Modeling the Interaction of Dodecylphosphocholine Micelles with the Anticoccidial Peptide PW2 Guided by NMR Data

Abstract
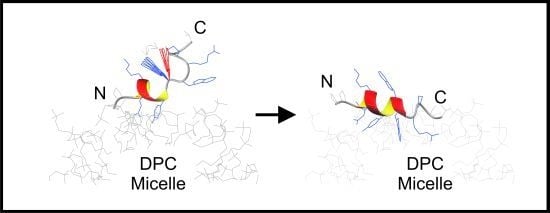
:
1. Introduction
2. Results
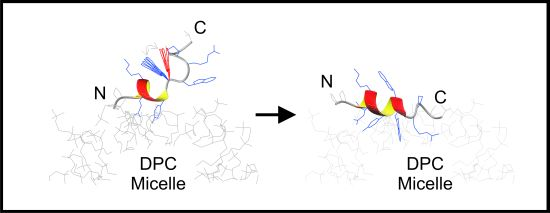
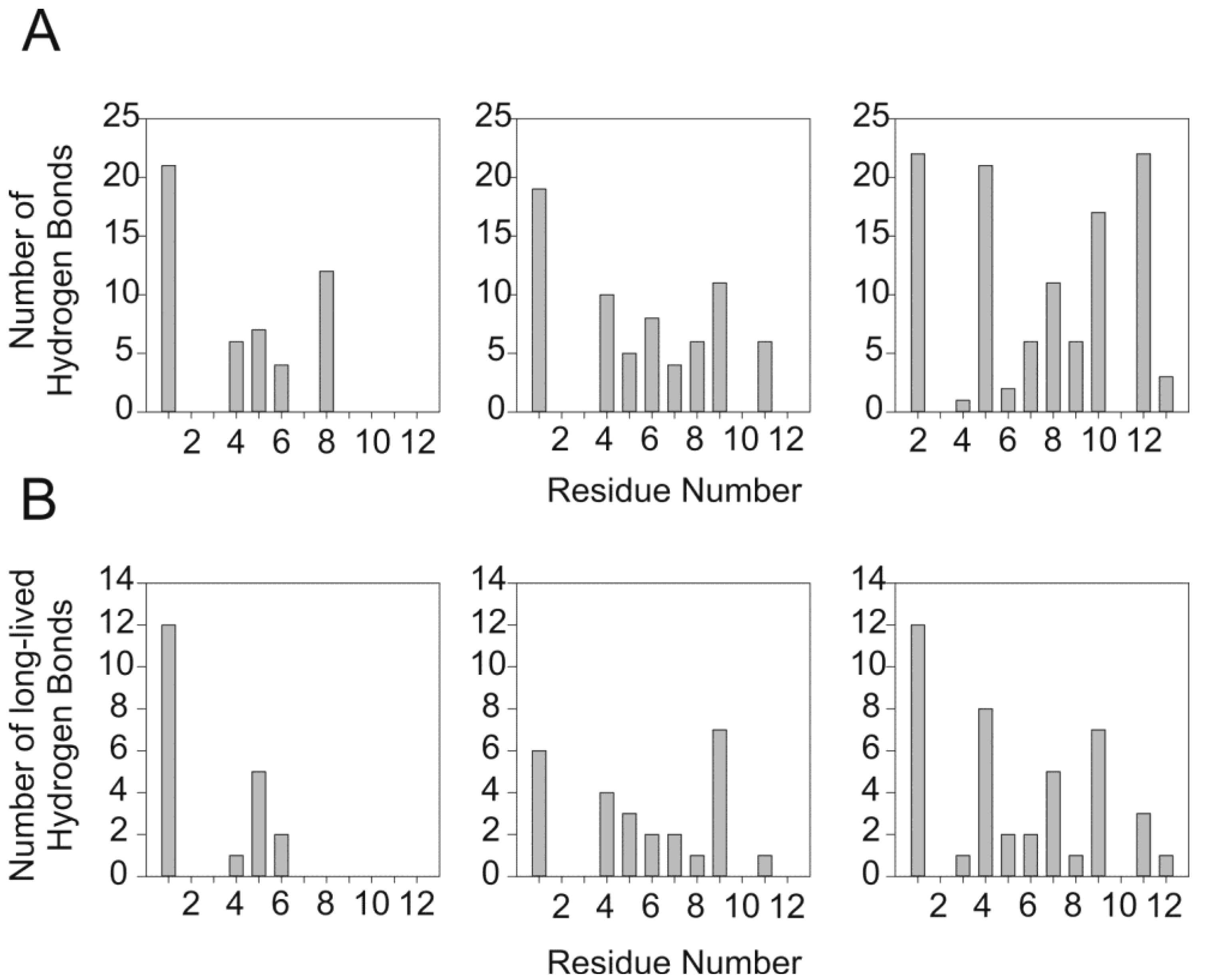
2.1. Restrained MD Simulation of PW2 Interacting with a Pre-Built DPC Micelle
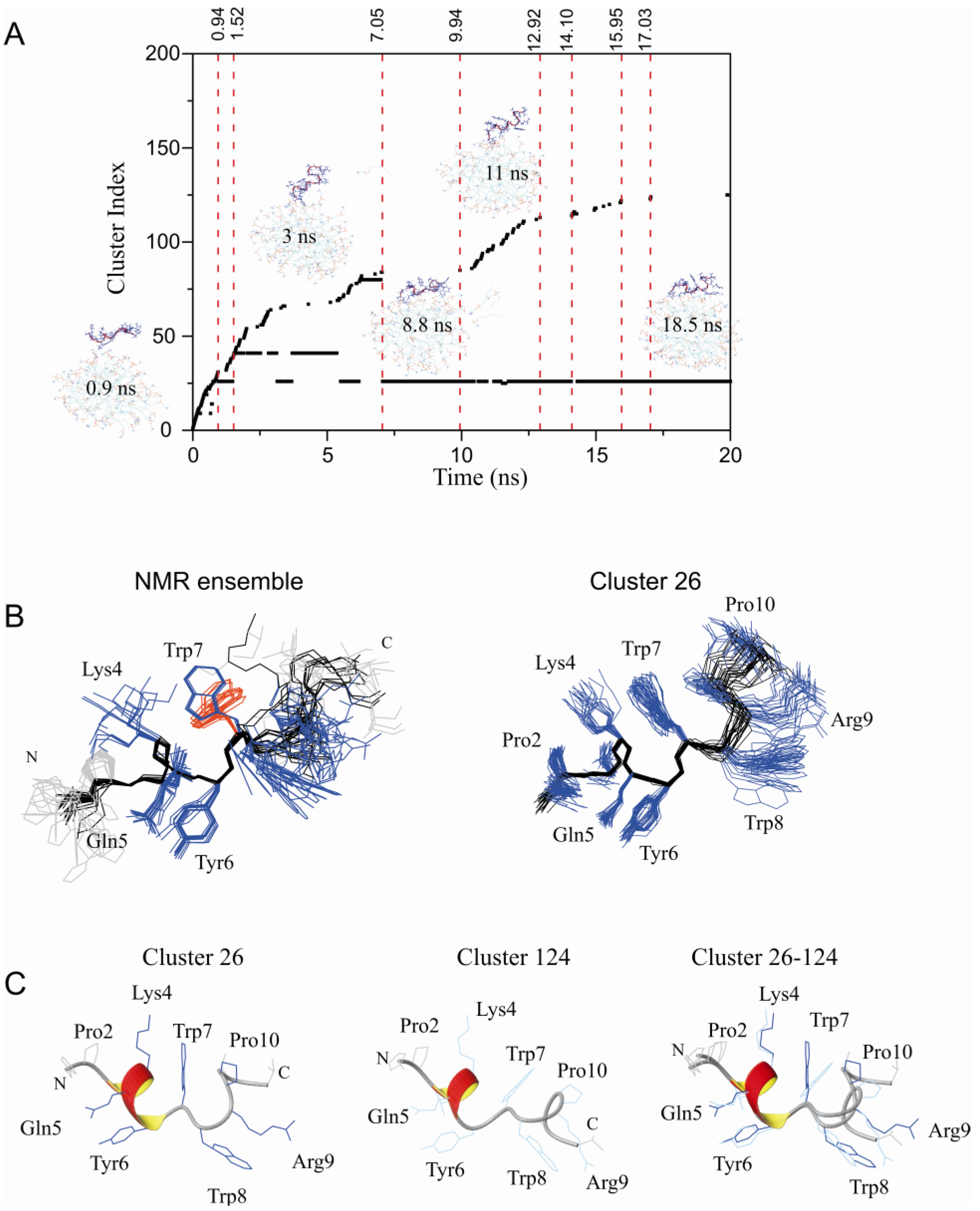
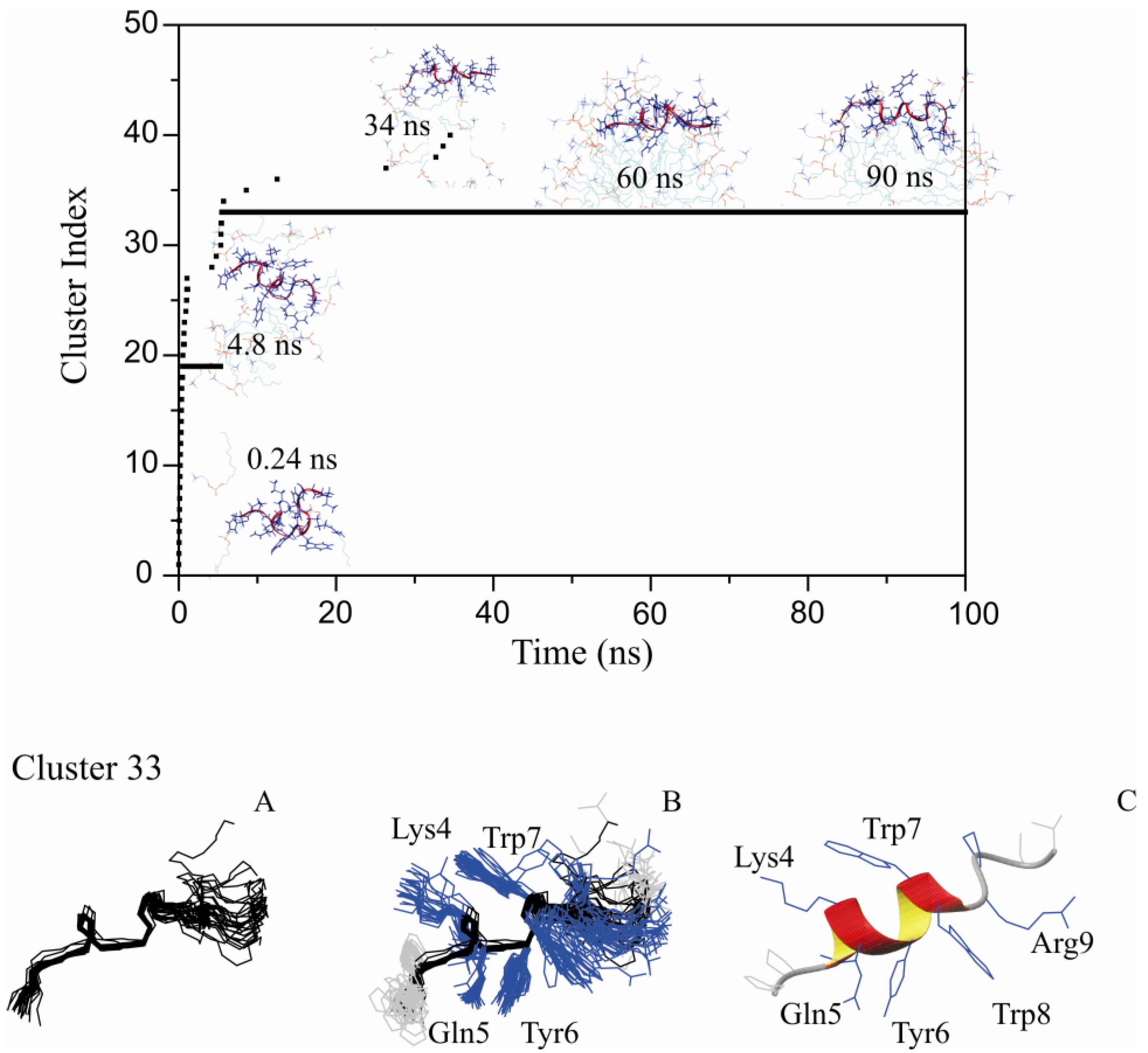
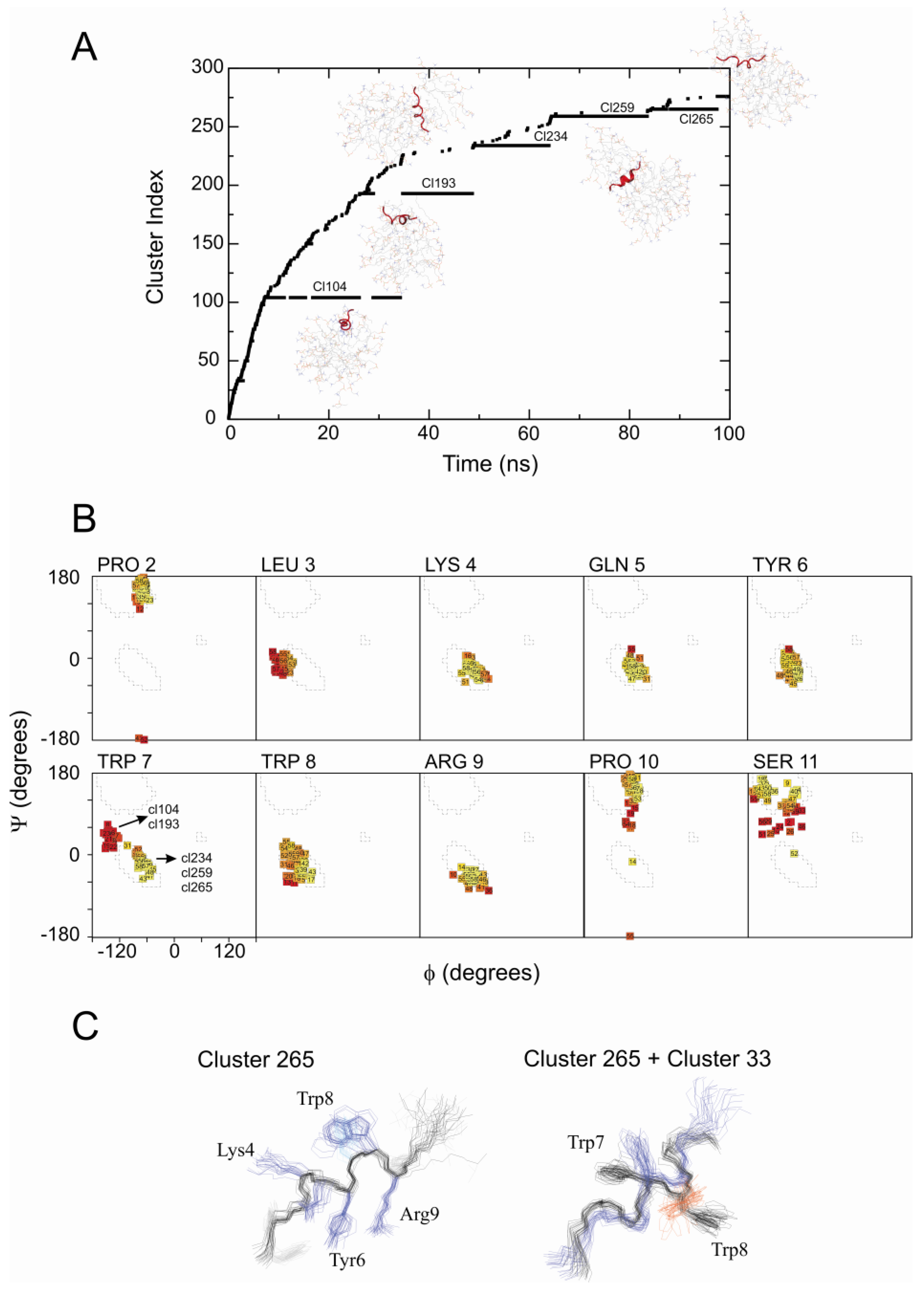
2.2. Restrained MD Simulation of PW2 Interacting with free-DPC during the Spontaneous Micellization Showed the Stabilization of a Tighter Micelle-Bound Conformation
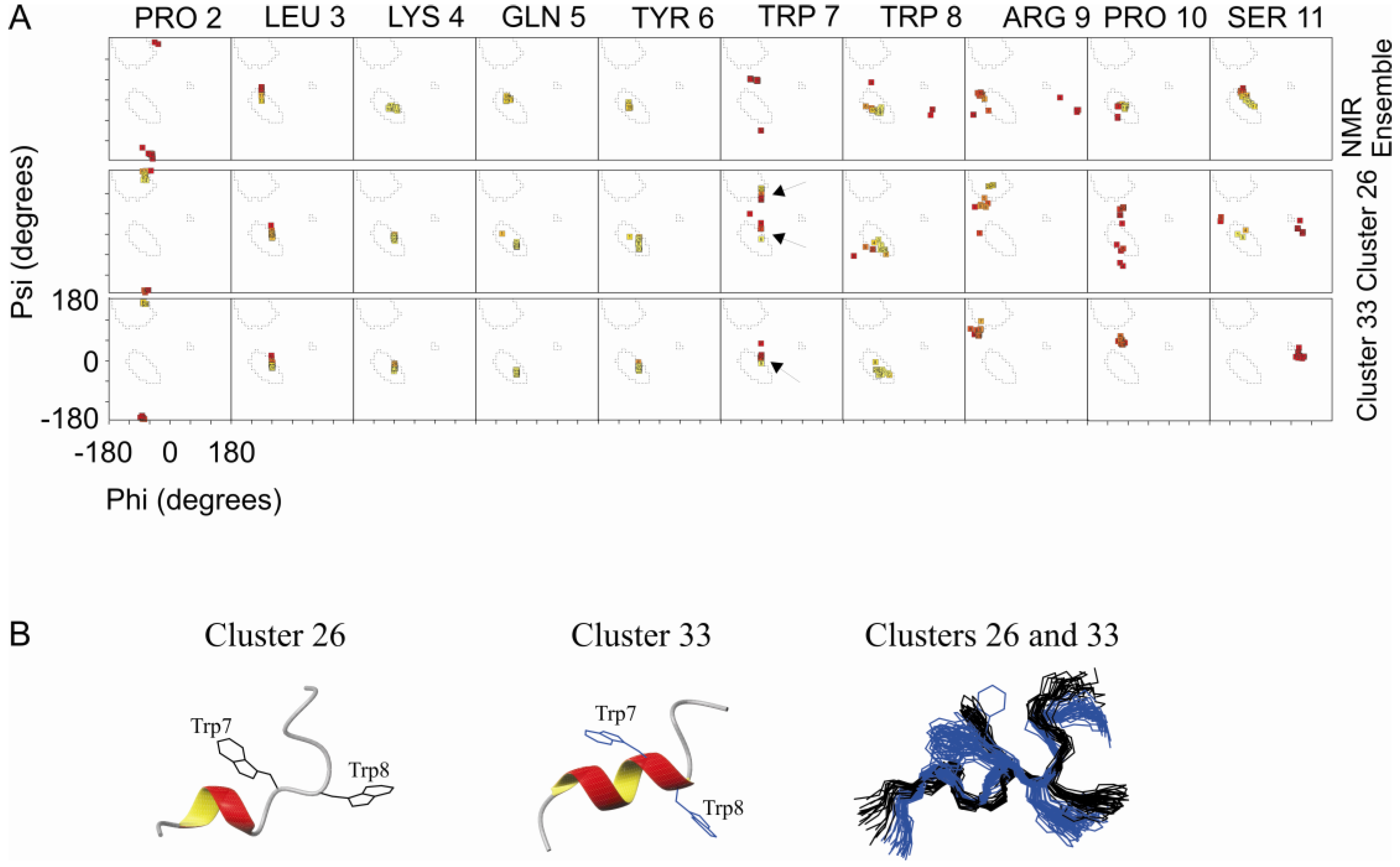
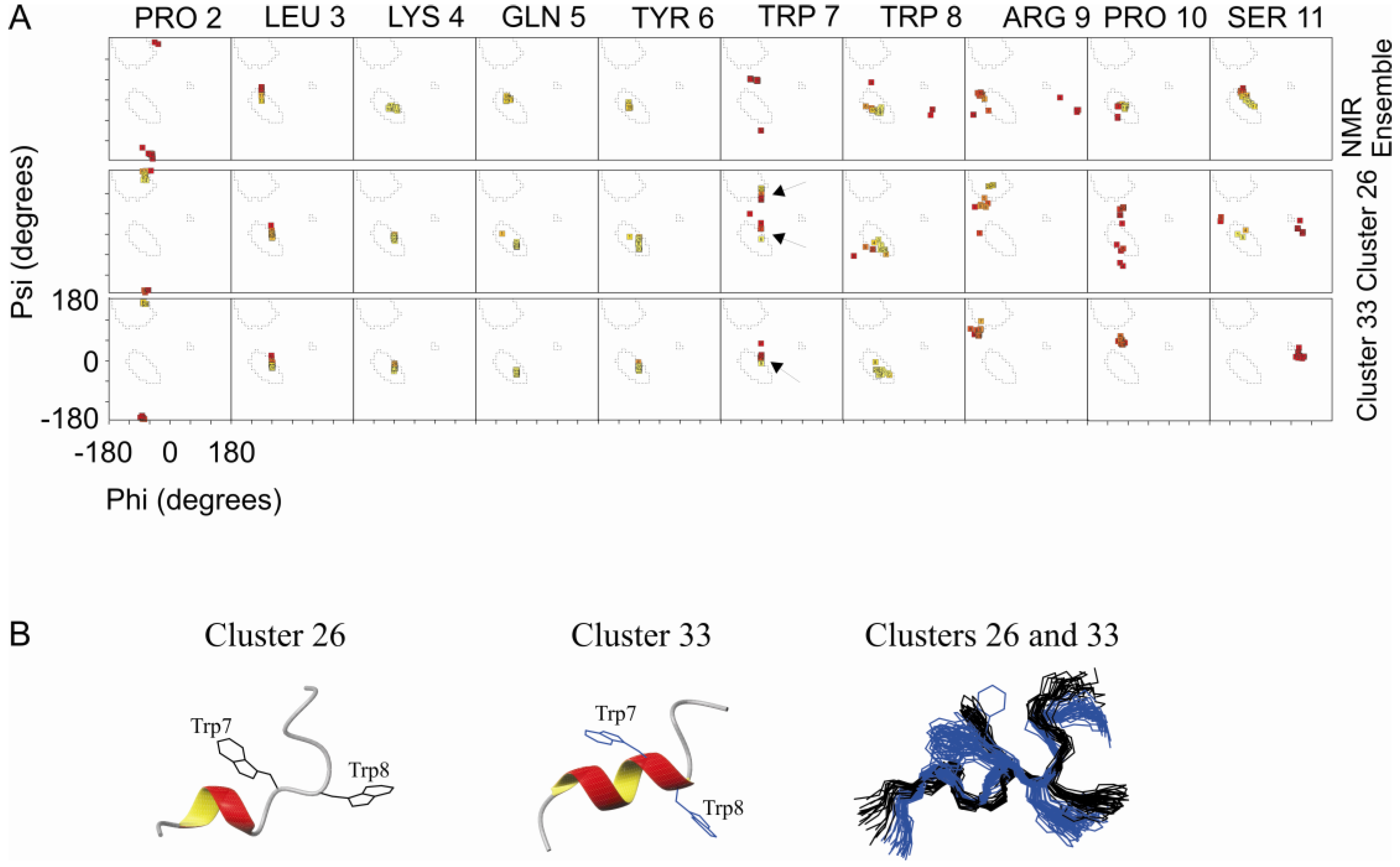
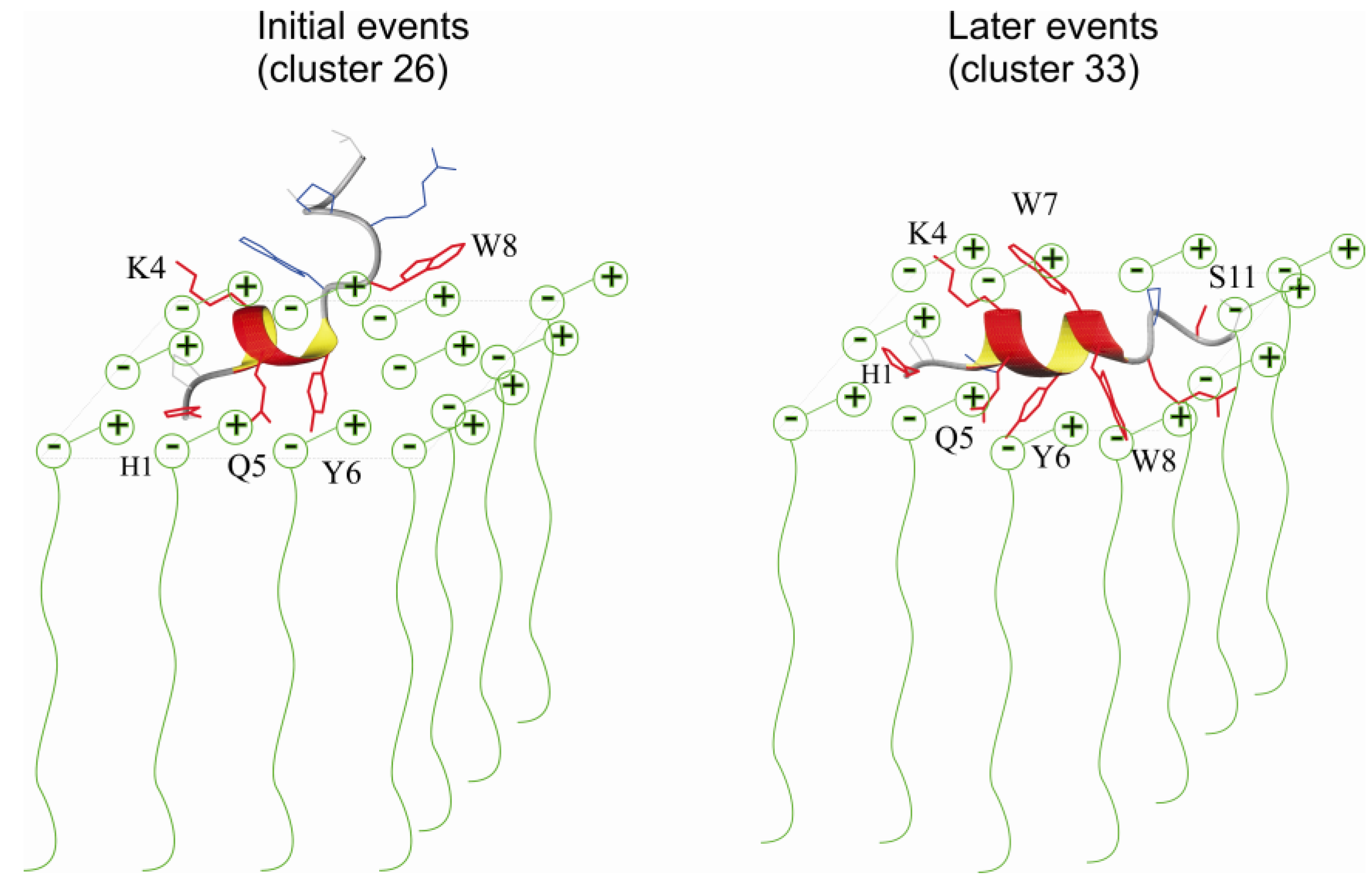
2.3. Comparison of Clusters 26 and 33
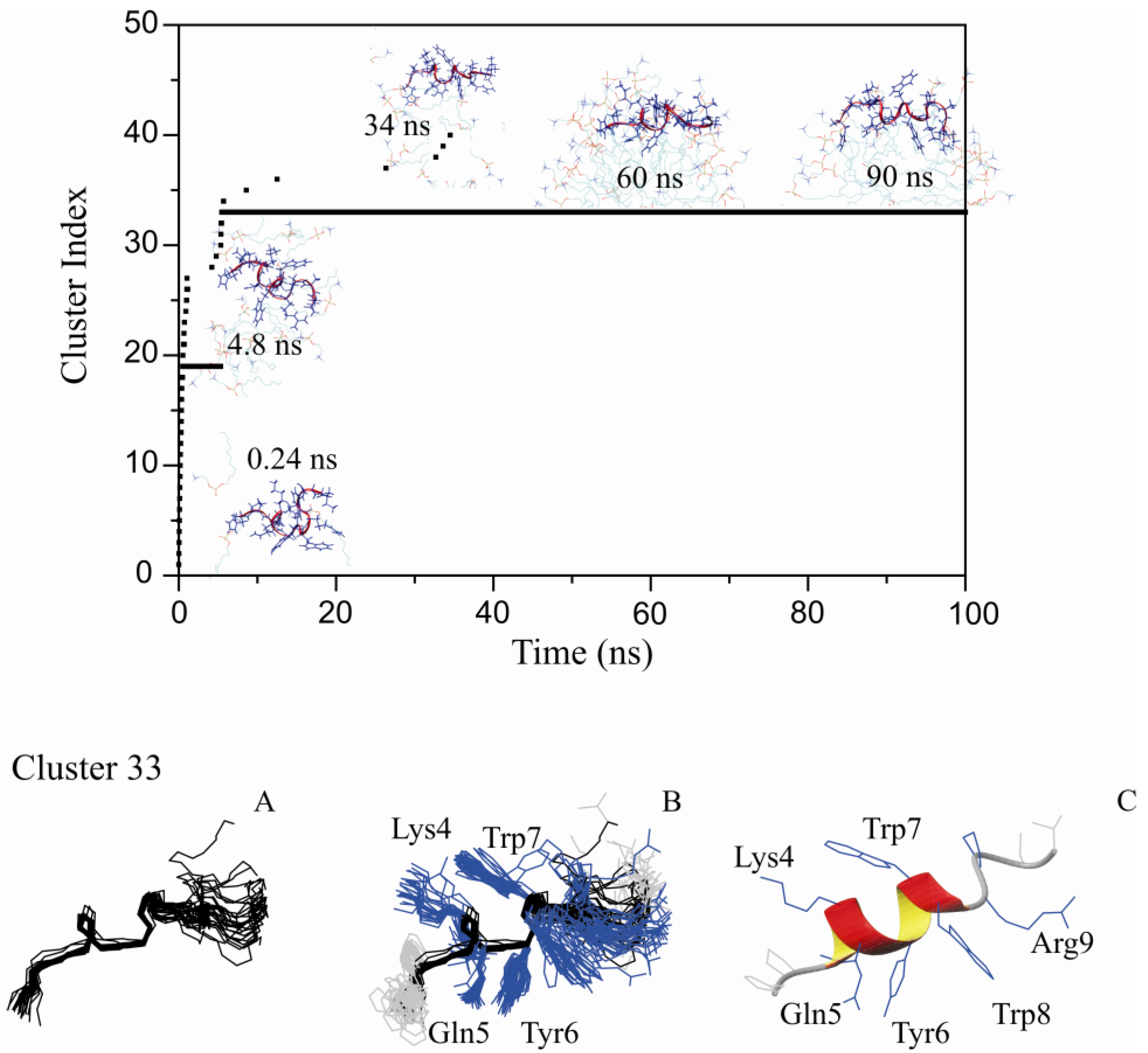
2.4. The Unrestrained MD Simulation of PW2 Interacting with Free-DPC Molecules during the Spontaneous Micellization was also Able to Probe the Bound State

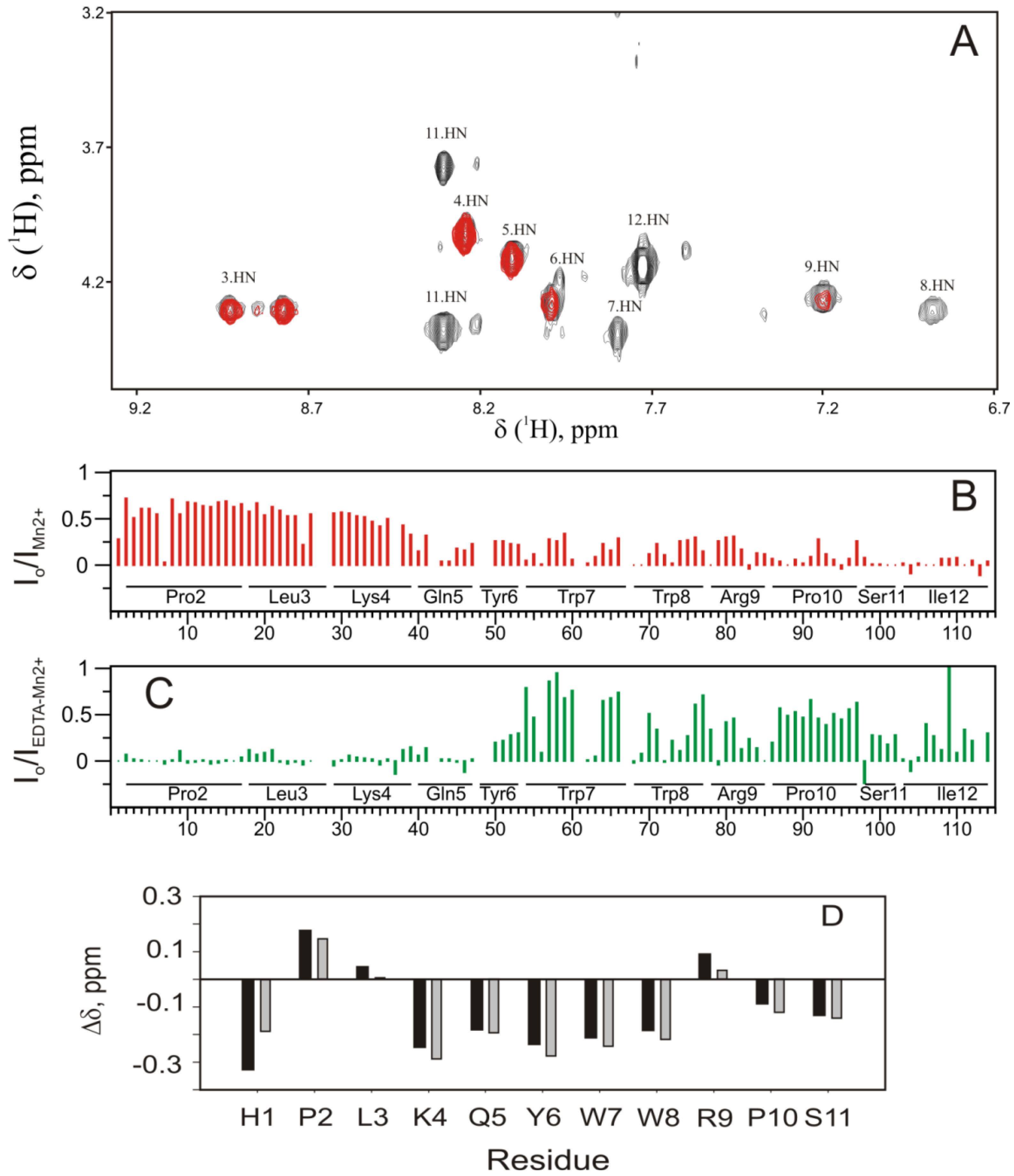
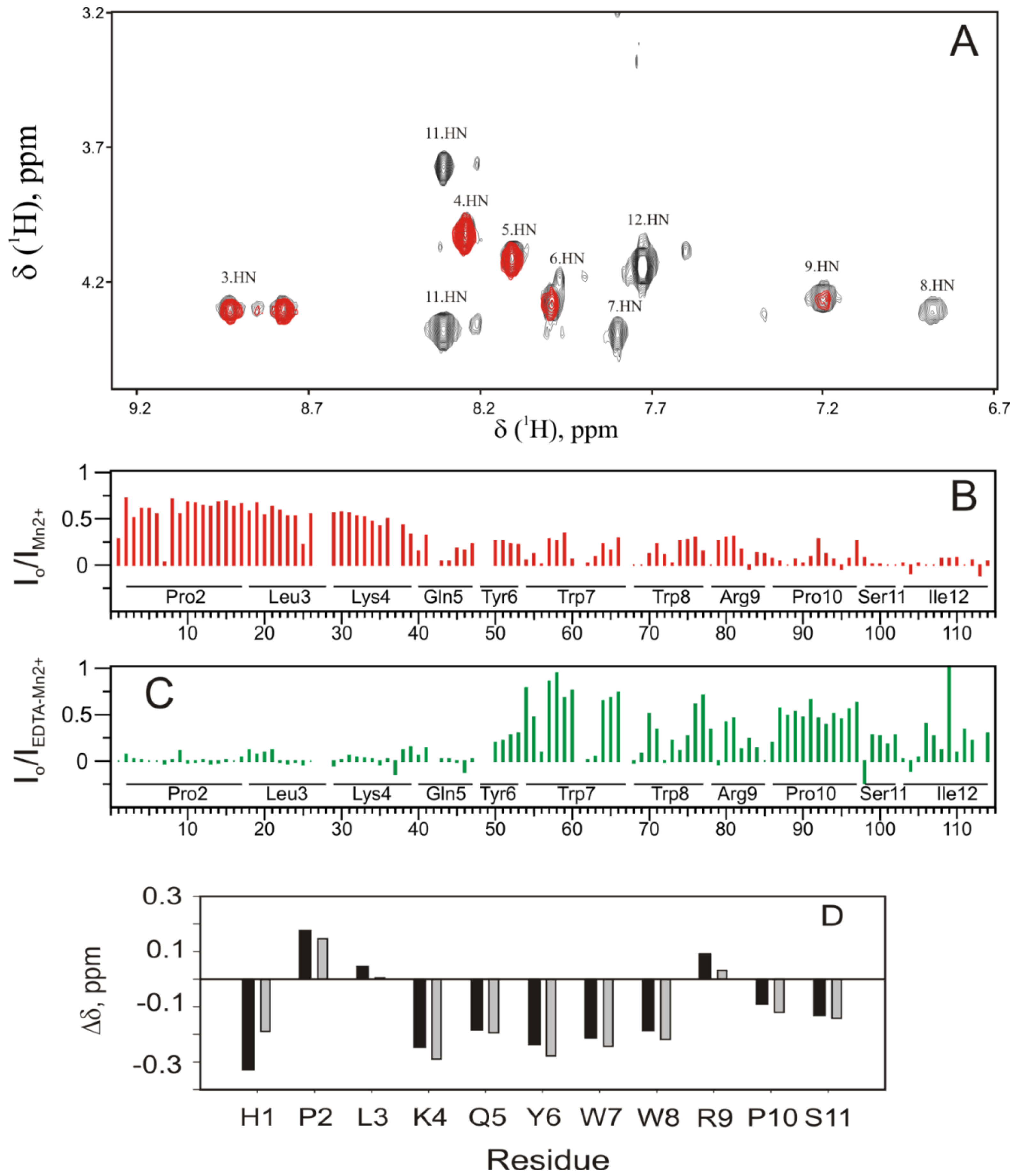
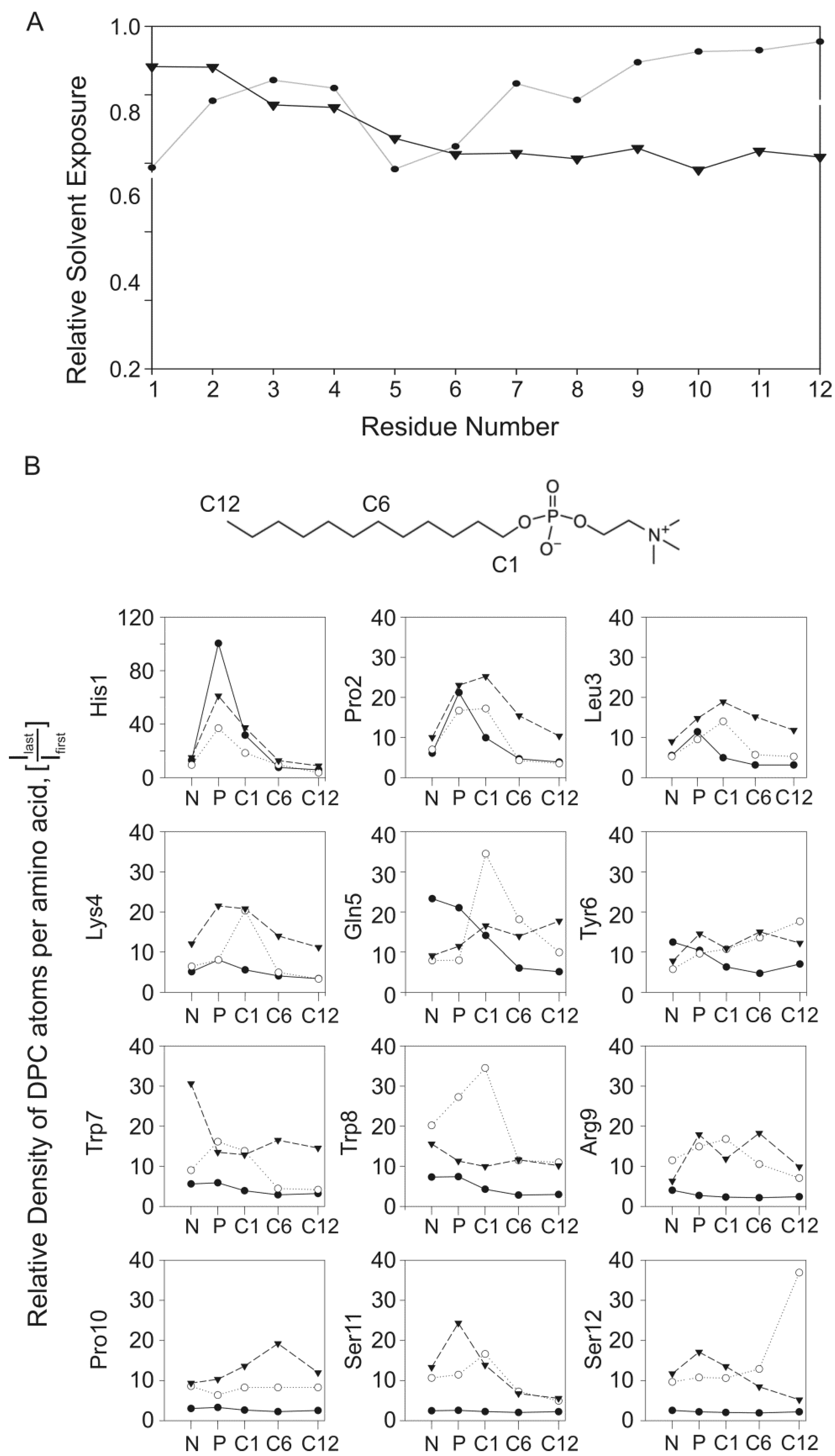
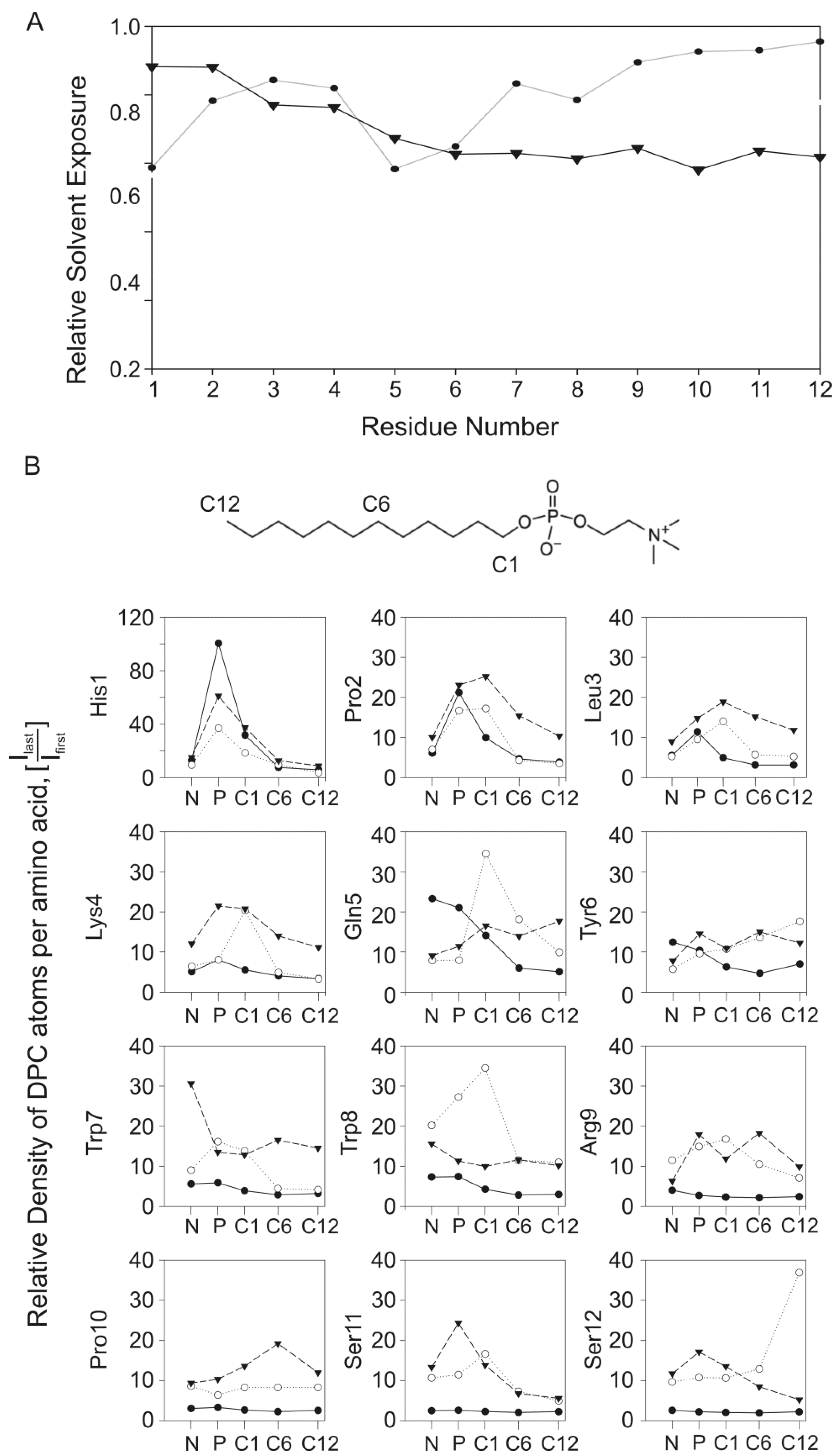
2.5. Experimental Results Are in Agreement with the MD Simulations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D (m2/s) | s.d. | |
|---|---|---|
| a PW2 free | 2.63 × 10−1° | ± 3 × 10−12 |
| b DPCm | 2.62 × 10−11 | ± 4 × 10−13 |
| c PW2 + DPCm | 2.37 × 10−11 | ± 6 × 10−13 |
| d PW2 + DPCm | 2.34 × 10−11 | ± 5 × 10−13 |
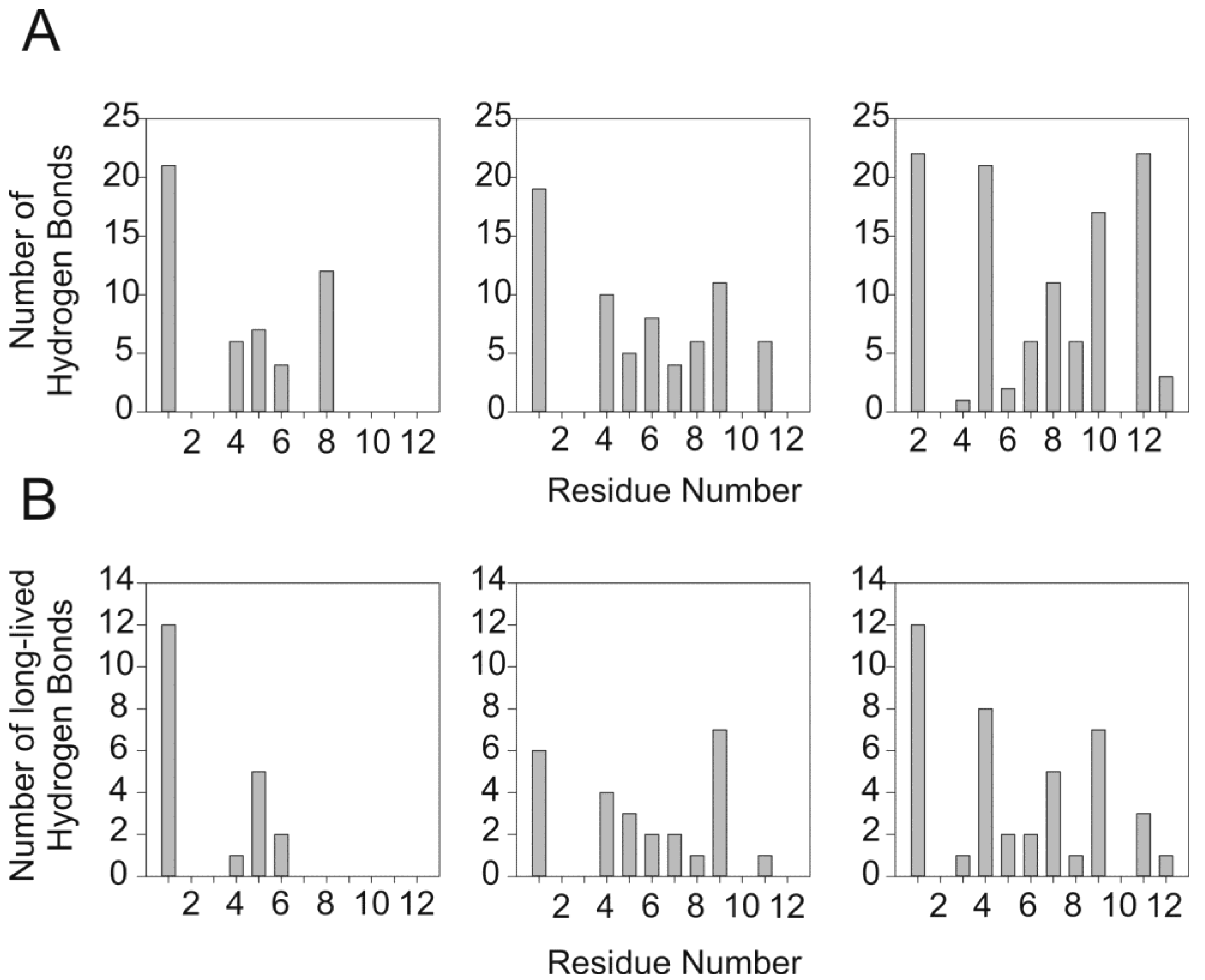
2.6. Analysis and Quantification of the Interaction of PW2 with DPC
3. Discussion

3.1. Interaction Forces
3.2. Discrete Conformational States of PW2
4. Experimental
4.1. Chemistry Procedures
4.1.1. Preparation of the Peptide Sample
4.1.2. Peptide Assignment
4.1.3. DPC Assignment
4.1.4. Analysis of the PW2/DPC Interaction by the Paramagnetic Relaxation Enhancement (PRE) Effect of Mn2+ Ions in Solution
4.1.5. Measurement of the Translational Diffusion Coefficient
4.1.6. Tendency to Helix-Chemical Shift Difference
4.2. Molecular Dynamics Simulation
4.2.1. General Setup
4.2.2. NMR Distance and Dihedral Restraints
4.2.3. Model Setup of PW2 Interacting with a DPC Micelle
4.2.4. Restrained/Unrestrained MD Simulations of PW2 in the Presence of free-DPC Molecules with Spontaneous Micelle Formation
4.2.5. Analysis of MD Simulations
4.2.5.1. Clustering Analysis
4.2.5.2. Density Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Bulet, P.; Stocklin, R.; Menin, L. Anti-microbial peptides: From invertebrates to vertebrates. Immunol. Rev. 2004, 198, 169–184. [Google Scholar] [CrossRef]
- Thomma, B.P.H.J.; Cammue, B.P.A.; Thevissen, K. Mode of action of plant defensins suggests therapeutic potential. Curr. Drug Targets Infect. Disord. 2003, 3, 1–8. [Google Scholar]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Nicolas, P.; Mor, A. Peptides as weapons against microorganisms in the chemical defense system of vertebrates. Annu. Rev. Microbiol. 1995, 49, 277–304. [Google Scholar] [CrossRef]
- Wu, M.H.; Maier, E.; Benz, R.; Hancock, R.E.W. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry 1999, 38, 7235–7242. [Google Scholar] [CrossRef]
- Lohner, K.; Staudegger, E.; Prenner, E.J.; Lewis, R.N.A.H.; Kriechbaum, M.; Degovics, G.; McElhaney, R.N. Effect of staphylococcal δ-lysin on the thermotropic phase behavior and vesicle morphology of dimyristoylphosphatidylcholine lipid bilayer model membranes. differential scanning calorimetric, 31 p nuclear magnetic resonance and fourier transform infrared. Biochemistry 1999, 38, 16514–16528. [Google Scholar] [CrossRef]
- Da Silva, A.; Kawazoe, U.; Freitas, F.F.T.; Gatti, M.S.V; Dolder, H.; Schumacher, R.I.; Juliano, M.A.; da Silva, M.J.; Leite, A. Avian anticoccidial activity of a novel membrane-interactive peptide selected from phage display libraries. Mol. Biochem. Parasitol. 2002, 120, 53–60. [Google Scholar] [CrossRef]
- Cruzeiro-Silva, C.; Gomes-Neto, F.; Tinoco, L.W.; Cilh, E.M.; Barros, P.V.R.; Lapido-Loureiro, P.A.; Bisch, P.M.; Almeida, F.C.L.; Valente, A.P. Structural biology of membrane-acting peptides: Conformational plasticity of anticoccidial peptide PW2 probed by solution NMR. Biochim. Biophys. Acta Biomembr. 2007, 1768, 3182–3192. [Google Scholar] [CrossRef]
- Villali, J.; Kern, D. Choreographing an enzyme’s dance. Curr. Opin. Chem. Biol. 2010, 14, 636–643. [Google Scholar] [CrossRef]
- Valente, A.P.; Miyamoto, C.A.; Almeida, F.C.L. Implications of protein conformational diversity for binding and development of new biological active compounds. Curr. Med. Chem. 2006, 13, 3697–3703. [Google Scholar] [CrossRef]
- Henzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef]
- Mukrasch, M.D.; Markwick, P.; Biernat, J.; von Bergen, M.; Bernado, P.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M.; Blackledge, M. Highly populated turn conformations in natively unfolded Tau protein identified from residual dipolar couplings and molecular simulation. J. Am. Chem. Soc. 2007, 129, 5235–5243. [Google Scholar] [CrossRef]
- Tinoco, L.W.; da Silva, A.; Leite, A.; Valente, A.P.; Almeida, F.C.L. NMR structure of PW2 bound to SDS micelles—A tryptophan-rich anticoccidial peptide selected from phage display libraries. J. Biol. Chem. 2002, 277, 36351–36356. [Google Scholar]
- Tinoco, L.W.; Gomes-Neto, F.; Valente, A.P.; Almeida, F.C.L. Effect of micelle interface on the binding of anticoccidial PW2 peptide. J. Biomol. NMR 2007, 39, 315–322. [Google Scholar] [CrossRef]
- Mierke, D.F.; Kurz, M.; Kessler, H. Peptide flexibility and calculations of an ensemble of molecules. J. Am. Chem. Soc. 1994, 116, 1042–1049. [Google Scholar] [CrossRef]
- Bonvin, A.; Boelens, R.; Kaptein, R. Time-Averaged and ensemble-averaged direct noe restraints. J. Biomol. NMR 1994, 4, 143–149. [Google Scholar]
- Epand, R.M.; Vogel, H.J. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta Biomembr. 1999, 1462, 11–28. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Patist, A.; Kanicky, J.R.; Shukla, P.K.; Shah, D.O. Importance of micellar kinetics in relation to technological processes. J. Colloid Interface Sci. 2002, 245, 1–15. [Google Scholar] [CrossRef]
- Marrink, S.J.; Tieleman, D.P.; Mark, A.E. Molecular dynamics simulation of the kinetics of spontaneous micelle formation. J. Phys. Chem. B 2000, 104, 12165–12173. [Google Scholar] [CrossRef]
- Bond, P.J.; Cuthbertson, J.M.; Deol, S.S.; Sansom, M.S.P. MD simulations of spontaneous membrane protein/detergent micelle formation. J. Am. Chem. Soc. 2004, 126, 15948–15949. [Google Scholar] [CrossRef]
- Galloux, M.; Libersou, S.; Alves, I.D.; Marquant, R.; Salgado, G.F.; Rezaei, H.; Lepault, J.; Delmas, B.; Bouaziz, S.; Morellet, N. NMR structure of a viral peptide inserted in artificial membranes: A view on the early steps of the birnavirus entry process. J. Biol. Chem. 2010, 285, 19409–19421. [Google Scholar] [CrossRef]
- Cook, G.a; Opella, S.J. Secondary structure, dynamics, and architecture of the p7 membrane protein from hepatitis C virus by NMR spectroscopy. Biochim. Biophys. Acta 2011, 1808, 1448–1453. [Google Scholar] [CrossRef]
- Respondek, M.; Madl, T.; Gobl, C.; Golser, R.; Zangger, K. Mapping the orientation of helices in micelle-bound peptides by paramagnetic relaxation waves. J. Am. Chem. Soc. 2007, 129, 5228–5234. [Google Scholar] [CrossRef]
- Wüthrich, K. NMR of Proteins and Nucleic Acids; Wiley-Interscience: New York, NY, USA, 1986; Volume 1. [Google Scholar]
- Pardi, A.; Wagner, G.; Wüthrich, K. Protein conformation and proton nuclear-magnetic-resonance chemical shifts. Eur. J. Biochem. FEBS 1983, 137, 445–454. [Google Scholar] [CrossRef]
- Schwarzinger, S.; Kroon, G.J.; Foss, T.R.; Wright, P.E.; Dyson, H.J. Random coil chemical shifts in acidic 8 M urea: Implementation of random coil shift data in NMRView. J. Biomol. NMR 2000, 18, 43–48. [Google Scholar] [CrossRef]
- Merutka, G.; Dyson, H.J.; Wright, P.E. “Random coil” 1H chemical shifts obtained as a function of temperature and trifluoroethanol concentration for the peptide series GGXGG. J. Biomol. NMR 1995, 5, 14–24. [Google Scholar] [CrossRef]
- Bonvin, A.; Brunger, A.T. Do NOE distances contain enough information to assess the relative populations of multi-conformer structures? J. Biomol. NMR 1996, 7, 72–76. [Google Scholar]
- Daura, X.; Antes, I.; van Gunsteren, W.F.; Thiel, W.; Mark, A.E. The effect of motional averaging on the calculation of NMR-derived structural properties. Proteins Struct. Funct. Genet. 1999, 36, 542–555. [Google Scholar] [CrossRef]
- Wennerstrom, H.; Lindman, B.; Soderman, O.; Drakenberg, T.; Rosenholm, J.B. C-13 Magnetic-Relaxation in micellar solutions - influence of aggregate motion on t1. J. Am. Chem. Soc. 1979, 101, 6860–6864. [Google Scholar] [CrossRef]
- Bax, A.; Davis, D.G. Mlev-17-Based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Resonance 1985, 65, 355–360. [Google Scholar]
- Piotto, M.; Saudek, V.; Sklenar, V. Gradient-Tailored excitation for single-quantum nmr-spectroscopy of aqueous-solutions. J. Biomol. NMR 1992, 2, 661–665. [Google Scholar] [CrossRef]
- Marion, D.; Wuthrich, K. Application of phase sensitive two-dimensional correlated spectroscopy (cosy) for measurements of H-1-H-1 spin-spin coupling-constants in proteins. Biochem. Biophys. Res. Commun. 1983, 113, 967–974. [Google Scholar] [CrossRef]
- Bax, A.; Summers, M.F. H-1 and C-13 assignments from sensitivity-enhanced detection of heteronuclear multiple-bond connectivity by 2d multiple quantum Nmr. J. Am. Chem. Soc. 1986, 108, 2093–2094. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. Nmrpipe—A multidimensional spectral processing system based on unix pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar]
- Johnson, B.A.; Blevins, R.A. Nmr View—A computer-program for the visualization and analysis of nmr data. J. Biomol. NMR 1994, 4, 603–614. [Google Scholar] [CrossRef]
- Pelta, M.D.; Morris, G.A.; Stchedroff, M.J.; Hammond, S.J. A one-shot sequence for high-resolution diffusion-ordered spectroscopy. Magn. Resonance Chem. 2002, 40, S147–S152. [Google Scholar] [CrossRef]
- Li, H.; Robertson, A.D.; Jensen, J.H. Very fast empirical prediction and rationalization of protein pK(a) values. Proteins Struct. Funct. Bioinforma. 2005, 61, 704–721. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Vanderspoel, D.; Vandrunen, R. Gromacs—A message-passing parallel molecular-dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Berger, O.; Edholm, O.; Jahnig, F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 1997, 72, 2002–2013. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tiradorives, J. The opls potential functions for proteins - energy minimizations for crystals of cyclic-peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Tieleman, D.P.; MacCallum, J.L.; Ash, W.L.; Kandt, C.; Xu, Z.T.; Monticelli, L. Membrane protein simulations with a united-atom lipid and all-atom protein model: Lipid-Protein interactions, side chain transfer free energies and model proteins. J. Phys. Condens. Matter 2006, 18, S1221–S1234. [Google Scholar] [CrossRef]
- Hermans, J.; Berendsen, H.J.C.; Vangunsteren, W.F.; Postma, J.P.M. A consistent empirical potential for water-protein interactions. Biopolymers 1984, 23, 1513–1518. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle—An analytical version of the shake and rattle algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Torda, A.E.; Scheek, R.M.; Vangunsteren, W.F. Time-Dependent distance restraints in molecular-dynamics simulations. Chem. Phys. Lett. 1989, 157, 289–294. [Google Scholar] [CrossRef]
- Brunger, A.T.; Adams, P.D.; Clore, G.M.; DeLano, W.L.; Gros, P.; Grosse-Kunstleve, R.W.; Jiang, J.S.; Kuszewski, J.; Nilges, M.; Pannu, N.S.; et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998, 54, 905–921. [Google Scholar]
- Tieleman, D.P.; van der Spoel, D.; Berendsen, H.J.C. Molecular dynamics simulations of dodecylphosphocholine micelles at three different aggregate sizes: Micellar structure and chain relaxation. J. Phys. Chem. B 2000, 104, 6380–6388. [Google Scholar]
- Schwieters, C.D.; Clore, G.M. The VMD-XPLOR visualization package for NMR structure refinement. J. Magn. Resonance 2001, 149, 239–244. [Google Scholar] [CrossRef]
- Koradi, R.; Billeter, M.; Wuthrich, K. Molmol: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 51–55. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Rullmann, J.A.C.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar]
- Sample Availability: Samples of the PW2 is available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gomes-Neto, F.; Valente, A.P.; Almeida, F.C.L. Modeling the Interaction of Dodecylphosphocholine Micelles with the Anticoccidial Peptide PW2 Guided by NMR Data. Molecules 2013, 18, 10056-10080. https://doi.org/10.3390/molecules180810056
Gomes-Neto F, Valente AP, Almeida FCL. Modeling the Interaction of Dodecylphosphocholine Micelles with the Anticoccidial Peptide PW2 Guided by NMR Data. Molecules. 2013; 18(8):10056-10080. https://doi.org/10.3390/molecules180810056
Chicago/Turabian StyleGomes-Neto, Francisco, Ana Paula Valente, and Fabio C. L. Almeida. 2013. "Modeling the Interaction of Dodecylphosphocholine Micelles with the Anticoccidial Peptide PW2 Guided by NMR Data" Molecules 18, no. 8: 10056-10080. https://doi.org/10.3390/molecules180810056