Halogen Bonding in (Z)-2-Iodocinnamaldehyde


Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
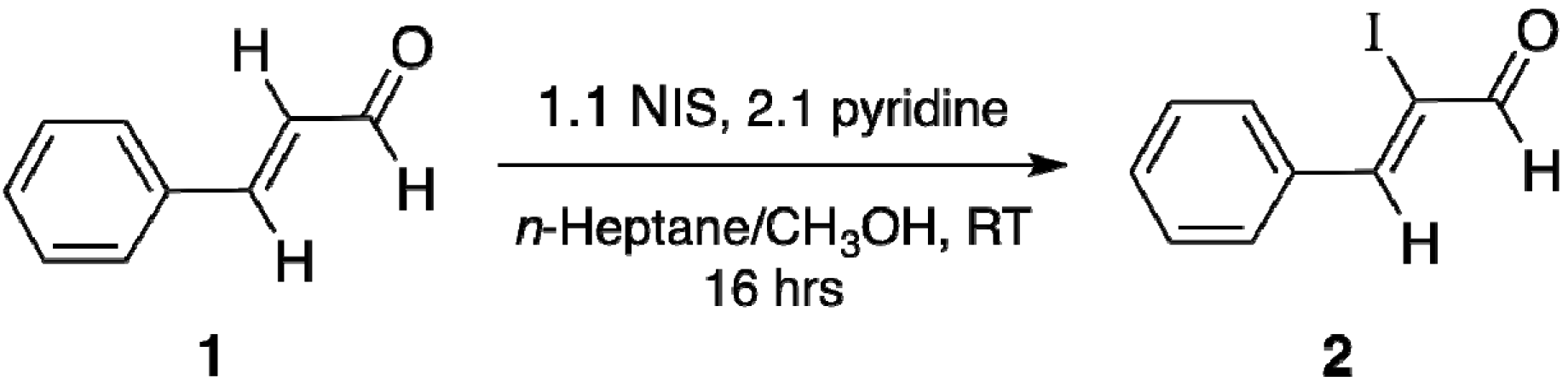
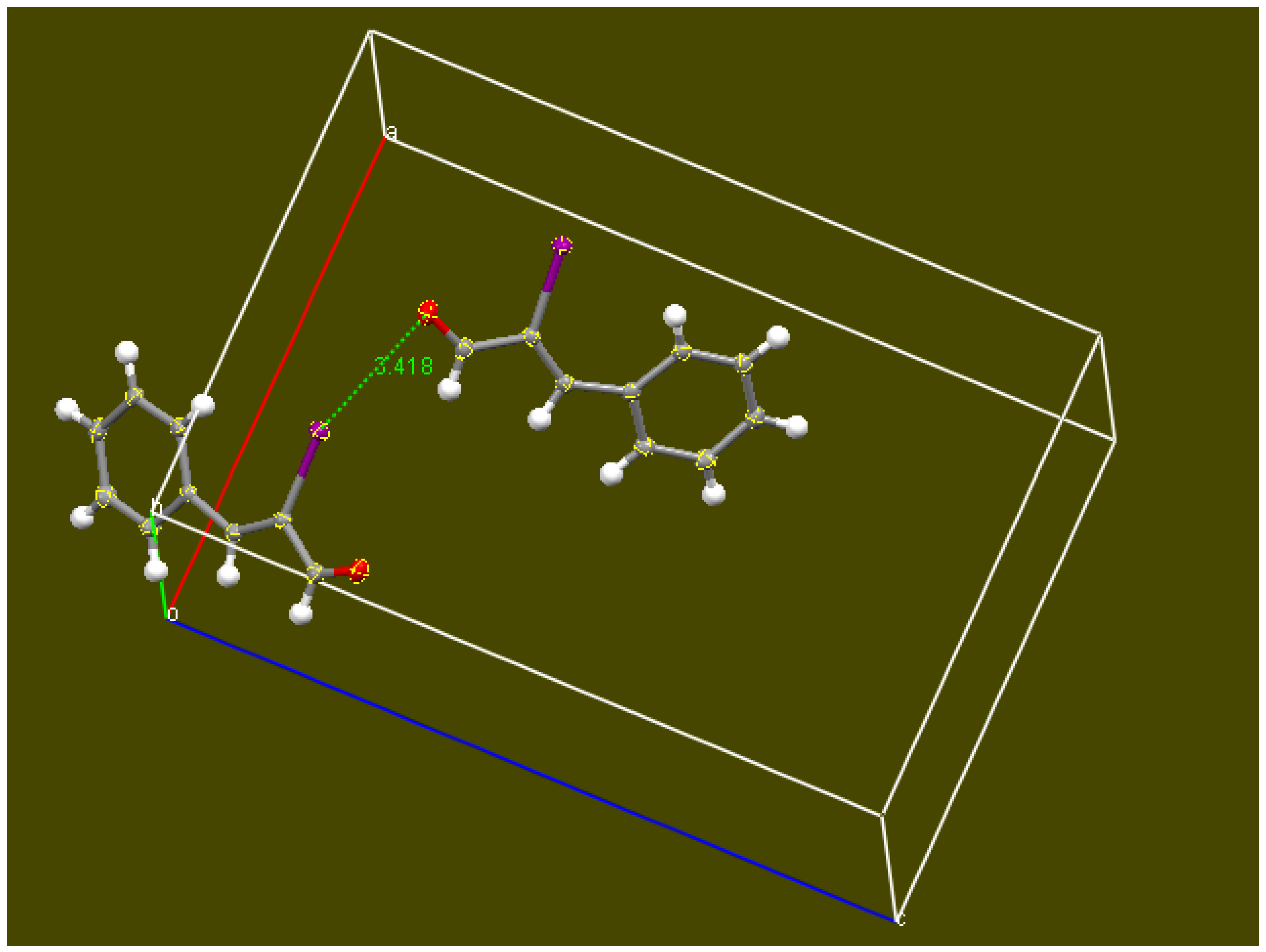
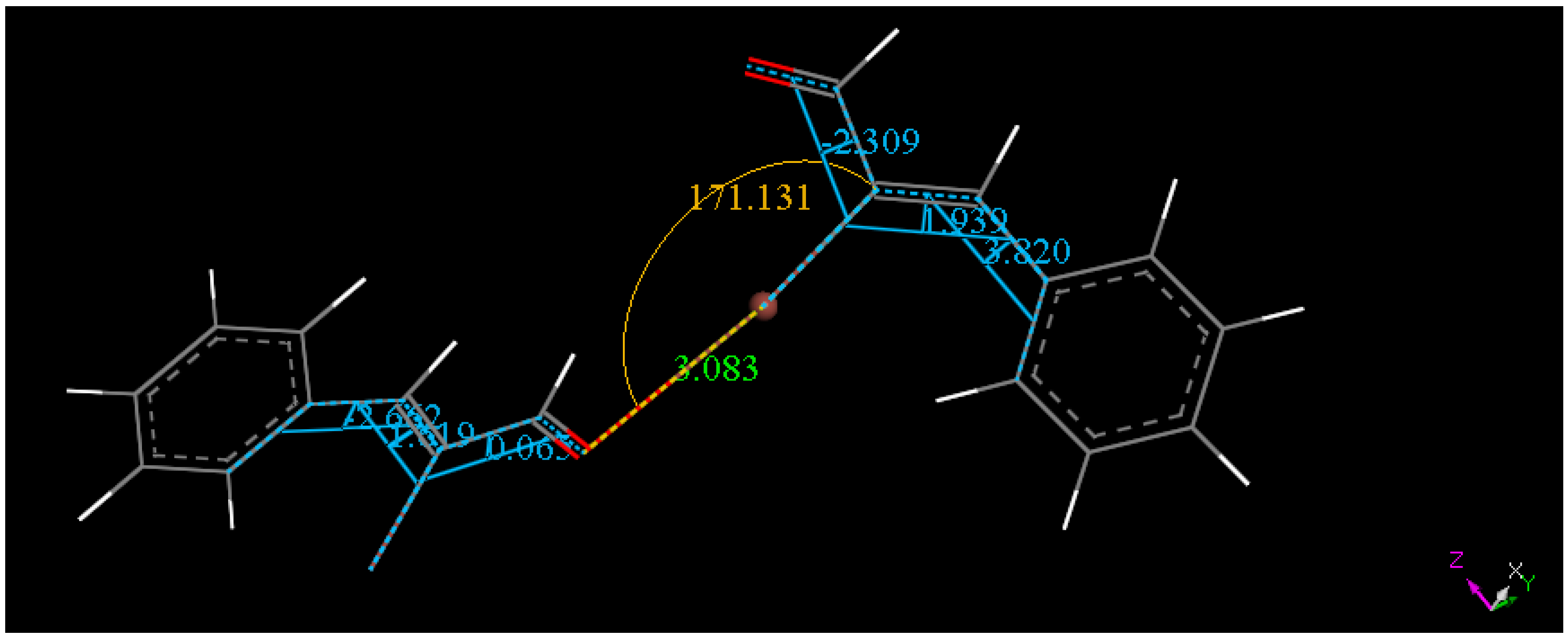
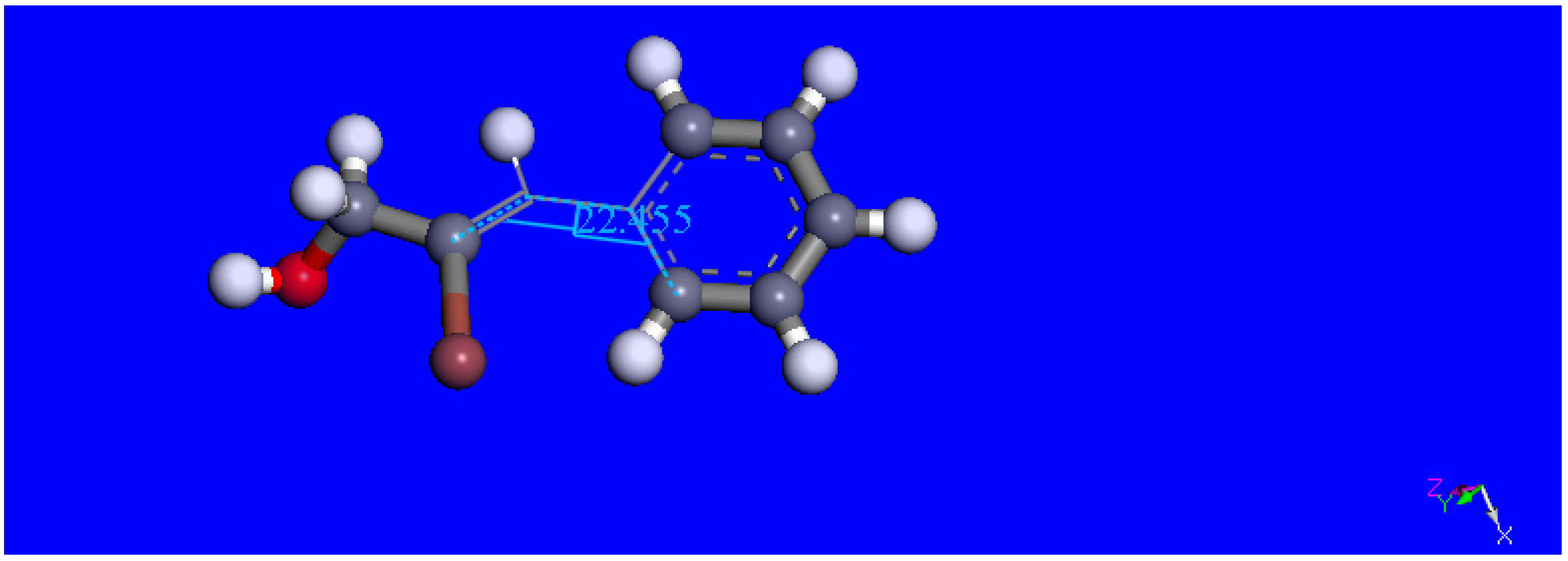
2.2. Single Crystal X-ray Diffraction Study
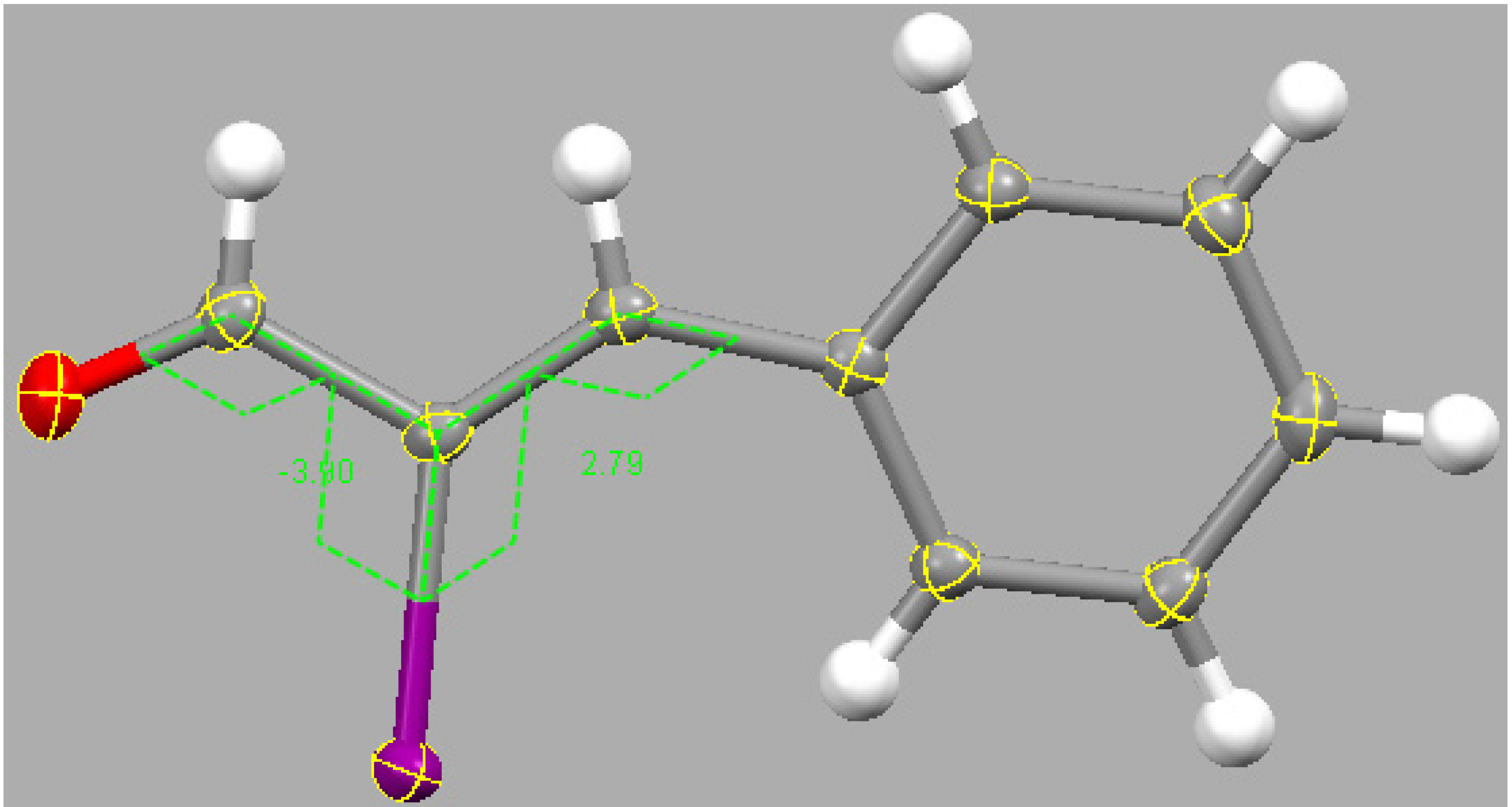
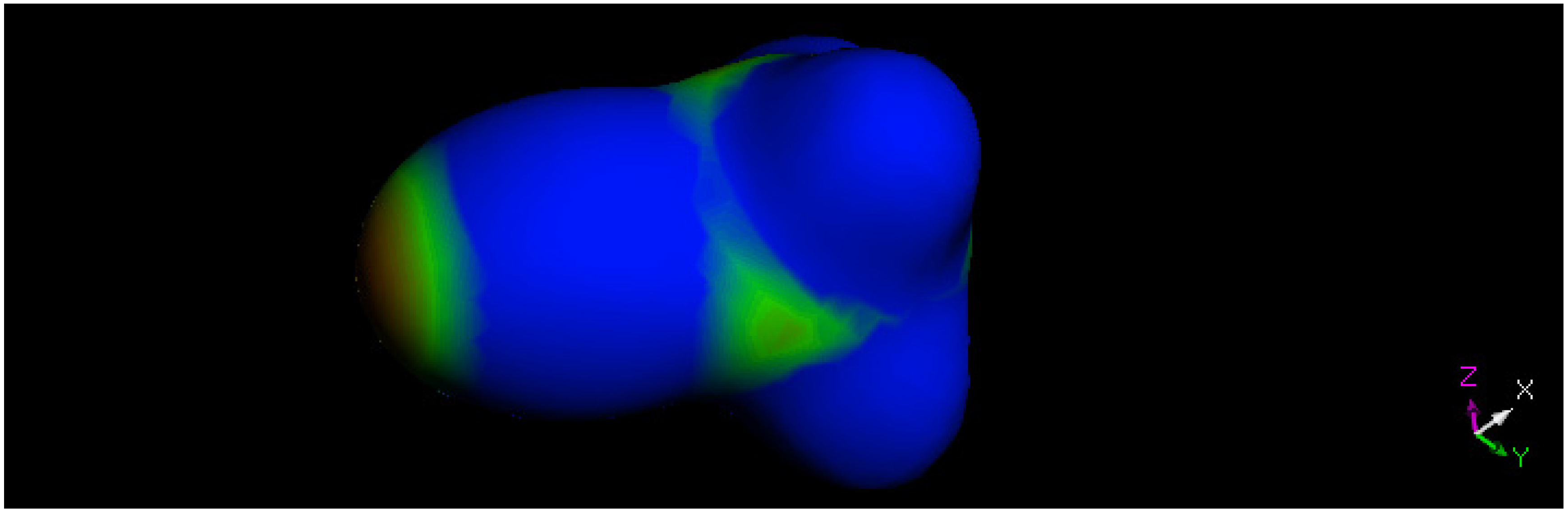
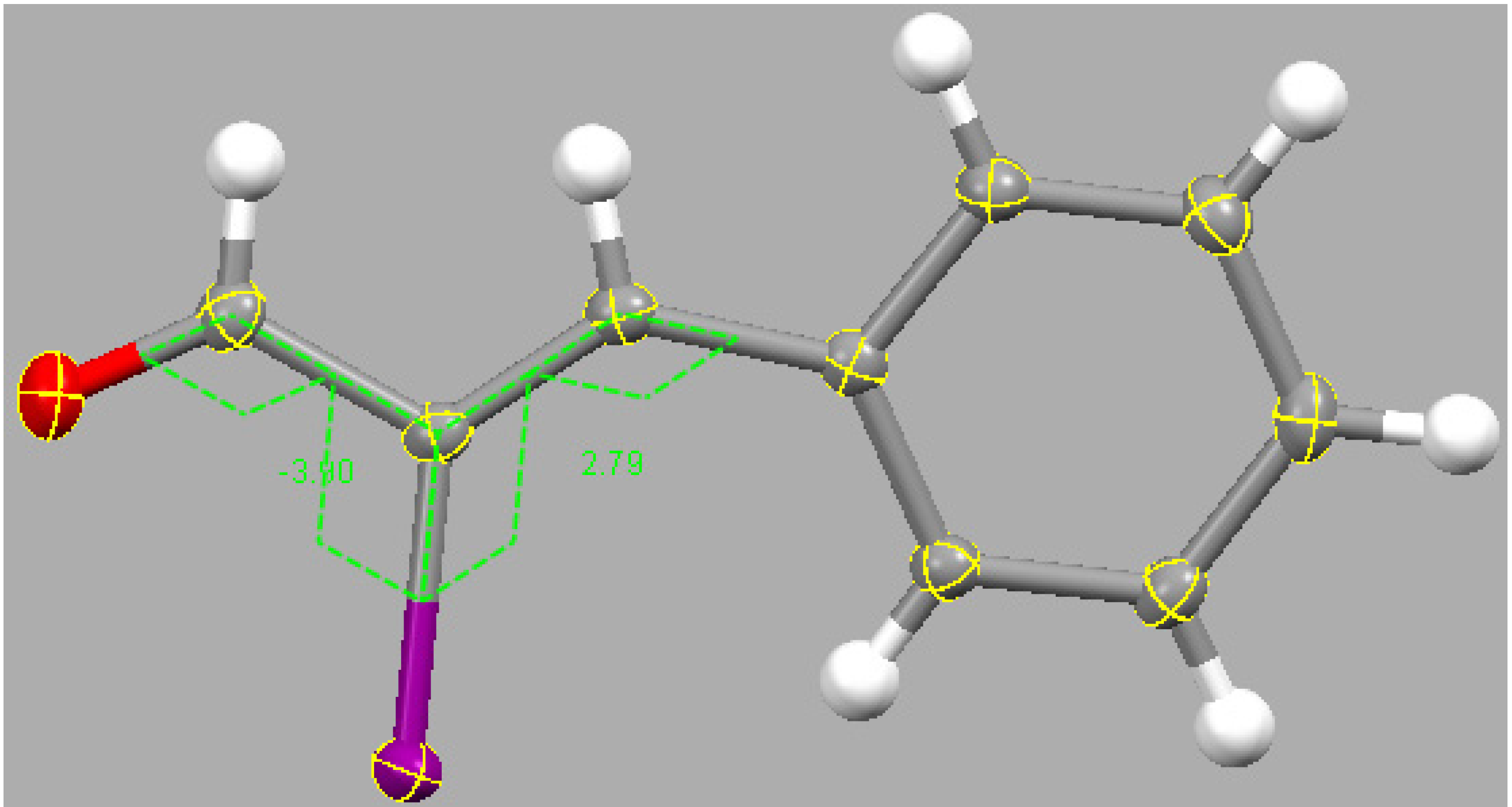
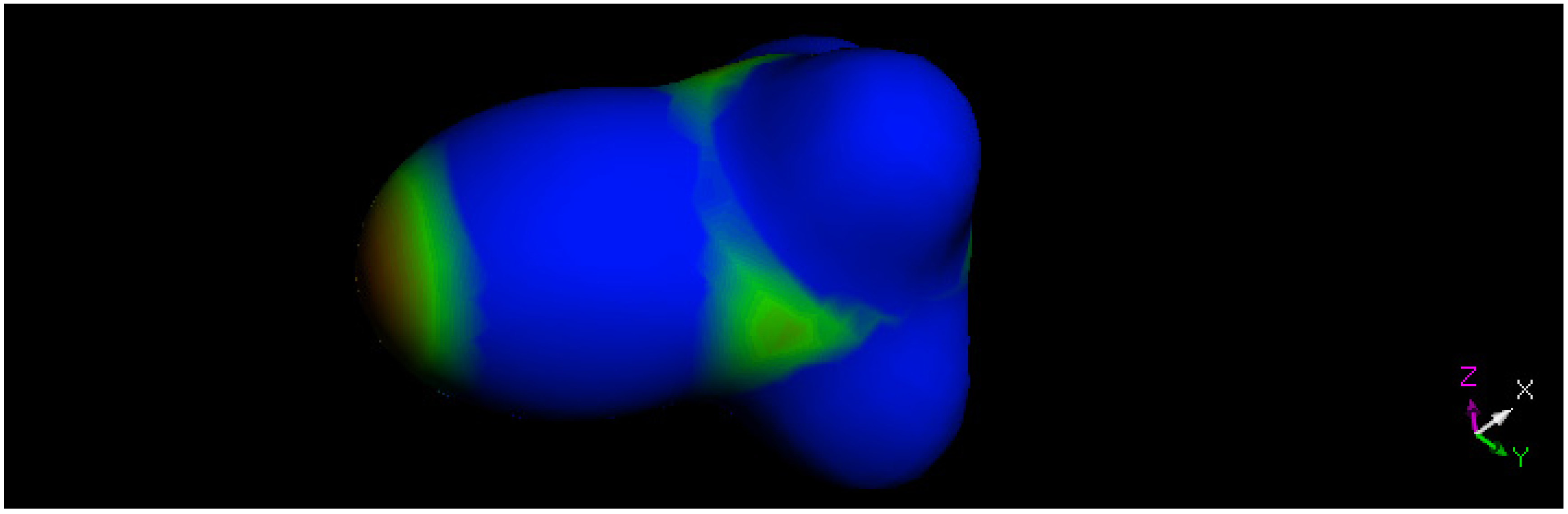
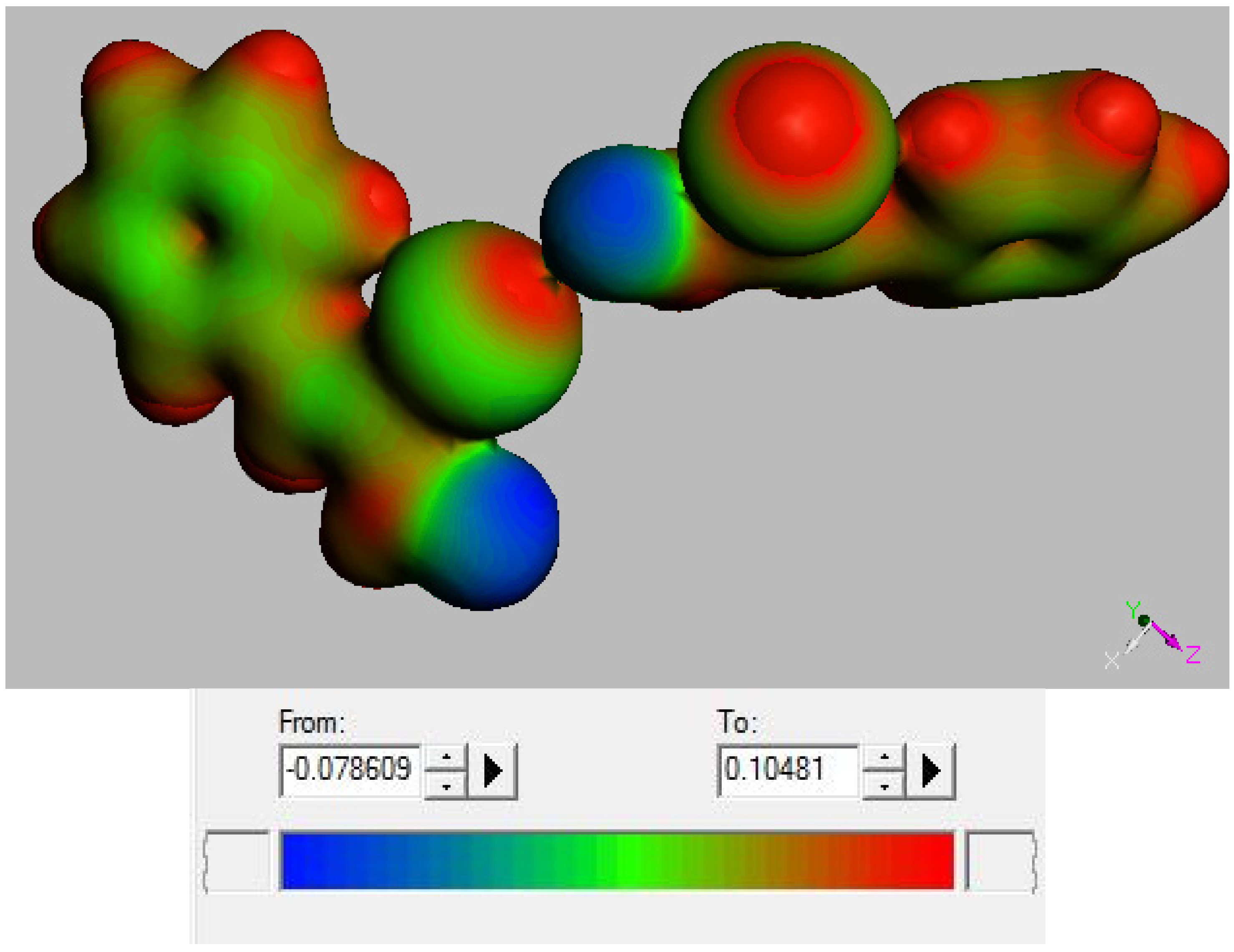
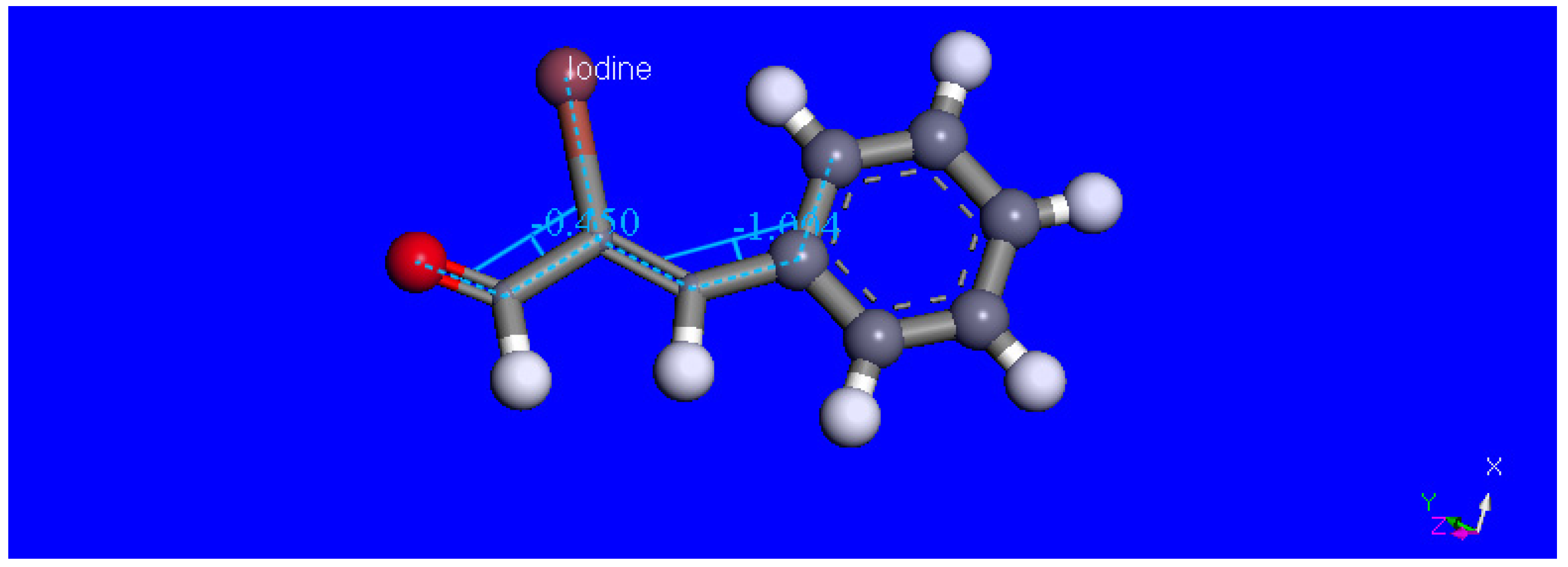
2.3. Theoretical Study

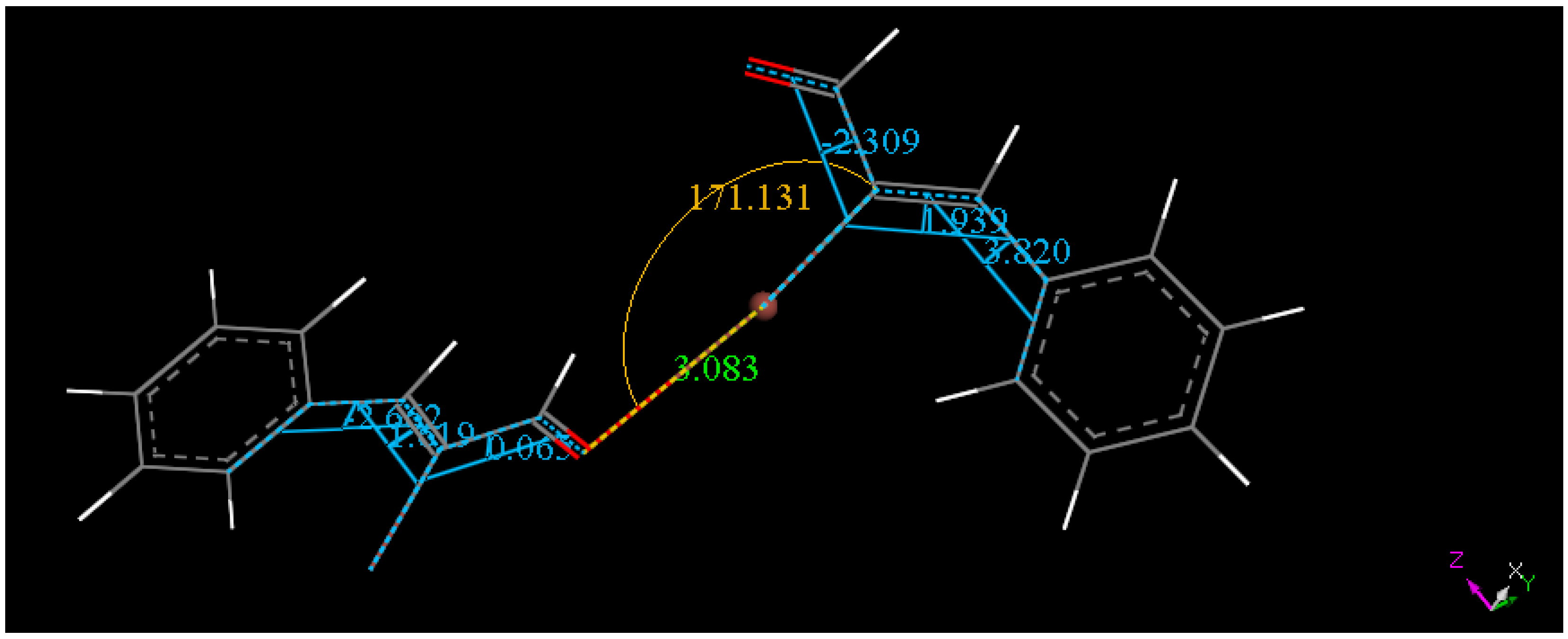
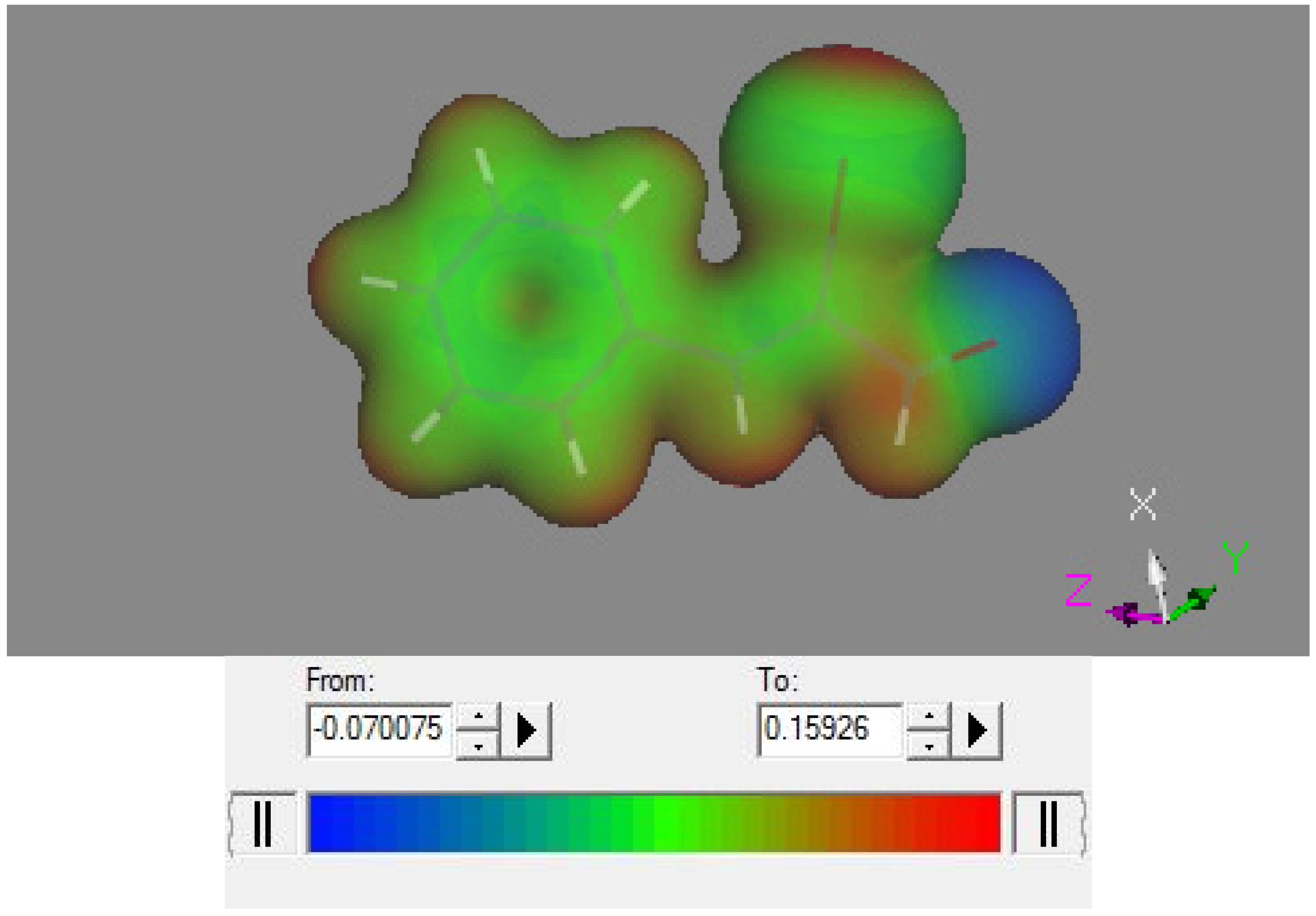



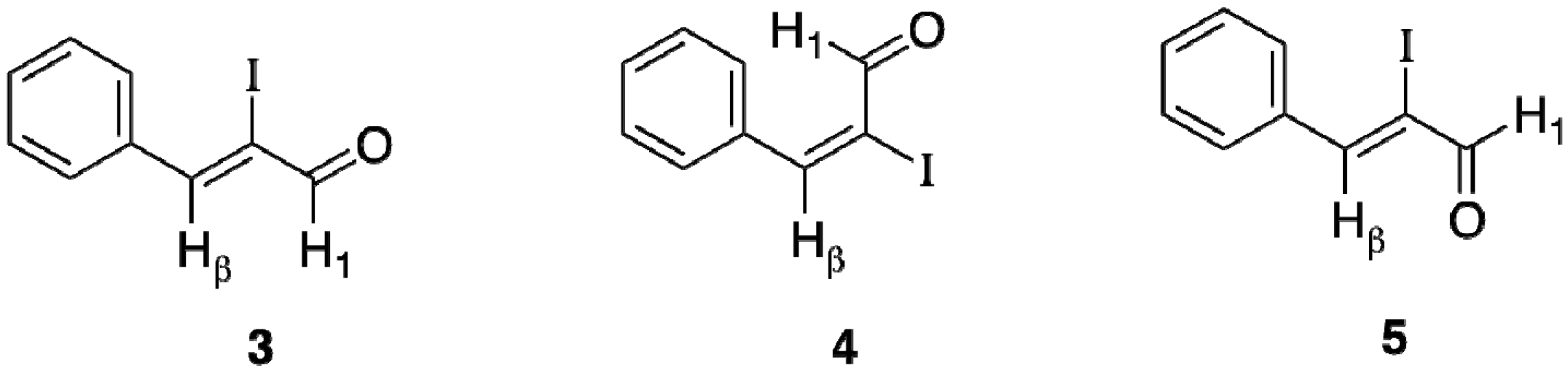
3. Experimental
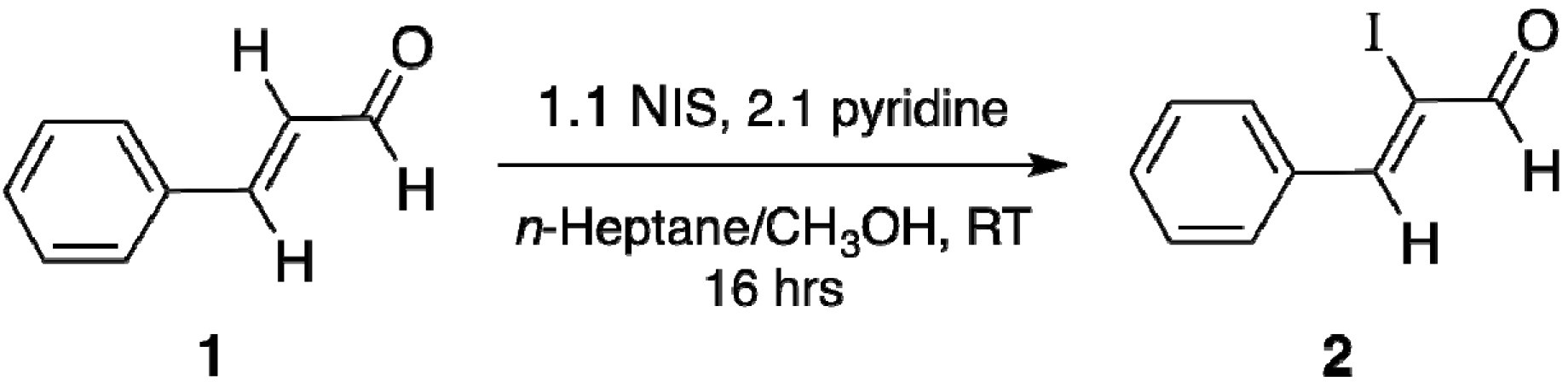
3.1. Chemistry
Synthesis Procedure of (Z)-2-iodo-3-phenylpropenal (2).
3.2. X-ray Diffraction Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical formula | C9H7INO |
|---|---|
| Crystal color | colorless |
| Formula weight | 258.05 |
| Crystal System | Orthorhombic |
| Space group | P bca |
| Temperature K | 125(2) |
| Wavelength (Å) | 0.71073 |
| a (Å) | 11.4372(6) |
| b (Å) | 8.6736(5) |
| c (Å) | 16.8274(9) |
| Volume (Å3) | 1669.31(16) |
| Z, density (mg/mm3) | 8, 2.053 |
| Absorption coefficient | 3.772 |
| Crystal size (mm) | 0.25 × 0.17 × 0.10 |
| θ range data collection | 2.42, 30.03 |
| Limiting índices | −16,16/−12.12/−23,23 |
| Data collected /unique | 24123, 2436 |
| Max, min. Transmission | 0.45/0.70 |
| Refinement method | F2 |
| Refined data / parameters | 2057/128 |
| Goodness-of-fit on F2 | 1.026 |
| Final R, Rw [I > 2sigma(I)] | 0.0181/0.0408 |
3.3. DFT Study
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References and Notes
- Hong, A.-W.; Cheng, T.-H.; Raghukumar, V.; Sha, C.-K. An expedient route to Montanine-type Amaryllidaceae alkaloids: Total syntheses of (−)-brunsvigine and (−)-manthine. J. Org. Chem. 2008, 73, 7580–7585. [Google Scholar] [CrossRef]
- Ohshima, T.; Xu, Y.; Takita, R.; Shibasaki, M. Enantioselective total synthesis of (−)-strychnine: Development of a highly practical catalytic asymmetric carbon–carbon bond formation and domino cyclization. Tetrahedron 2004, 60, 9569–9588. [Google Scholar] [CrossRef]
- Begum, L.; Box, J.M.; Drew, M.G.B.; Harwood, L.M.; Humphreys, J.L.; Lowes, D.J.; Morris, G.A.; Redon, P.M.; Walker, F.M.; Whitehead, R.C. Difluorinated analogues of shikimic acid. Tetrahedron 2003, 59, 4827–4841. [Google Scholar] [CrossRef]
- Jeong, L.S.; Yoo, S.J.; Lee, K.M.; Koo, M.J.; Choi, W.J.; Kim, H.O.; Moon, H.R.; Lee, M.Y.; Park, J.G.; Lee, S.K.; et al. Design, Synthesis, and biological evaluation of fluoroneplanocin A as the novel mechanism-based inhibitor of S-adenosylhomocysteine hydrolase. J. Med. Chem. 2003, 46, 201–203. [Google Scholar] [CrossRef]
- Bamba, M.; Nishikawa, T.; Isobe, M. Stereoelectronic and steric control in chiral cyclohexane synthesis toward (−)-tetrodotoxin. Tetrahedron 1998, 54, 6639–6650. [Google Scholar] [CrossRef]
- Negishi, E. Novel and selective α-substitution of ketones and other carbonyl compounds based on Pd-catalyzed cross coupling of α,β-unsaturated carbonyl derivatives containing α-halogen or α-metal groups. J. Organomet. Chem. 1999, 576, 179–194. [Google Scholar] [CrossRef]
- Stille, J.K. The palladium-catalyzed cross-coupling reactions of organotin reagents with organic electrophiles. Angew. Chem. 1986, 25, 508–524. [Google Scholar] [CrossRef]
- Heck, R.F. Palladium reagents in organic synthesis. Org. React. 1982, 27, 345–390. [Google Scholar]
- Suzuki, A.; Miyaura, N. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Scott, T.L.; Söderberg, B.C.G. Palladium-catalyzed synthesis of 1,2-dihydro-4(3H)-carbazolones. Formal total synthesis of murrayaquinone A. Tetrahedron 2003, 59, 6323–6332. [Google Scholar] [CrossRef]
- Chudzinski, M.G.; Taylor, M.S. Correlations between computation and experimental thermodynamics of halogen Bonding. J. Org. Chem. 2012, 77, 3483–3491. [Google Scholar] [CrossRef]
- McIntosh, J.M. Reaction of Iodine azide with cyclopentenone and cyclohexenone. Can J. Chem. 1971, 49, 3045–3047. [Google Scholar] [CrossRef]
- Jirkovsky, I. Studies on Enaminoketones. Can. J. Chem. 1974, 52, 55–65. [Google Scholar] [CrossRef]
- Smith, A.B.; Branca, S.J.; Guaciaro, M.A.; Wovkulich, P.M.; Korn, A. 2-Hydroxymethyl-2-cyclopentenone. Org. Synth. Coll. Vol. VII 1990, 271–274. [Google Scholar]
- Kim, T.H.; Asakura, J.; Asaka, Y. Reaction of enones with iodine in the presence of Ce(IV). Synthesis of 2-iodovinyl ketones. Chem. Express 1990, 5, 221–224. [Google Scholar]
- Johnson, C.R.; Adams, J.P.; Braun, M.P.; Senanayake, C.B.; Wovkulich, P.M.; Uskokovic, M.R. Direct α-iodination of cycloalkenones. Tetrahedron Lett. 1992, 33, 917–918. [Google Scholar] [CrossRef]
- Bovonsombat, P.; Angara, G.J.; McNelis, E. Concerning the formations of α-iodoenones. Tetrahedron Lett. 1994, 35, 6787–6790. [Google Scholar] [CrossRef]
- Djuardi, E.; Bovonsombat, P.; McNelis, E. Formations of α-Iodoenones by iodine and catalytic amounts of amines. Synth. Commun. 1997, 27, 2497–2503. [Google Scholar] [CrossRef]
- Sha, C.K.; Huang, S.J. Synthesis of β-substituted α-iodocycloalkenones. Tetrahedron Lett. 1995, 36, 6927–6928. [Google Scholar]
- Roush, W.R.; Barda, D.A. Second generation synthesis of the quartromicin spirotetronic acid subunits via a Claisen rearrangement-intramolecular aldol sequence. Tetrahedron Lett. 1997, 38, 8785–8788. [Google Scholar] [CrossRef]
- Benhida, R.; Blanchard, P.; Fourrey, J.-L. A mild and effective iodination method using iodine in the presence of bis-(trifluoroacetoxy)iodobenzene. Tetrahedron Lett. 1998, 39, 6849–6852. [Google Scholar] [CrossRef]
- Krafft, M.E.; Cran, J.W. A convenient protocol for the α-iodination of α,β-unsaturated carbonyl compounds with I2 in an aqueous medium. Synlett 2005, 1263–1266. [Google Scholar] [CrossRef]
- Bowman, W.R.; Bridge, C.F.; Cloonan, M.O.; Leach, D.C. Synthesis of heteroarenes via radical cyclisation onto nitriles. Synlett 2001, 765–768. [Google Scholar]
- Bovonsombat, P.; Rujiwarangkul, R.; Bowornkiengkai, T.; Leykajarakul, J. a-Bromination of linear enals and cyclic enones. Tetrahedron Lett. 2007, 48, 8607–8610. [Google Scholar] [CrossRef]
- The Chemistry of Enones, Part 1; Patai, S.S.; Rappoport, Z. (Eds.) Wiley: New York, NY, USA, 1989.
- Kirchner, M.T.; Dieter Blaser, D.; Roland Boese, R.; Thakur, T.S.; Desiraju, G.R. Weak C-H···O hydrogen bonds in anisaldehyde, Salicylaldehyde and cinnamaldehyde. Acta Cryst. 2011, C67, o387–o390. [Google Scholar]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen bonding in supramolecular chemistry. Angew. Chem. Int. Ed. 2008, 47, 6114–6127. [Google Scholar] [CrossRef]
- Halogen Bonding: Fundamentals and Applications, Structure and Bonding; Metrangolo, P.; Resnati, G. (Eds.) Springer: Berlin, Germany, 2008; Volume 126.
- Liljefors, T.; Allinger, N.L. Conformational analysis. CXII. Conformations, Energies, And electronic absorption spectra of α,β-unsaturated aldehydes and ketones. J. Am. Chem. Soc. 1976, 98, 2745–2749. [Google Scholar] [CrossRef]
- Estok, G.K.; Dehn, J.S. Electric moments of some unsaturated carbonyl compounds. J. Am. Chem. Soc. 1955, 77, 4769–4770. [Google Scholar] [CrossRef]
- Suzuki, M.; Kozima, K. Microwave spectra, barrier heights to internal rotation of methyl group, and dipole moments for conjugated chain compounds: Methacrolein. Mol. Spectrosc. 1971, 38, 314–321. [Google Scholar] [CrossRef]
- Hsu, S.L.; Flygare, W.H. Microwave spectrum and the barrier to internal rotation of the methyl group in trans- crotonaldehyde. Chem. Phys. Lett. 1969, 4, 317–319. [Google Scholar] [CrossRef]
- Forster, P.D.; Rao, V.M.; Curl, R.F., Jr. Microwave spectrum of Methyl Vinyl Ketone. J. Chem. Phys. 1965, 43, 1064–1066. [Google Scholar] [CrossRef]
- Forbes, W.F.; Shilton, R. Electronic spectra and molecular dimensions. III.1 Steric Effects in Methyl-substituted α,β-Unsaturated Aldehydes. J. Am. Chem. Soc. 1959, 81, 786–790. [Google Scholar] [CrossRef]
- Chen, Y.; Shibata, M.; Rajeswaran, M.; Srikrishnan, T.; Dugar, S.; Pandey, R.K. Utility of Japp-Klingemann reaction for the preparation of 5-carboxy-6-chloroindole via Fischer indole protocol. Tetrahedron Lett. 2007, 48, 2353–2356. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXTL, PC version 5.1 An integrated system for solving, refining and displaying crystal structures from diffraction data, Bruker Analytical X-ray Systems; Karlsruhe, 2000.
- Dmol3 Software, Accelrys, Inc.: San Diego, CA, USA, 2012.
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Hanke, F.; Dyer, M.S.; Bjork, J.; Persson, M. Structure and stability of weakly chemisorbed ethane adsorbed on low index Cu surfaces: Performance of density functionals with van der Waals interactions. J. Phys. Condensed Matter, 2012; 42, 424217–424225. [Google Scholar]
- Sample Availability: Samples of the compound (Z)-2-iodo-3-phenylpropenal (2) are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bovonsombat, P.; Caruso, F.; Jdaydani, A.; Rossi, M. Halogen Bonding in (Z)-2-Iodocinnamaldehyde. Molecules 2013, 18, 8712-8724. https://doi.org/10.3390/molecules18088712
Bovonsombat P, Caruso F, Jdaydani A, Rossi M. Halogen Bonding in (Z)-2-Iodocinnamaldehyde. Molecules. 2013; 18(8):8712-8724. https://doi.org/10.3390/molecules18088712
Chicago/Turabian StyleBovonsombat, Pakorn, Francesco Caruso, Andrew Jdaydani, and Miriam Rossi. 2013. "Halogen Bonding in (Z)-2-Iodocinnamaldehyde" Molecules 18, no. 8: 8712-8724. https://doi.org/10.3390/molecules18088712