Total Synthesis, Cytotoxic Effects of Damnacanthal, Nordamnacanthal and Related Anthraquinone Analogues

Abstract
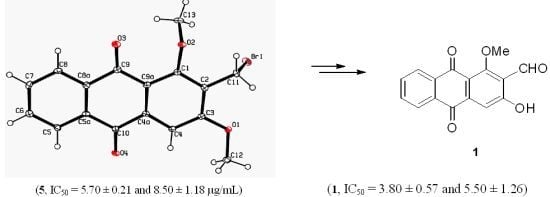
:
1. Introduction
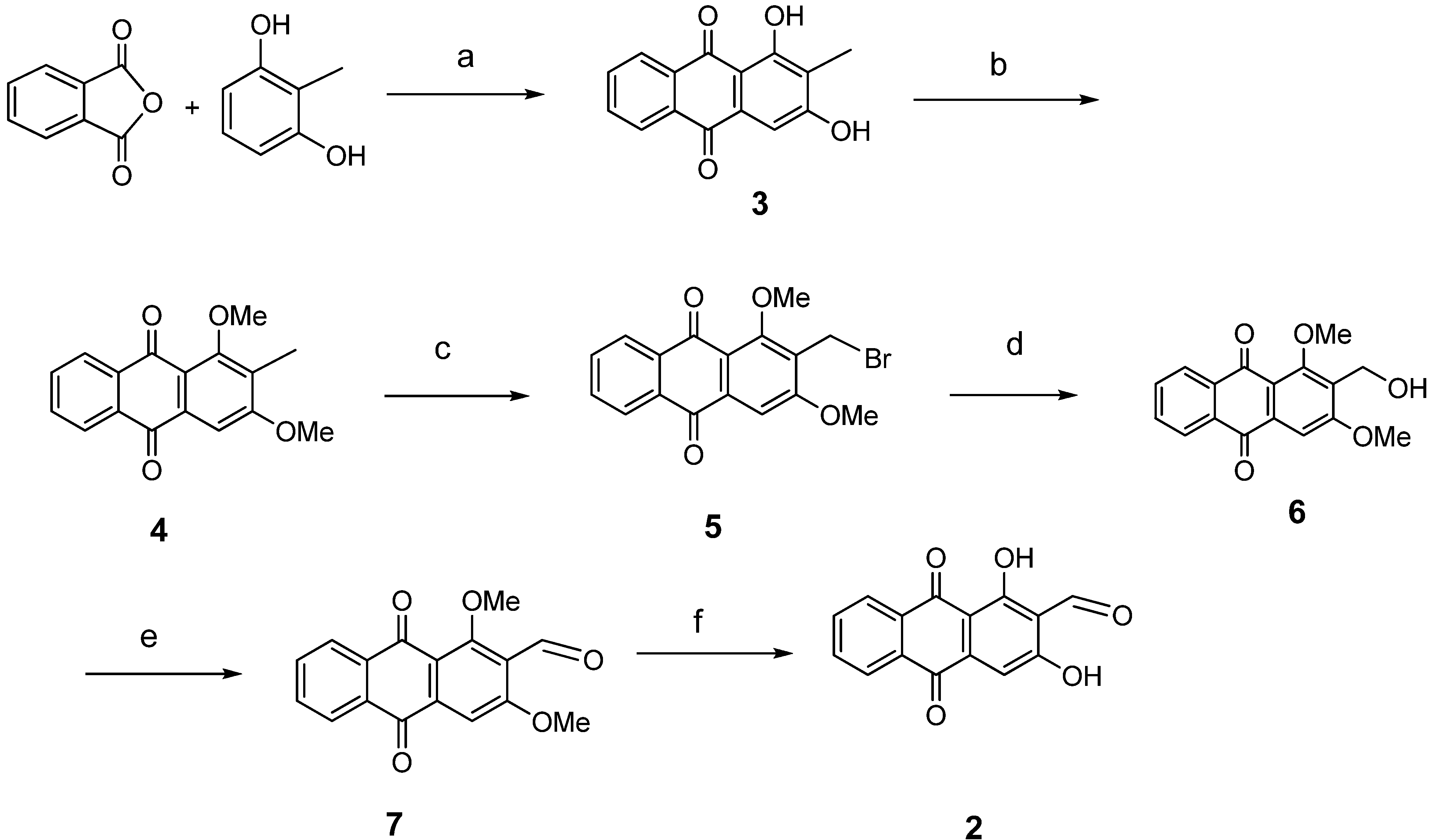
2. Results and Discussion
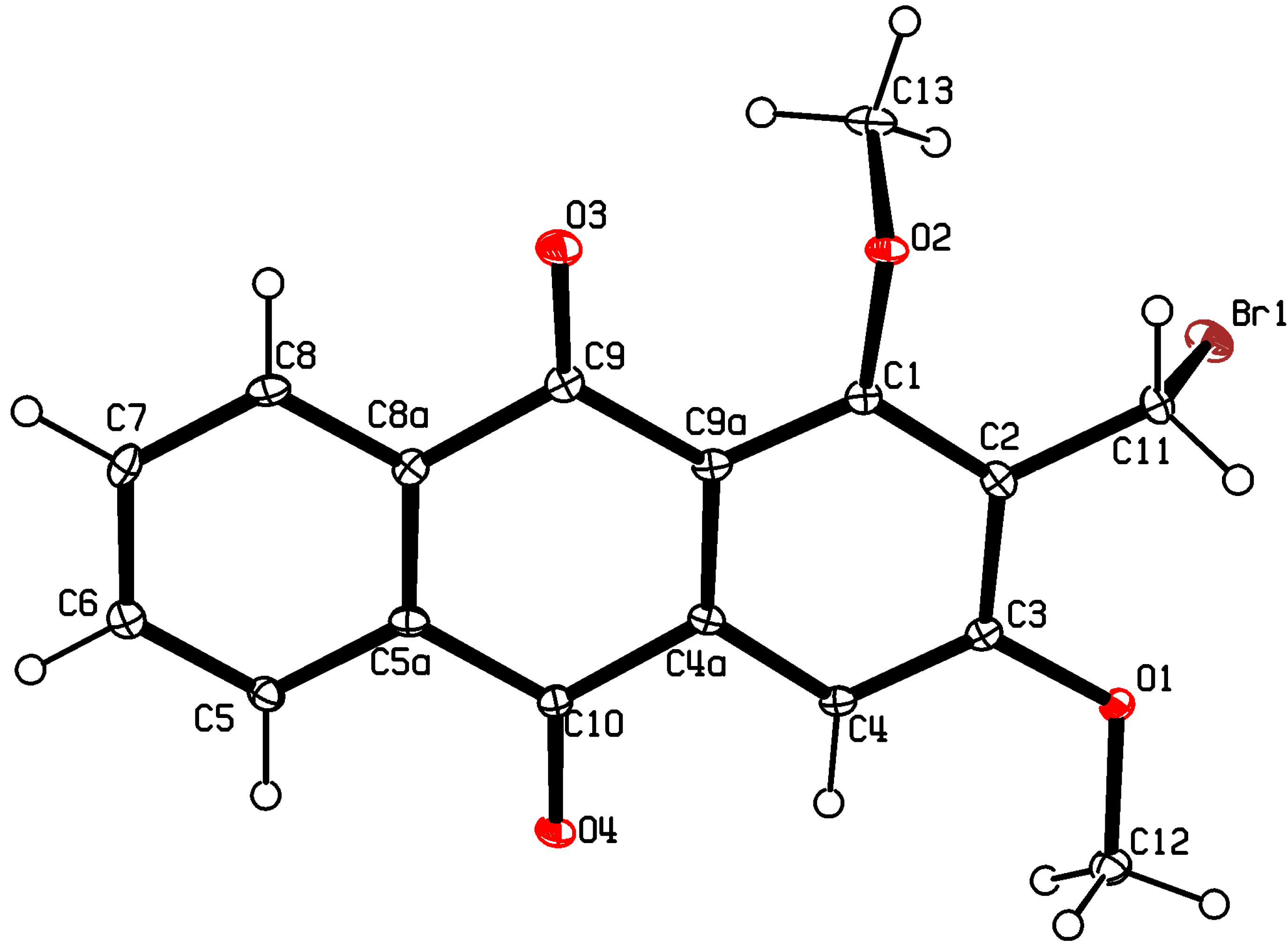
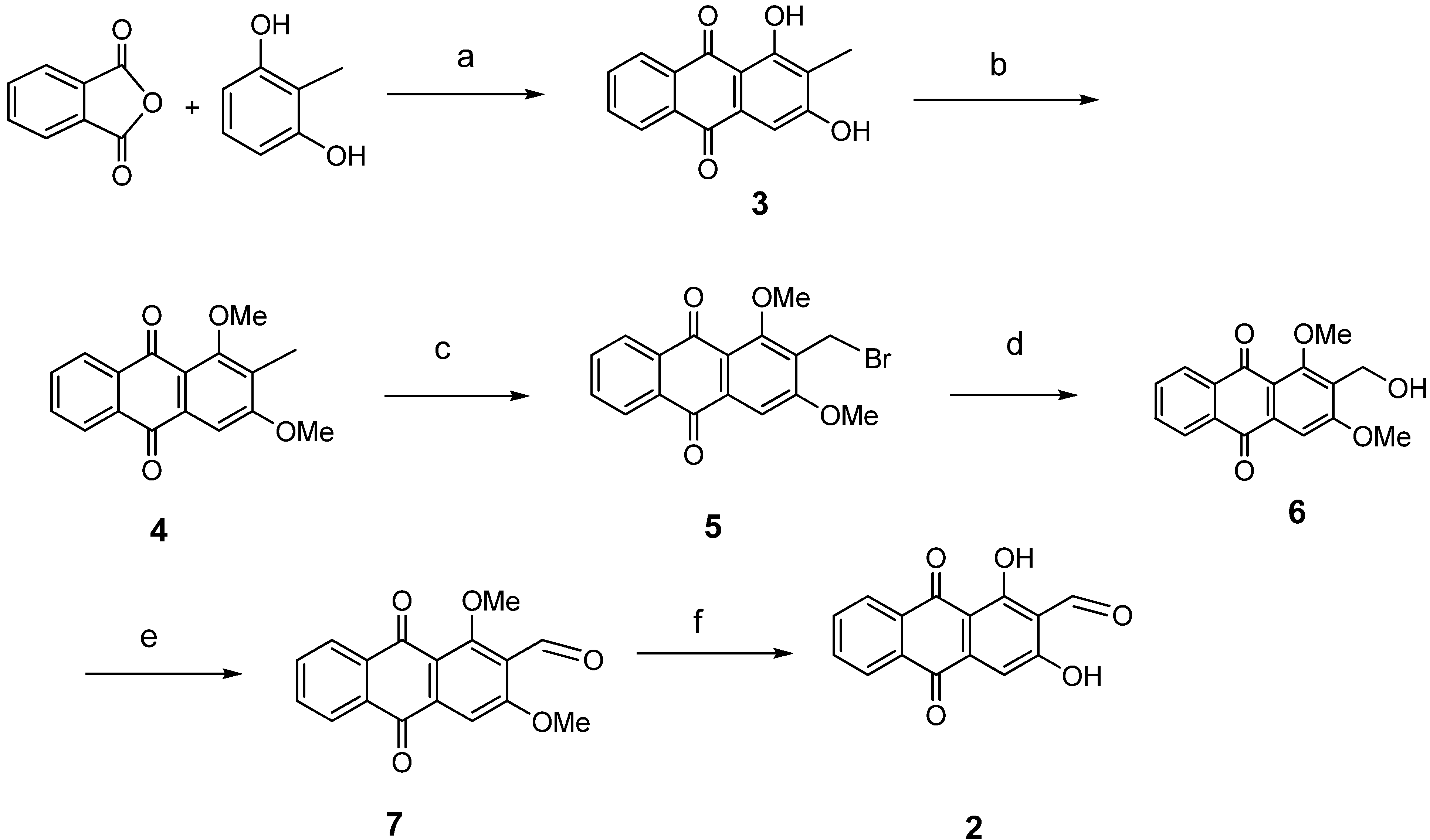
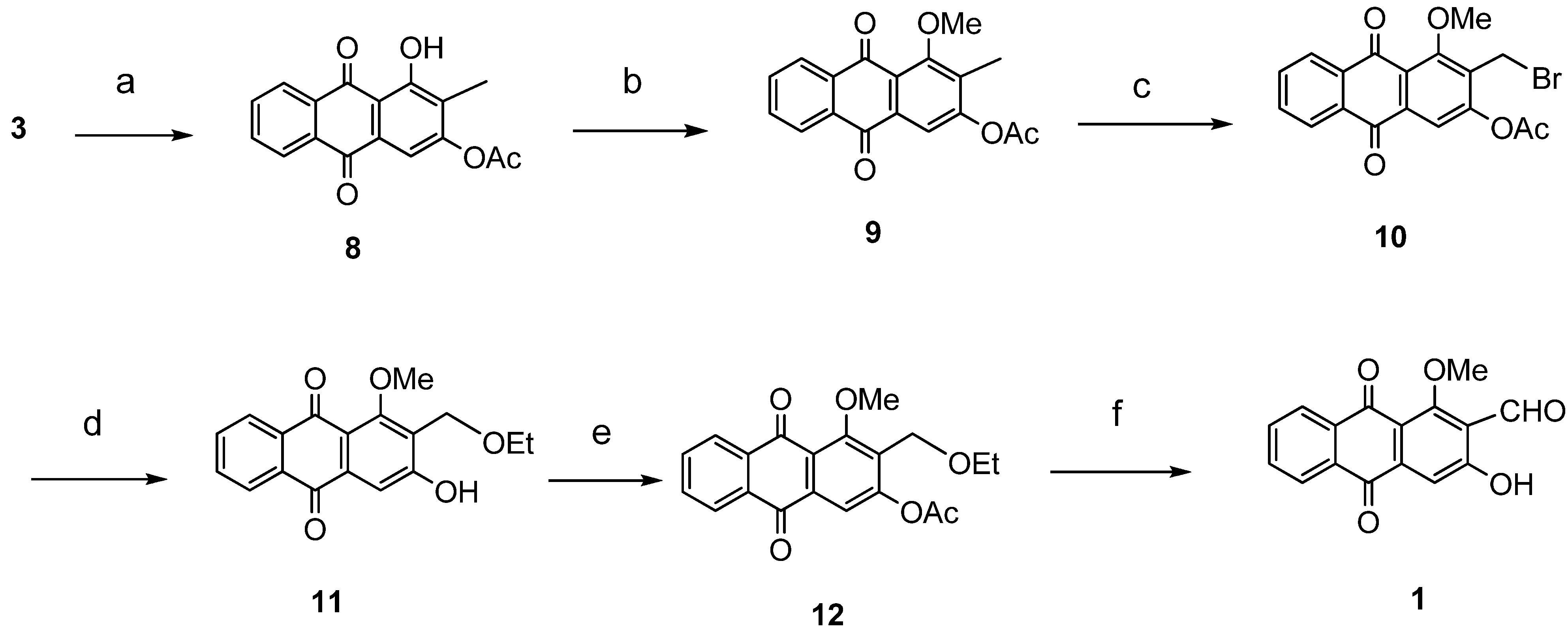
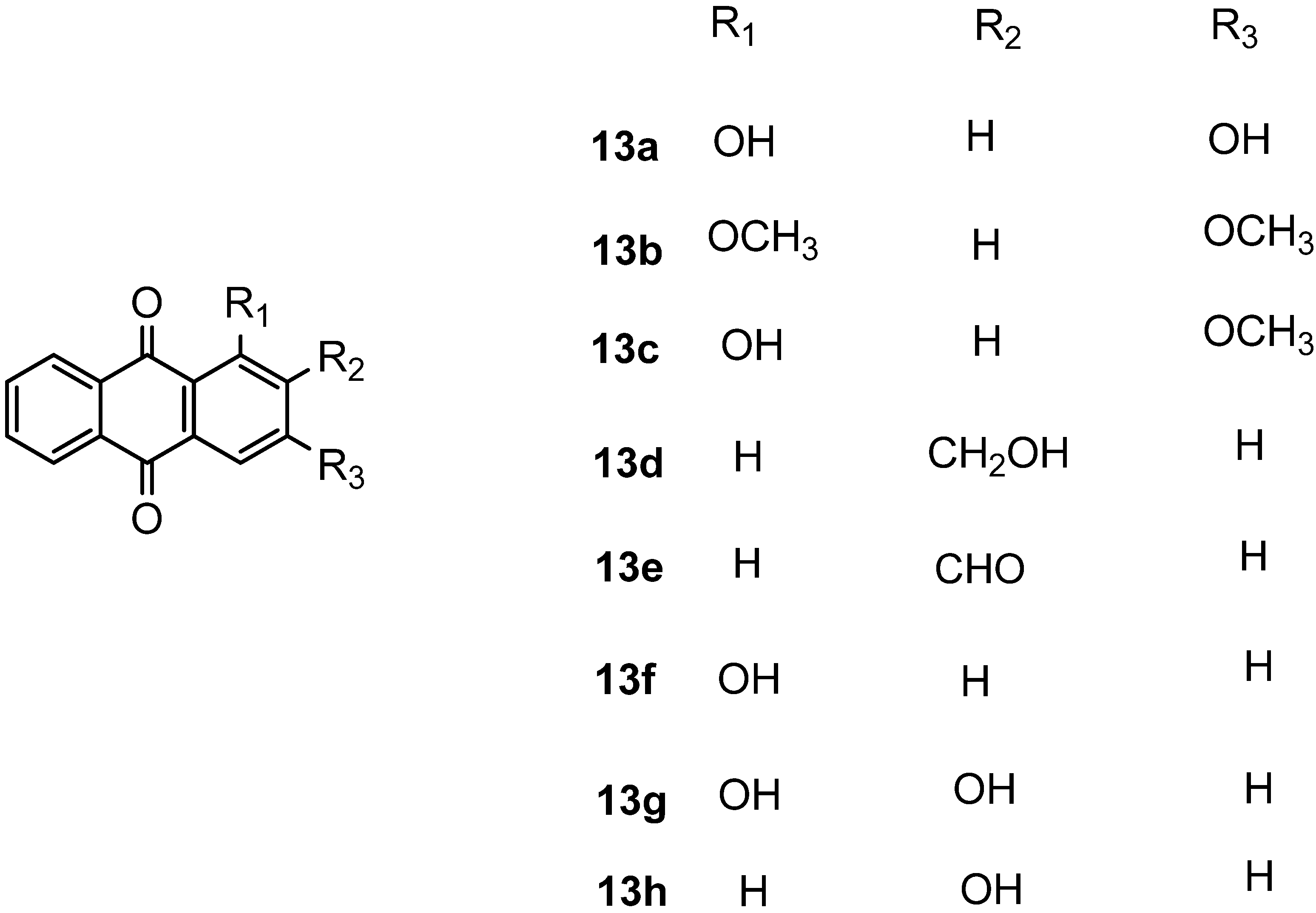

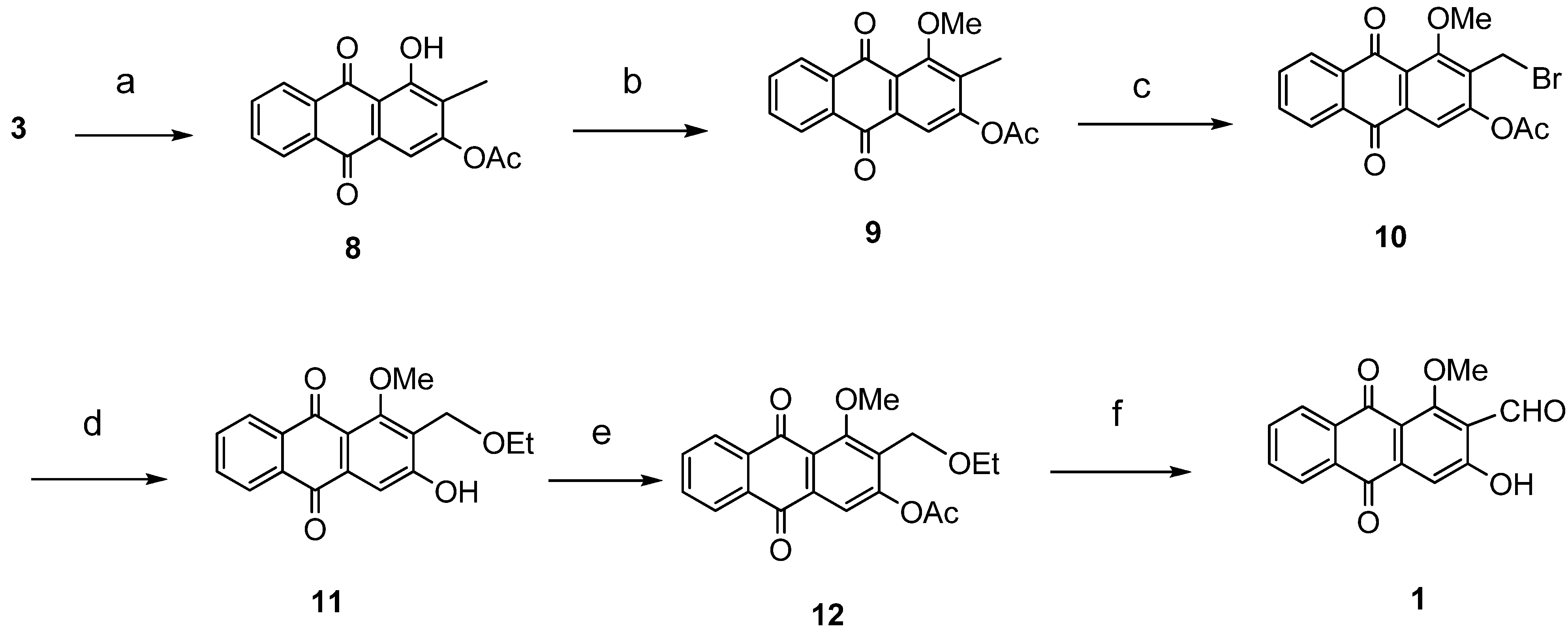
{kind=link}
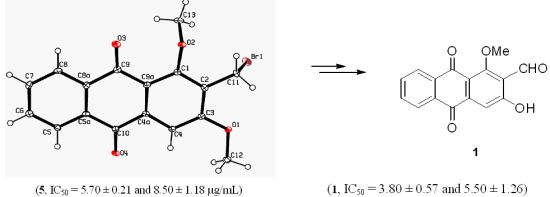
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | IC50 value (μM) | ||
|---|---|---|---|
| Chemical Structure | MCF-7 | K562 | |
| 1 |  | 13.48 ± 2.02 | 19.50 ± 4.47 |
| 2 |  | 60.07 ± 6.46 | 32.46 ± 8.73 |
| 3 |  | 90.78 ± 1.49 | 100.71 ± 2.80 |
| 4 |  | 37.01 ± 13.82 | 87.81 ± 9.17 |
| 5 |  | 15.83 ± 0.58 | 23.61 ± 3.28 |
| 6 |  | 43.54 ± 0.48 | 47.62 ± 7.24 |
| 7 |  | 43.96 ± 3.42 | 49.66 ± 2.48 |
| 8 |  | 28.38 ± 7.94 | 69.59 ± 1.62 |
| 9 |  | 59.35 ± 8.23 | 46.77 ± 3.81 |
| 10 |  | 39.69 ± 6.24 | 54.90 ± 7.11 |
| 11 |  | 59.13 ± 11.59 | 42.06 ± 6.39 |
| 12 |  | 43.62 ± 5.35 | 89.36 ± 3.83 |
  | |||
| 13a |  | 82.08 ± 1.46 | 60.42 ± 5.33 |
| 13b |  | 24.25 ± 2.46 | 22.01 ± 3.54 |
| 13c |  | 87.80 ± 2.01 | 64.96 ± 1.57 |
| 13d |  | 49.58 ± 12.61 | 50.84 ± 14.12 |
| 13e |  | 43.64 ± 2.12 | 46.61 ± 12.37 |
| 13f |  | 45.98 ± 2.95 | 107.14 ± 0.54 |
| 13g |  | 63.84 ± 12.46 | 70.54 ± 1.56 |
| 13h |  | 52.50 ± 2.63 | 94.17 ± 1.88 |
| Doxorubicin |  | 0.94 ± 0.20 | 0.24 ± 0.07 |
3. Experimental
3.1. General
| Empirical formula | C17H13O4Br |
| Formula weight | 361.18 |
| Temperature | 100(2) K |
| Wavelength | 0.71073 Å |
| Crystal system | Monoclinic |
| Space group | P2(1)/c |
| Unit cell dimensions | a = 4.7401(8)Å, α = 90° |
| b = 17.364(3)Å, β = 94.543(2)° | |
| c = 16.825(3)Å, γ = 90° | |
| Volume | 1380.5(4) Å3 |
| Z | 4 |
| Calculated density | 1.738 Mgm−3 |
| Absorption coefficient | 2.994 mm−1 |
| F(000) | 728 |
| Crystal size | 0.20 × 0.10 × 0.10 mm |
| θ range for data collection | 1.69–28.26° |
| Limiting indices | −6 ≤ h ≤6, −14 ≤ k ≤23, −22 ≤ l ≤ 16 |
| Reflections collected | 3404 |
| Independent reflections | 2522 [R (int) = 0.0494] |
| Completeness to θ = 26.00º | 99.40% |
| Absorption correction | Multi-scan |
| Max. and min. transmission | 0.5973 and 0.7457 |
| Refinement method | Full-matrix least-squares on F2 |
| Data / restraints / parameters | 3404/0/201 |
| Goodness-of-fit on F2 | 1.038 |
| Final R indices [I>2σ(I)] | R1 = 0.0417, wR2 = 0.0984 |
| R indices (all data) | R1 = 0.0633, wR2 = 0.1124 |
| Largest difference peak and hole | 0.521 and −0.782 e.Å−3 |
3.2. General Procedure for Anthraquinone Synthesis
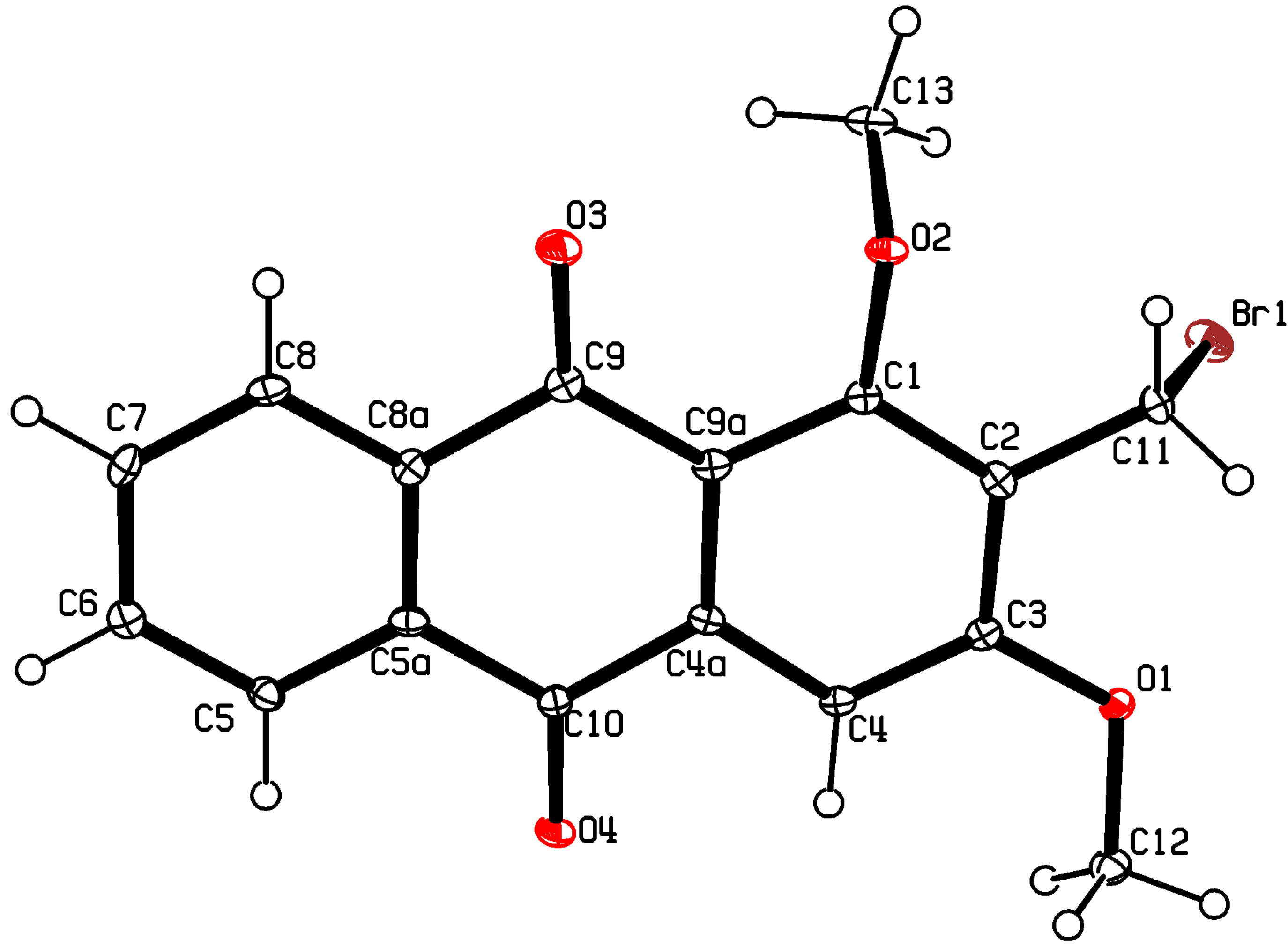
3.3. X-ray Crystallography of Compound 5
4. Cytotoxicity Assay
4.1. Preparation of Compounds
4.2. Preparation of Cell Lines
4.3. Cell Viability Assay
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Murphy, G.P.; Lawrence, W.J.; Lenhard, R.E. American Society Textbook of Clinical Oncology, 2nd ed.; American Cancer Society: Atlanta, GA, USA, 1995. [Google Scholar]
- Doroshow, J.H. Anthracyclines and anthracenediones. In Cancer Chemotherapy and Biotherapy: Principles and Practice, 2nd ed.; Chabner, B.A., Longo, D.L., Eds.; Lippincott-Raven Publishers: Philadelphia, PA, USA, 1996; pp. 409–434. [Google Scholar]
- Faulds, D.; Balfour, J.A.; Chrisp, P.; Langtry, H.D. Mitoxantrone, a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in the chemotherapy of cancer. Drugs 1991, 3, 400–449. [Google Scholar]
- Kosalec, I.; Kremer, D.; Locatelli, M.; Epifano, F.; Genovese, S.; Carlucci, G.; Randic, M.; Koncic, Z.M. Anthraquinone profile, antioxidant and antimicrobial activity of bark extracts of Rhamnus alaternus, R. fallax, R. intermedia and R. pumila. Food Chem. 2013, 136, 335–341. [Google Scholar] [CrossRef]
- Shi, Y.Q.; Fukai, T.; Sakagami, H.; Kuroda, J.; Miyaoka, R.; Tamura, M.; Yoshida, N.; Nomura, T. Cytotoxic and DNA damage-inducing activities of low molecular weight phenols from rhubarb. Anticancer Res. 2001, 21, 2847–2853. [Google Scholar]
- Sittie, A.A.; Lemmich, E.; Olsen, C.E.; Hviid, L.; Kharazmi, A.; Nkrumah, F.K.; Christensen, S.B. Structure-activity studies: in vitro antileishmanial and antimalarial activities of anthraquinones from Morinda lucida. Planta Med. 1999, 65, 259–261. [Google Scholar] [CrossRef]
- Rath, G.; Ndonzao, M.; Hostettmann, K. Antifungal anthraquinones from Morinda lucida. Int. J. Pharmacogol. 1995, 33, 107–114. [Google Scholar] [CrossRef]
- Younos, C.; Rolland, A.; Fleurentin, J.; Lanhers, M.; Misslin, R.; Mortier, F. Analgesic and behavioral effects of Morinda citrifolia. Planta Med. 1990, 56, 430–434. [Google Scholar] [CrossRef]
- Adwankar, M.K.; Chitnis, M.P. In vivo Anti-Cancer Activity of RC-18. Chemotherapy 1982, 28, 291–293. [Google Scholar] [CrossRef]
- Koumaglo, K.; Gbeassor, M.; Nikabu, O.; de Souza, C.; Werner, W. Effects of three compounds extracted from Morinda lucida on Plasmodium falciparum. Planta Med. 1992, 58, 533–434. [Google Scholar] [CrossRef]
- Tripathi, Y.B.; Sharma, M.; Manickam, M. Rubiadin, a new antioxidant from Rubia cordifolia. Indian J. Biochem. Biophys. 1997, 34, 302–306. [Google Scholar]
- Chang, P.; Chen, C. Isolation and Characterization of Antitumor Anthraquinones from Morinda umbellate. (Taipei). Chin. Pharm. J. 1995, 47, 347–353. [Google Scholar]
- Ismail, N.H.; Ali, A.M.; Aimi, N.; Kitajima, M.; Takayama, H.; Lajis, N.H. Anthraquinones from Morinda elliptica. Phytochemistry 1997, 45, 1723–1725. [Google Scholar] [CrossRef]
- Di Marco, A.; Gaetani, M.; Orezzi, P.; Scarpinato, B. M.; Silvestrini, R.; Soldati, M.; Dasdia, T.; Valentini, L. Daunomycin, A New Antibiotic of the Rhodomycin Group. Nature 1964, 201, 706–707. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, M.C.; Lee, B.J.; Park, D.H.; Hong, S.H.; Um, J.Y. Anti-Inflammatory Activity of Chrysophanol through the Suppression of NF-κB/Caspase-1 Activation in Vitro and in Vivo. Molecules 2010, 15, 6436–6451. [Google Scholar]
- Preobrazhenskaya, M.N.; Shchekotikhin, A.E.; Shtil, A.A.; Huang, H.S. Antitumor Anthraquinone Analogues for Multidrug Resistant Tumor Cells. J. Med. Sci. 2006, 26, 1–4. [Google Scholar]
- Teng, C.H.; Won, S.J.; Lin, C.N. Design, synthesis and cytotoxic effect of hydroxy- and 3-alkylaminopropoxy-9,10-anthraquinone derivatives. Bioorg. Med. Chem. 2005, 13, 3439–3435. [Google Scholar] [CrossRef]
- Jin, G.Z.; Song, G.Y.; Zheng, X.G.; Kim, Y.; Sok, D.E.; Ahn, B.Z. 2-(1-Oxyalkyl)-1,4-dioxy-9,10-anthraquinone: synthesis and Evaluation of Antitumor Activity. Arch. Pharm. Res. 1998, 21, 198–206. [Google Scholar] [CrossRef]
- Cairns, D.; Michalitsi, E.; Jenkins, T.C.; Mackay, S.P. Molecular Modelling and Cytotoxicity of Substituted Anthraquinones as Inhibitors of Human Telomerase. Bioorg. Med. Chem. 2002, 10, 803–817. [Google Scholar] [CrossRef]
- Murdock, K.C.; Child, R.C.; Fabio, P.F.; Angier, R.B.; Wallace, R.E.; Durr, F.E.; Chella, R.V. Antitumor Agents.1.1,4-Bis[(Aminoalkyl)amino]-9,19-Anthracenediones. J. Med. Chem. 1979, 22, 1024–1030. [Google Scholar] [CrossRef]
- Chang, P.; Lee, K.H.; Shingu, T.; Hirayama, T.; Hall, I.H.; Huang, H.C. Antitumor agents 50. Morindaparvin-A, a New Antileukemic Anthraquinone, and Alizarin-1-methyl ether from Morinda parvifolia, and the Antileukemic Activity of the Related Derivatives. J. Nat. Prod. 1982, 45, 206–210. [Google Scholar] [CrossRef]
- Ali, A.M.; Ismail, N.H.; Mackeen, M.M.; Yazan, L.S.; Mohammad, S.M.; Ho, A.S.H.; Lajis, N.H. Antiviral, Cytotoxic and Antimicrobial Activities of Anthraquinones isolated from the roots of Morinda elliptca. Pharm. Biol. 2000, 38, 298–301. [Google Scholar]
- Alitheen, N.B.; Mashitoh, A.R.; Yeap, S.K.; Shuhaimi, M.; Manaf, A.A.; Lajis, N.H. Cytotoxic effect of damnacanthal, nordamnacanthal, zerumbone and betulinic acid isolated from Malaysian plant sources. Int. Food Res. J. 2010, 17, 711–719. [Google Scholar]
- Noorjahan, B.M.A.; Ali, A.M.; Yeap, S.K.; Suhaimi, M.; Lajis, N.H.; Mashitoh, A.R.; Ho, W.Y.; Ismail, N.H. Cytotoxicity and Immunomodulatory Effects of Damnacanthal and Nordamnacanthal Isolated from Roots of Morinda elliptica. J. Agrobiotec. 2010, 1, 29–42. [Google Scholar]
- Hirose, Y. Syntheses of Damnacanthal, Damnacanthol, Norjuzunal and Norjuzunol, the Coloring Matters of Damnacanthus Spp. Chem. Pharm. Bull. 1960, 8, 417–426. [Google Scholar] [CrossRef]
- Roberts, J.L.; Rutledge, P.S.; Trebilcock, M.J. Experiments Directed Towards the Synthesis of Anthracyclinones.1 Synthesis of 2-formylmethoxyanthraquinones. Aust. J. Chem. 1977, 30, 1553–1560. [Google Scholar] [CrossRef]
- Roberts, J.L.; Rutledge, P.S. Experiments Directed Towards the Synthesis of Anthracyclinones. 111 P-Formylation of Hydroxyanthraquinonesby the Claisen Rearrangement. Aust. J. Chem. 1977, 30, 1743–1760. [Google Scholar] [CrossRef]
- Saha, K.; Lam, K.W.; Abas, F.; Hamzah, A.S.; Stanslas, J.; Hui, L.S.; Lajis, N.H. Synthesis of damnacanthal, a naturally occurring 9,10-anthraquinone and its analogues, and its biological evaluation against five cancer cell lines. Med. Chem. Res. 2013, 22, 2093–2104. [Google Scholar] [CrossRef]
- Wei, B.L.; Wu, S.H.; Chung, M.I.; Won, S.J.; Lin, C.N. Synthesis and cytotoxic effect of 1,3-dihydroxy-9,10-anthrquinone derivatives. Eur. J. Med. Chem. 2000, 35, 1089–1098. [Google Scholar] [CrossRef]
- Horii, Z.; Hakusui, H.; Momose, T. Synthetic Studies on Anthracyclinones. VII. Synthesis of Bisanhydroaklavinone. Chem. Pharm. Bull. 1968, 16, 1262–1265. [Google Scholar] [CrossRef]
- Castonguay, A.; Brassard, P. Total synthesis of (+)-averufin and (+)-bipolarin. Can. J. Chem. 1977, 55, 1324–1332. [Google Scholar] [CrossRef]
- Ma, H.M.; Liu, Z.Z.; Chen, S.Z. Regioselective Bromination of 3, 4-Dimethoxytoluene with N-Bromosuccinimide. Chin. Chem. lett. 2003, 14, 371–374. [Google Scholar]
- Abraham, S.A.; Waterhouse, D.N.; Mayer, L.D.; Cullis, P.R.; Madden, T.D.; Bally, M.B. The liposomal formulation of doxorubicin. Meth. Enzymol. 2005, 391, 71–97. [Google Scholar] [CrossRef]
- Iyer, M.; Tseng, Y.J.; Senese, C.L.; Liu, J.; Hopfinger, A.J. Prediction and Mechanistic Interpretation of Human Oral Drug Absorption Using MI-QSAR Analysis. Mol. Pharm. 2007, 4, 218–231. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Le, J.; Abraham, M.H.; Hersey, A.; Eddershaw, P.J.; Luscombe, C.N.; Butina, D.; Beck, G.; Sherborne, B.; Cooper, I.; Platts, J.A. Evaluation of human intestinal absorption data foruse in QSAR studies and quantitative relationship obtained with the Abraham descriptors. J. Pharm. Sci. 2001, 90, 749–784. [Google Scholar] [CrossRef]
- Saha, K.; Lajis, N.H.; Abas, F.; Naji, N.A.; Hamzah, A.S.; Shaari, K. Rearrangement in Lewis Acid-Catalyzed Friedel-Crafts Conditions: Evidence of Competitive Initial Protonation and Acylation. Aust. J. Chem. 2008, 61, 821–825. [Google Scholar] [CrossRef]
- Mossman, T. Rapid calorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Meth. 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–13h are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Akhtar, M.N.; Zareen, S.; Yeap, S.K.; Ho, W.Y.; Lo, K.M.; Hasan, A.; Alitheen, N.B. Total Synthesis, Cytotoxic Effects of Damnacanthal, Nordamnacanthal and Related Anthraquinone Analogues. Molecules 2013, 18, 10042-10055. https://doi.org/10.3390/molecules180810042
Akhtar MN, Zareen S, Yeap SK, Ho WY, Lo KM, Hasan A, Alitheen NB. Total Synthesis, Cytotoxic Effects of Damnacanthal, Nordamnacanthal and Related Anthraquinone Analogues. Molecules. 2013; 18(8):10042-10055. https://doi.org/10.3390/molecules180810042
Chicago/Turabian StyleAkhtar, Muhammad Nadeem, Seema Zareen, Swee Keong Yeap, Wan Yong Ho, Kong Mun Lo, Aurangzeb Hasan, and Noorjahan Banu Alitheen. 2013. "Total Synthesis, Cytotoxic Effects of Damnacanthal, Nordamnacanthal and Related Anthraquinone Analogues" Molecules 18, no. 8: 10042-10055. https://doi.org/10.3390/molecules180810042