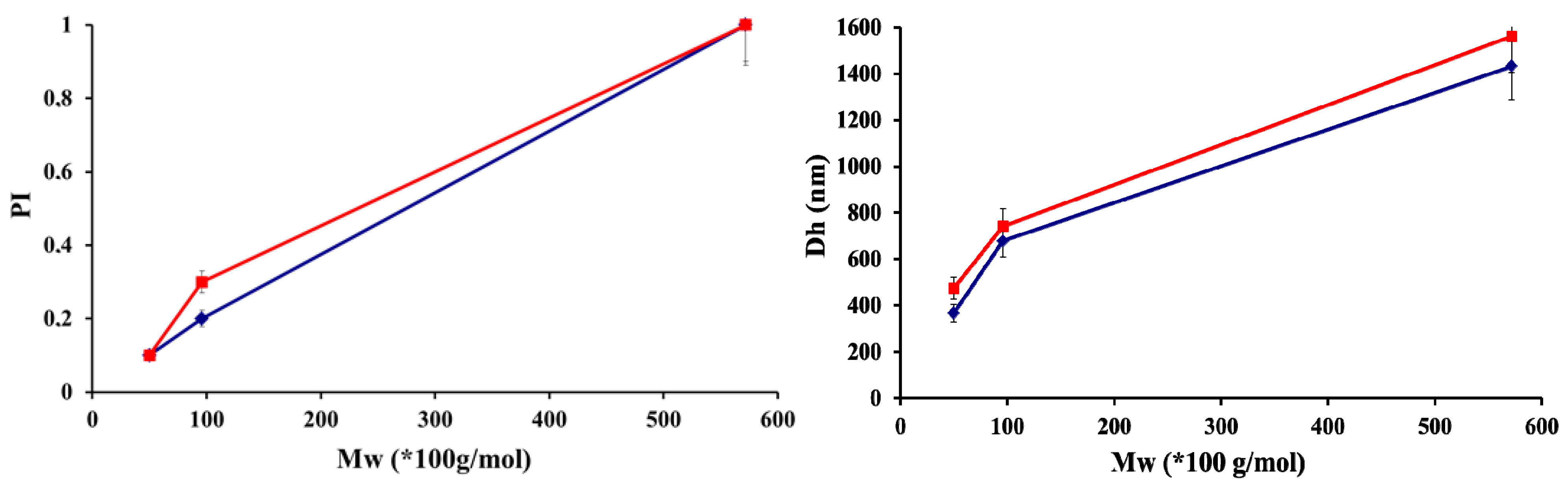
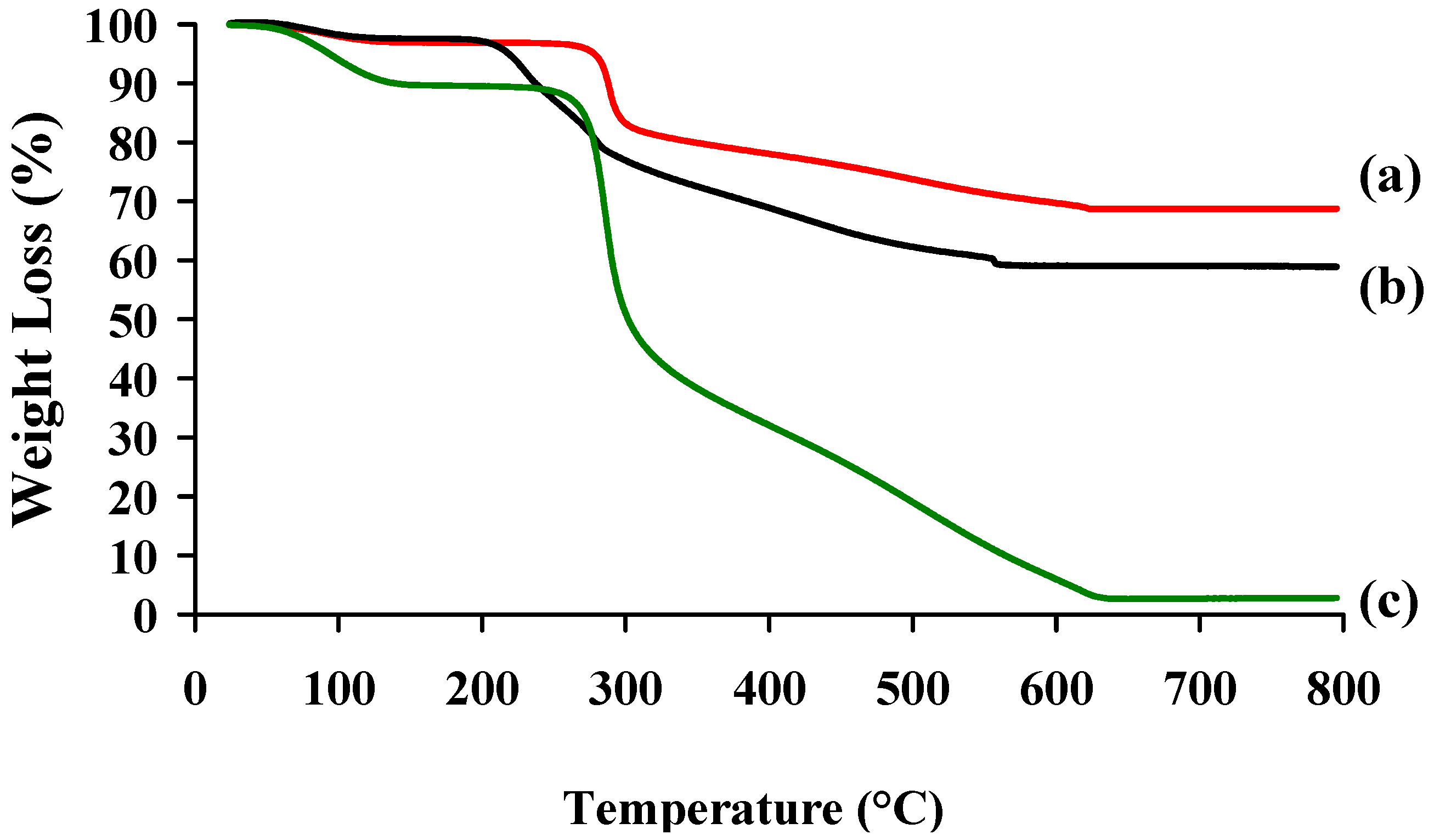
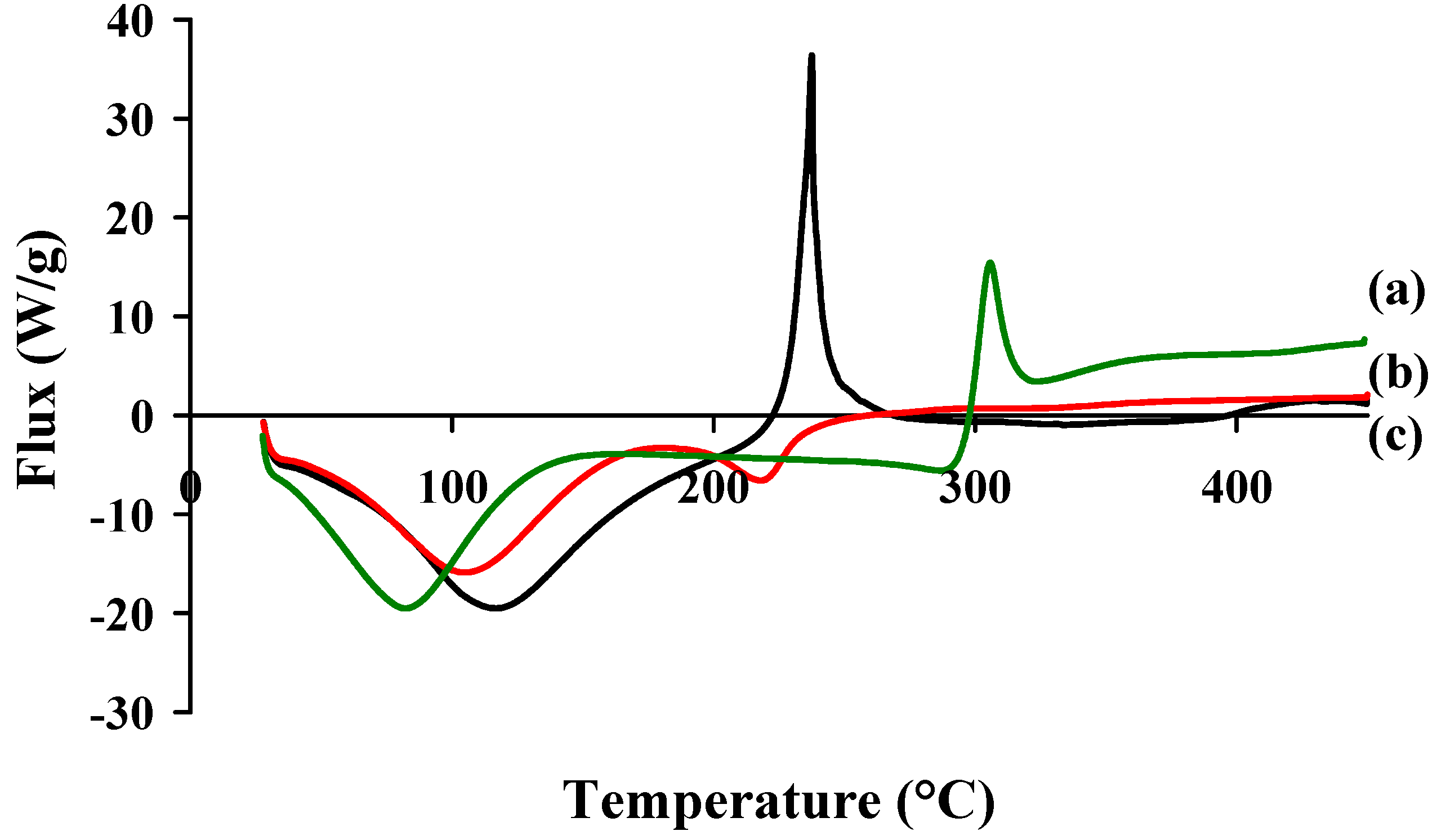
2.2. Formation of Polyelectrolyte Complexes
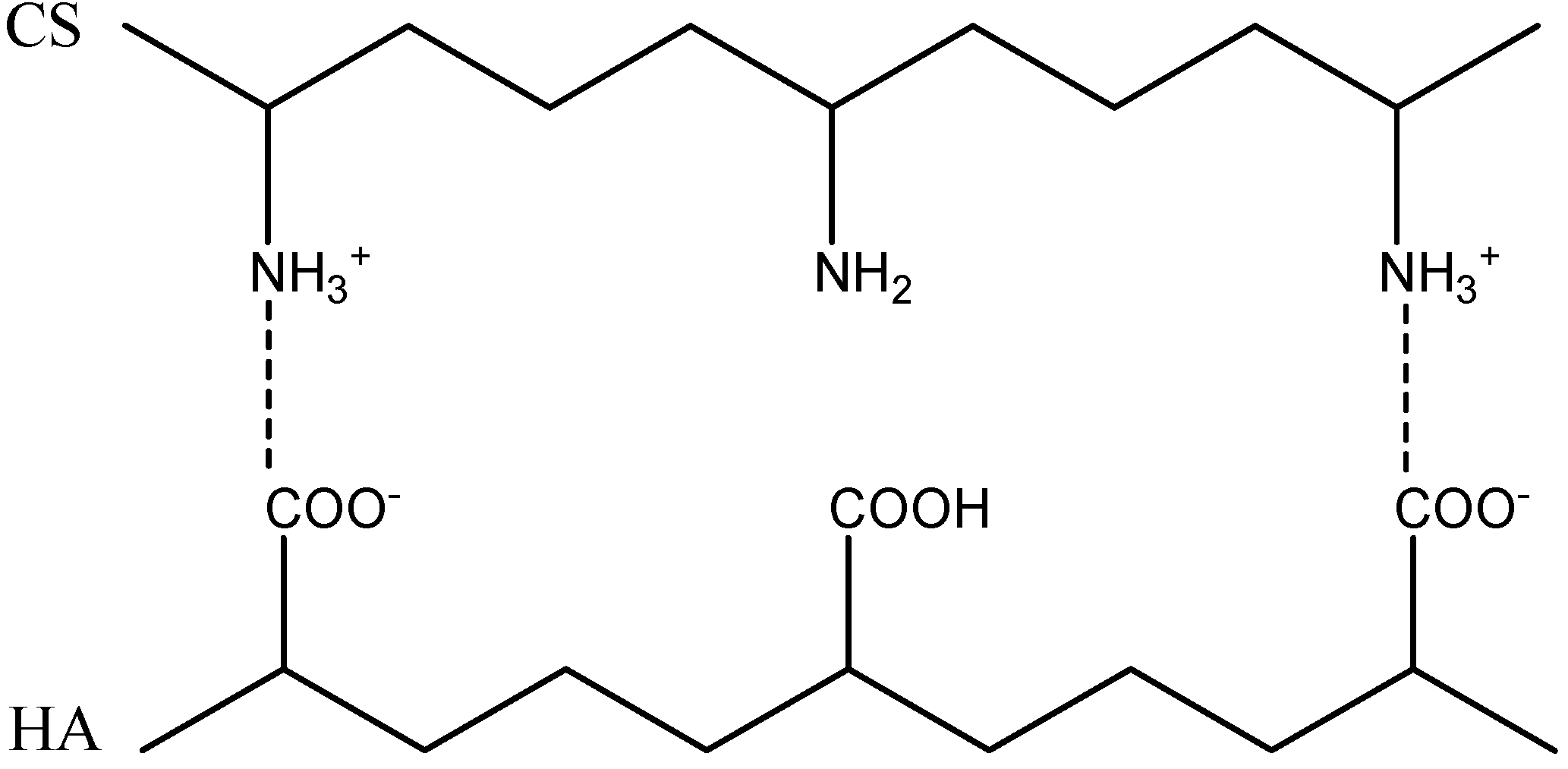

| CS-HA | Ratio (n+/n−) | Solid Content (%) | Size(nm) | PI | Zeta potential (mV) | Appearance |
|---|---|---|---|---|---|---|
| 0.5 | 1100 | 1 | Precipitation | |||
| Water | 1.5 | 6 | 350 | 0.1 | 30 ± 0.31 | Medium turbidity |
| 2.5 | 3 | 290 | 0.1 | 35 ± 0.27 | Medium turbidity | |
| 3.5 | 1 | 271 | 0.1 | 47 ± 0.13 | Low turbidity | |
| 0.5 | 1220 | 1 | Precipitation | |||
| 1.5 | 5 | 632 | 0.1 | 31 ± 0.19 | Medium turbidity | |
| PBS | 2.5 | 3 | 424 | 0.2 | 48 ± 0.23 | Medium turbidity |
| 3.5 | 1 | 418 | 0.1 | 49 ± 1.83 | Low turbidity |

2.3. Colloidal Stability
| CS-HA In water | Ratio (n+/n−) = 1.5 | Ratio (n+/n−) = 2.5 | Ratio (n+/n−) = 3.5 | |||
|---|---|---|---|---|---|---|
| Time (days) | Size (nm) | PI | Size (nm) | PI | Size (nm) | PI |
| 0 | 350 | 0.1 | 291 | 0.1 | 270 | 0.1 |
| 7 | 332 | 0.1 | 256 | 0.1 | 265 | 0.1 |
| 14 | 280 | 0.2 | 233 | 0.1 | 204 | 0.1 |
| 30 | 181 | 0.3 | 204 | 0.2 | 112 | 0.2 |
| CS-HA In PBS | Ratio (n+/n−) = 1.5 | Ratio (n+/n−) = 2.5 | Ratio (n+/n−) = 3.5 | |||
| Time (days) | Size (nm) | PI | Size (nm) | PI | Size (nm) | PI |
| 0 | 632 | 0.1 | 424 | 0.2 | 418 | 0.1 |
| 7 | 621 | 0.1 | 410 | 0.2 | 368 | 0.1 |
| 14 | 588 | 0.1 | 376 | 0.2 | 340 | 0.1 |
| 30 | 332 | 0.3 | 243 | 0.2 | 254 | 0.2 |
2.4. IgA Sorption
| Time(h) | IgA input in PBS (µg/mL) | IgA input in water (µg/mL) | ||||
|---|---|---|---|---|---|---|
| 12 | 36 | 54 | 8 | 31 | 62 | |
| 2 | 97 | 93 | 91 | 95 | 93 | 89 |
| 4 | 99 | 99 | 96 | 99 | 95 | 90 |
| 6 | 100 | 99 | 98 | 100 | 99 | 95 |
| 16 | 100 | 100 | 100 | 100 | 100 | 100 |
| 24 | 100 | 100 | 100 | 100 | 100 | 100 |
3. Experimental
3.1. Materials
3.2. Chitosan Characterization
3.3. Preparation of Polyelectrolyte Solutions
3.4. Polyelectrolyte Complex Formation
 1, where R is the mixing molar charge ratio) by a one-shot addition of the polymer in default to the polymer in excess under magnetic stirring (750 rpm) at room temperature. The final particle dispersion volume was 30 mL at a solid content of 0.1 wt %. Under these conditions, the volume of excess polymer solution was always higher than the volume of the default polymer. Particles were separated from the polyelectrolyte mixture by centrifugation at 7 000× g and 20 °C for 10 min. The supernatant was removed and the pellet was re-suspended in deionized water or PBS.
1, where R is the mixing molar charge ratio) by a one-shot addition of the polymer in default to the polymer in excess under magnetic stirring (750 rpm) at room temperature. The final particle dispersion volume was 30 mL at a solid content of 0.1 wt %. Under these conditions, the volume of excess polymer solution was always higher than the volume of the default polymer. Particles were separated from the polyelectrolyte mixture by centrifugation at 7 000× g and 20 °C for 10 min. The supernatant was removed and the pellet was re-suspended in deionized water or PBS.3.5. Particle Solid Contents
3.6. Physicochemical Characterization of the Complex Dispersions
3.7. Antibody Sorption onto Colloidal PECs

4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Lankalapalli, S.; Kolapalli, V. Polyelectrolyte complexes: A review of their applicability in drug delivery technology. Indian J. Pharm. Sci. 2009, 71, 481–487. [Google Scholar] [CrossRef]
- Sun, W.; Mao, S.; Mei, D.; Kissel, T. Self-assembled polyelectrolyte nanocomplexes between chitosan derivatives and enoxaparin. Eur. J. Pharm. Biopharm. 2008, 69, 417–425. [Google Scholar] [CrossRef]
- Delair, T. Colloidal polyelectrolyte complexes of chitosan and dextran sulfate towards versatile nanocarriers of bioactive molecules. Eur. J. Pharm. Biopharm. 2010, 78, 10–18. [Google Scholar] [CrossRef]
- Dautzenberg, H. Polyelectrolyte complex formation in highly aggregating systems. 1. Effect of salt: polyelectrolyte complex formation in the presence of NaCl. Macromol 1997, 30, 7810–7815. [Google Scholar] [CrossRef]
- Schatz, C.; Lucas, J.M.; Viton, C.; Domard, A.; Pichot, C.; Delair, T. Formation and properties of positively charged colloids based on polyelectrolyte complexes of biopolymers. Langmuir 2004, 20, 7766–7778. [Google Scholar] [CrossRef]
- Rinaudo, M.; Domard, A. Chitin and Chitosan; Elsevier Applied Sciences: London, UK, 1989; pp. 71–86. [Google Scholar]
- Vila, A.; Sanchez, A.; Tobıo, M.; Calvo, P.; Alonso, M.J. Design of biodegradable particles for protein delivery. J. Control. Release 2002, 78, 15–24. [Google Scholar] [CrossRef]
- Dodane, V.; Vilivalam, V.D. Pharmaceutical application of chitosan. Pharm. Sci. Technol. Today 1998, 1, 246–253. [Google Scholar] [CrossRef]
- Kean, T.; Thanou, M. Biodegradation, biodistribution and tocixity of chitosan. Adv. Drug. Deliv. Rev. 2010, 62, 3–11. [Google Scholar] [CrossRef]
- Kima, S.J.; Yoona, S.G.; Leeb, K.B.; Parkb, Y.D.; Kima, S.I. Electrical sensitive behavior of a polyelectrolyte complex composed of chitosan/hyaluronic acid. Solid State Ionics 2003, 164, 199–204. [Google Scholar] [CrossRef]
- Chen, W.B.; Wang, L.F.; Chen, J.S.; Fan, S.Y. Characterization of polyelectrolyte complexes between chondroitin sulfate and chitosan in the solid state. J. Biomed. Mater. Res. Part A 2005, 75, 128–137. [Google Scholar]
- Sæther, H.V.; Holme, H.K.; Maurstad, G.; Smidsrød, O.; Stokke, B. Polyelectrolyte complex formation using alginate and chitosan. Carbohydr. Polym. 2008, 74, 813–821. [Google Scholar] [CrossRef]
- Zhao, Q.; Qian, J.; An, Q.; Gao, C.; Gui, Z.; Jin, H. Synthesis and characterization of soluble chitosan/sodium carboxymethyl cellulose polyelectrolyte complexes and the pervaporation dehydration of their homogeneous membranes. J. Membrane Sci. 2009, 333, 68–78. [Google Scholar] [CrossRef]
- Hugerth, A.; Caram-Lelham, N.; Sundeliir, L.O. The effect of charge density and conformation on the polyelectrolyte complex formation between carrageenan and chitosan. Carbohydr. Polym. 1997, 34, 149–156. [Google Scholar] [CrossRef]
- Arguelles-Monal, W.; Cabrera, G.; Peniche, C.; Rinaudo, M. Conductimetric study of the interpolyelectrolyte reaction betweenchitosan and polygalacturonic acid. Polym. 2000, 41, 2373–2378. [Google Scholar] [CrossRef]
- Liu, W.G.; Sun, S.J.; Cao, Z.Q.; Xin, Z.; Yao, K.D.; Lu, W.W.; Luk, K.D.K. An investigation on the physicochemical properties of chitosan/DNA polyelectrolyte complexes. Biomaterials 2005, 26, 2705–2711. [Google Scholar] [CrossRef]
- Garg, H.G.; Hales, C.A. Chemistry and Biology of Hyaluronan; Elsevier Science & Technology: London, UK, 2004. [Google Scholar]
- Prestwich, G.D. Hyaluronic acid-based clinical biomaterials derived for cell and molecule delivery in regenerative medicine. J. Control. Release 2011, 155, 193–199. [Google Scholar] [CrossRef]
- Yun, Y.H.; Goetz, D.J.; Yellen, P.; Chen, W. Hyaluronan micropheres for sustained gene delivery and site-specific targeting. Biomaterials 2004, 25, 147–157. [Google Scholar] [CrossRef]
- Eun, J.O.; Park, K.; Ki, S.K.; Jiseok, K.; Jeong, A.Y.; Hoffman, S.; Sei, K.H. Target specific and long-acting delivery of protein, peptide, and nucleotide therapeutics using hyaluronic acid derivatives. J. Control. Release 2010, 141, 2–12. [Google Scholar] [CrossRef]
- Yamane, S.; Iwasaki, N.; Majima, T.; Funakoshi, T.; Masuko, T.; Harada, K.; Minami, A.; Monde, K.; Nishimura, S. Feasibility of chitosan-based hyaluronic acid hybrid biomaterial for a novel scaffold in cartilage tissue engineering. Biomaterials 2005, 26, 611–619. [Google Scholar] [CrossRef]
- Hahna, S.K.; Hoffmanb, A.S. Preparation and characterization of biocompatible polyelectrolyte complex multilayer of hyaluronic acid and poly-l-lysine. Inter. J. Biol. Macromol. 2005, 37, 227–231. [Google Scholar] [CrossRef]
- Malay, Ö; Yalçın, D.; Batıgün, A.; Bayraktar, O. Characterization of silk fibroin/hyaluronic acid polyelectrolyte complex (PEC) films. J. Therm. Anal. Calorim. 2008, 94, 749–755. [Google Scholar] [CrossRef]
- Lu, H.; Zhao, H.; Wang, K.; Lv, L. Novel hyaluronic acid-chitosan nanoparticles as non-viral gene delivery vectors targeting osteoarthritis. Int. J. Pharm. 2011, 420, 358–365. [Google Scholar] [CrossRef]
- Duceppe, N.; Tabrizian, M. Factors influencing the transfection efficiency of ultra low molecular weight chitosan/hyaluronic acid nanoparticles. Coll. Surfaces A: Physicochem. Eng. Aspects 2005, 257–258, 85–88. [Google Scholar] [CrossRef]
- Verheul, R.; Slütter, B.; Bal, S.M.; Bouwstra, J.A.; Jiskoot, W.; Hennink, W.E. Covalently stabilized trimethyl chitosan-hyaluronic acid nanoparticles for nasal and intradermal vaccination. J. Controlled Release 2011, 156, 46–52. [Google Scholar] [CrossRef]
- Chua, P.-H.; Neoh, K.-G.; Kang, E.T.; Wu, W. Surface functionalization of titanium with hyaluronic acid/chitosan polyelectrolyte multilayers and RGD for promoting osteoblast functions and inhibiting bacterial adhesion. Biomaterials 2008, 29, 1412–1421. [Google Scholar] [CrossRef]
- Drogoz, A.; David, L.; Rochas, C.; Domard, A.; Delair, T. Polyelectrolyte complexes from polysaccharides: Formation and stoichiometry, monitoring. Langmuir 2007, 23, 10950–10958. [Google Scholar] [CrossRef]
- Miyazaki, T.; Yomota, C.; Okada, S. Ultrasonic depolymerization of hyaluronic acid. Polym. Degrad. Stab. 2001, 74, 77–85. [Google Scholar] [CrossRef]
- Sorlier, P.; Denuziere, A.; Viton, C.; Domard, A. Relation between the degree of acetylation and the electrostatic properties of chitin and chitosan. Biomacromolecules 2001, 2, 765–772. [Google Scholar] [CrossRef]
- Denuziere, A.; Ferrier, D.; Domard, A. Chitosan-Chondroitin sulfate and chitosan-hyaluronate PECs. Physico-chemical aspects. Carbohydr. Polym. 1996, 29, 317–323. [Google Scholar]
- Umerska, A.; Palucha, K.J.; Inkielewicz-Stępniaka, I.; Santos-Martineza, M.J.; Corrigana, O.I.; Medinaa, C.; Tajbera, L. Exploring the assembly process and properties of novel crosslinker-free hyaluronate-based polyelectrolyte complex nanocarriers. Int. J. Pharm. 2012, 436, 75–87. [Google Scholar] [CrossRef]
- Boddohi, S.; Moore, N.; Johnson, P.A.; Kipper, M.J. Polysaccharide-Based polyelectrolyte complex nanoparticles from chitosan, heparin, and hyaluronan. Biomacromolecules 2009, 10, 1402–1409. [Google Scholar] [CrossRef]
- Coimbra, P.; Alvers, P.; Valente, T.A.M.; Santos, R.; Correia, I.J.; Ferreira, P. Sodium hyaluronate/chitosan polyelectrolyte complex for dental pulp regeneration: Synthesis and characterization. Int. J. Biol. Macromol. 2011, 49, 573–579. [Google Scholar] [CrossRef]
- Bigucci, F.; Luppi, B.; Cerchiara, T.; Sorrenti, M.; Bettinetti, G.; Rodriguez, L. Chitosan/pectin polyelectrolyte complexes: Selection of suitable preparative conditions for colon-specific delivery of vancomycin. Eur. J. Pharm. Sci. 2008, 35, 435–441. [Google Scholar] [CrossRef]
- Sarmento, B.; Ribeiro, A.; Veiga, F.; Ferreira, D. Development and characterization of new insulin containing polysaccharide nanoparticles. Coll. Surfaces B Biointerfaces 2006, 53, 193–202. [Google Scholar] [CrossRef]
- Martins, A.F.; Pereira, A.G.B.; Fajardo, A.R.; Rubira, A.F. Characterization of polyelectrolytes complexes based on N,N,N-trimethyl chitosan/heparin prepared at different pH conditions. Carbohydr. Polym. 2011, 86, 1266–1272. [Google Scholar] [CrossRef]
- Vachoud, L.; Zydowicz, N.; Domard, A. Formation and characterisation of a physical chitin gel. Carbohydr. Res. 1997, 302, 169–177. [Google Scholar] [CrossRef]
- Schatz, C.; Domard, A.; Viton, C.; Pichot, C.; Delair, T. Versatile and efficient formation of colloids of biopolymer-based polyelectrolyte complexes. Biomacromolecules 2004, 5, 1882–1892. [Google Scholar] [CrossRef]
- Popa-Nita, S.; Lucas, J.-M.; Ladaviere, C.; David, L.; Domard, A. Mechanisms involved during the ultrasonically induced depolymerization of chitosan: Characterization and control. Biomacromolecules 2009, 10, 1203–1211. [Google Scholar] [CrossRef]
- Hirai, A.; Odami, H. Determination of degree of deacetylation of chitosan by 1H NMR spectroscopy. Polym. Bull. 1991, 26, 87–94. [Google Scholar] [CrossRef]
- Coombes, A.G.A.; Scholes, P.D.; Davies, M.C.; Illum, L.; Davis, S.S. Resorbable polymeric microspheres for drug-delivery- Production and simultaneous surface modification using Peo-Ppo surfactants. Biomaterials 1994, 15, 673–680. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Polexe, R.C.; Delair, T. Elaboration of Stable and Antibody Functionalized Positively Charged Colloids by Polyelectrolyte Complexation between Chitosan and Hyaluronic Acid. Molecules 2013, 18, 8563-8578. https://doi.org/10.3390/molecules18078563
Polexe RC, Delair T. Elaboration of Stable and Antibody Functionalized Positively Charged Colloids by Polyelectrolyte Complexation between Chitosan and Hyaluronic Acid. Molecules. 2013; 18(7):8563-8578. https://doi.org/10.3390/molecules18078563
Chicago/Turabian StylePolexe, Ramona C., and Thierry Delair. 2013. "Elaboration of Stable and Antibody Functionalized Positively Charged Colloids by Polyelectrolyte Complexation between Chitosan and Hyaluronic Acid" Molecules 18, no. 7: 8563-8578. https://doi.org/10.3390/molecules18078563