Synthesis and Luminescence Properties of Iridium(III) Azide- and Triazole-Bisterpyridine Complexes


Abstract
:1. Introduction
2. Results and Discussion
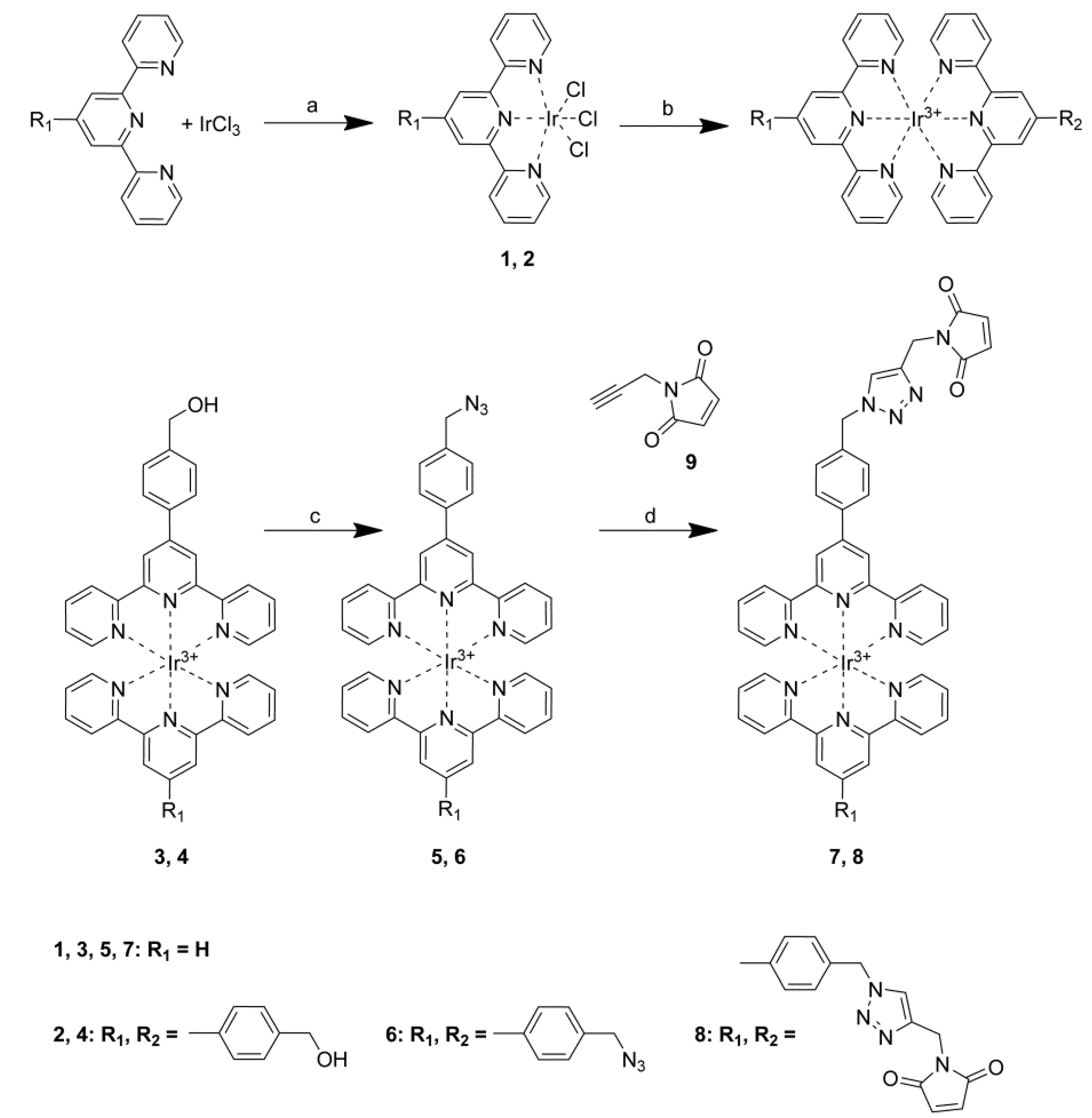
2.1. Synthesis of Iridium(III) Bisterpyridine Azide Complexes 5 and 6
2.2. Synthesis of 1-(Prop-2-ynyl)-1H-pyrrole-2,5-dione (9)
2.3. Click Chemistry Reaction
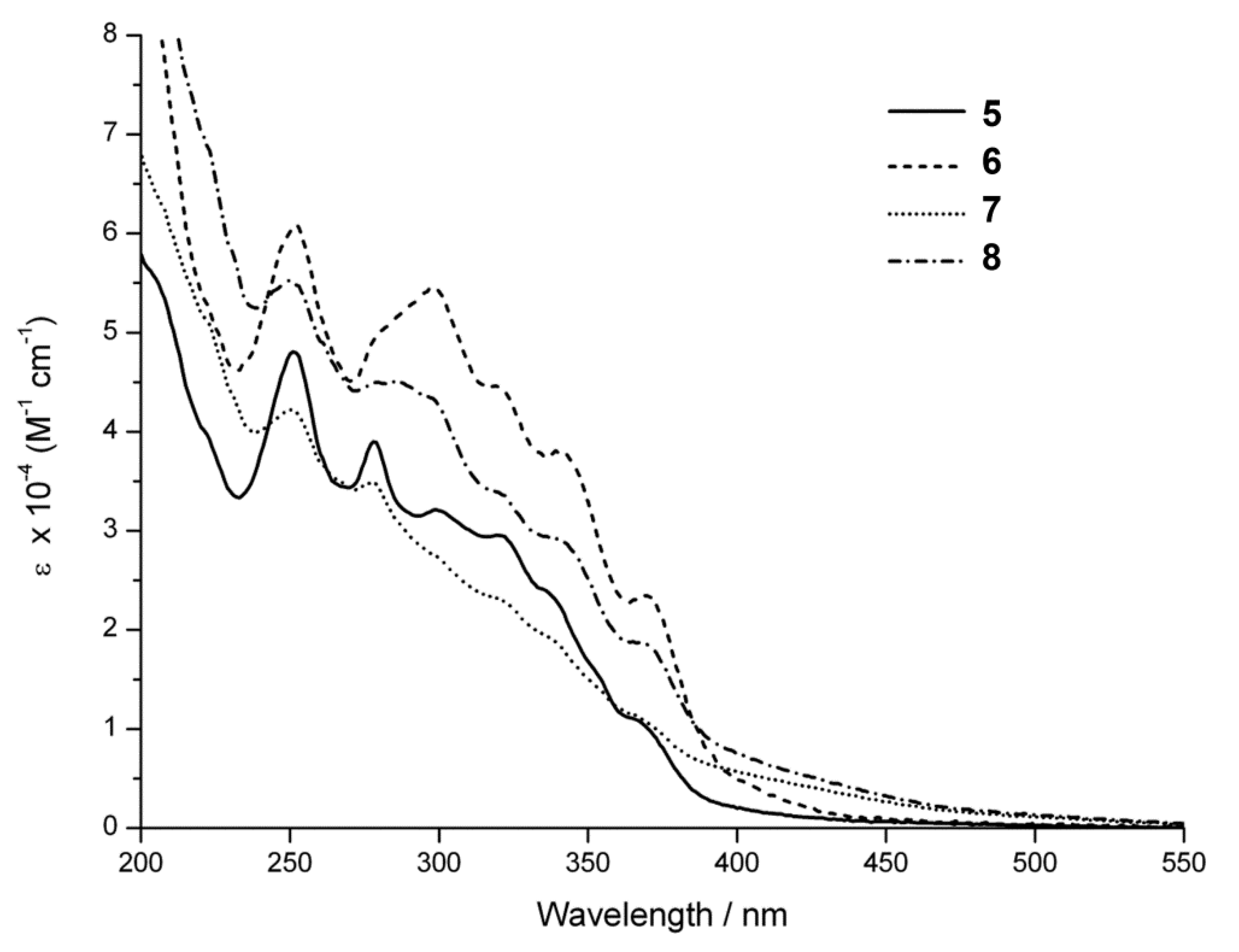
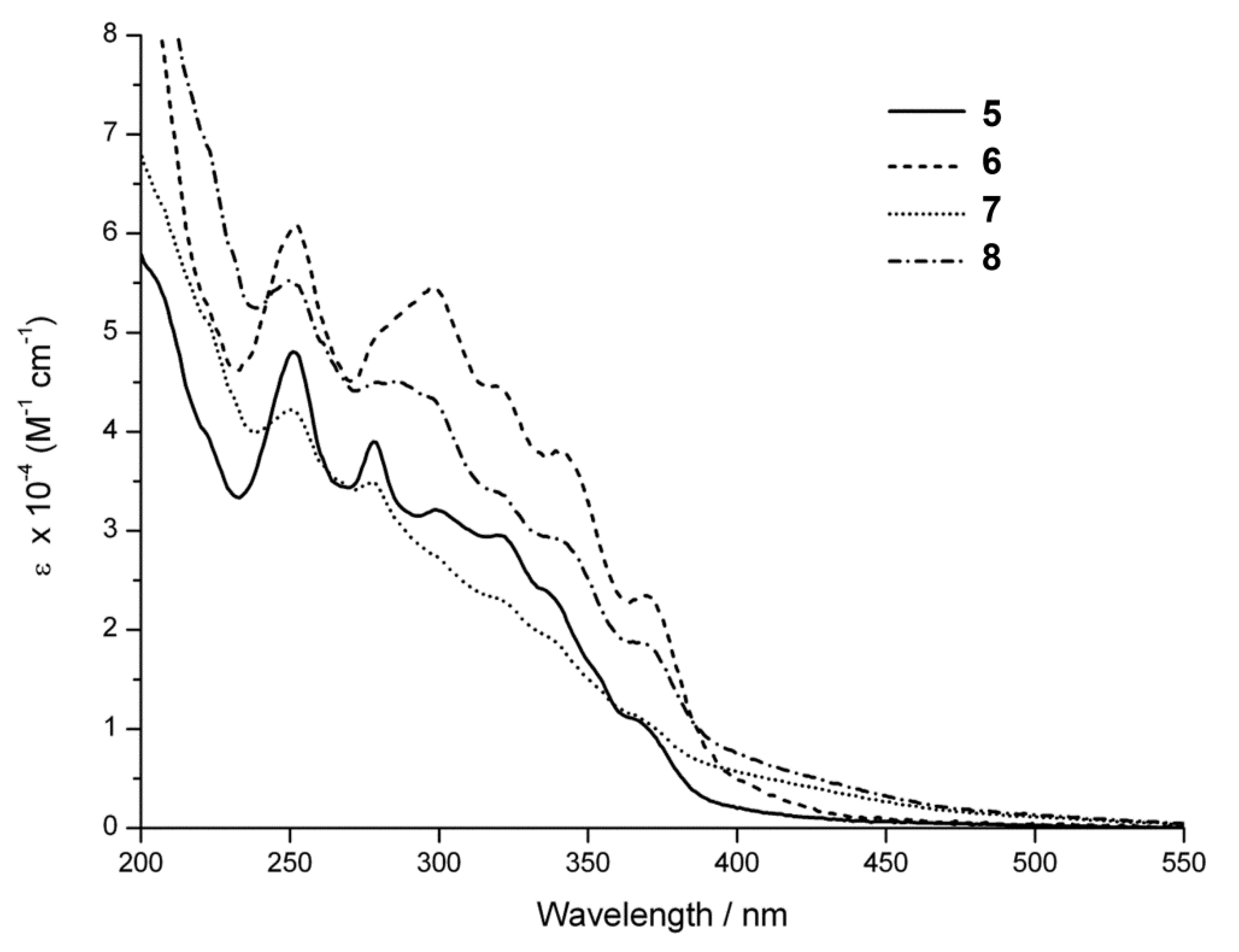
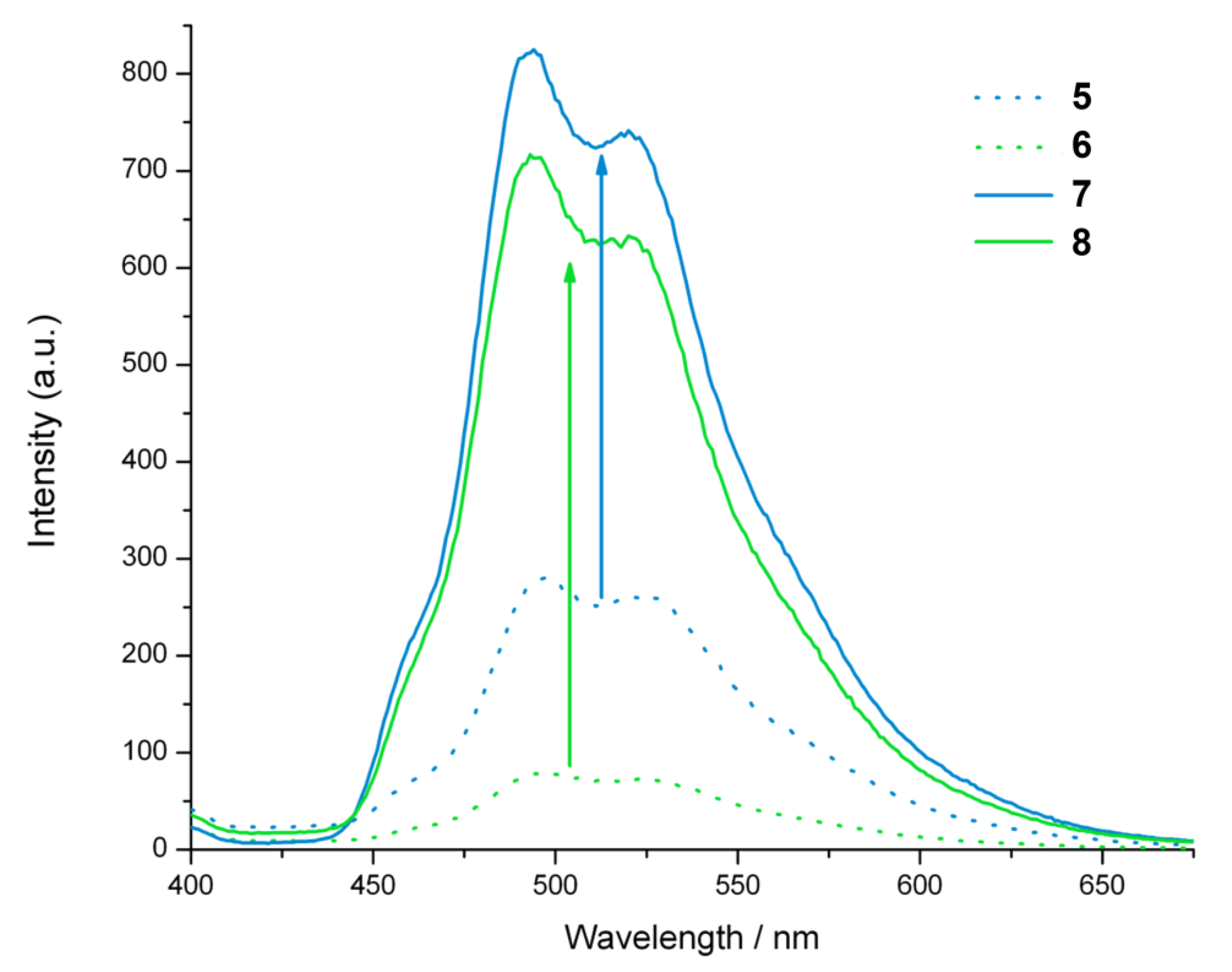
2.4. Steady-State Photophysical Characterization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| λmax/nm (ε/M−1cm−1) a | |
|---|---|
| 5 | 252 (47,900); 278 (39,000); 299 (32,100); 320 (29,500); 335 (24,200); 366 (10,900) |
| 6 | 252 (61,000); 298 (54,600); 320 (44,600); 341 (37,800); 370 (23,400) |
| 7 | 250 (42,200); 298 (34,800); 320 (23,200); 365 (11,400); 371 (26,300) |
| 8 | 251 (55,100); 286 (45,000); 320 (33,800); 341 (29,000); 370 (18,600) |
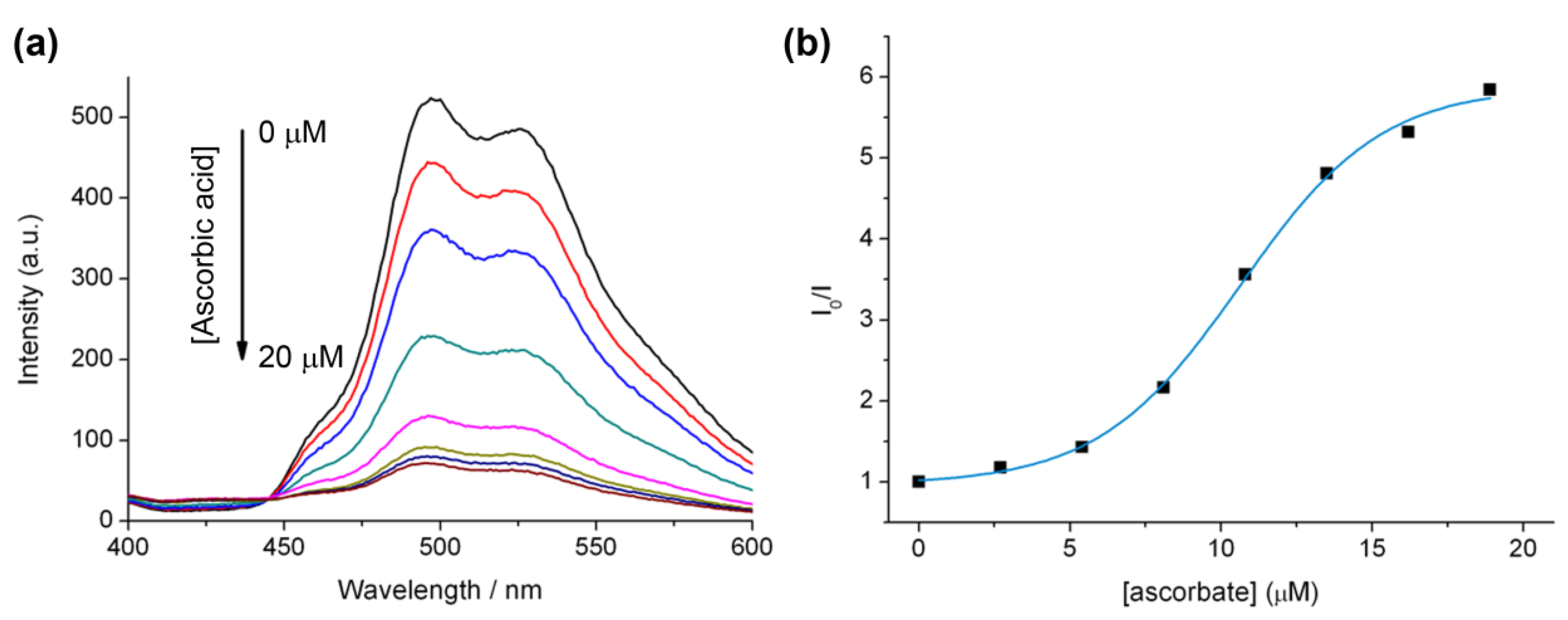
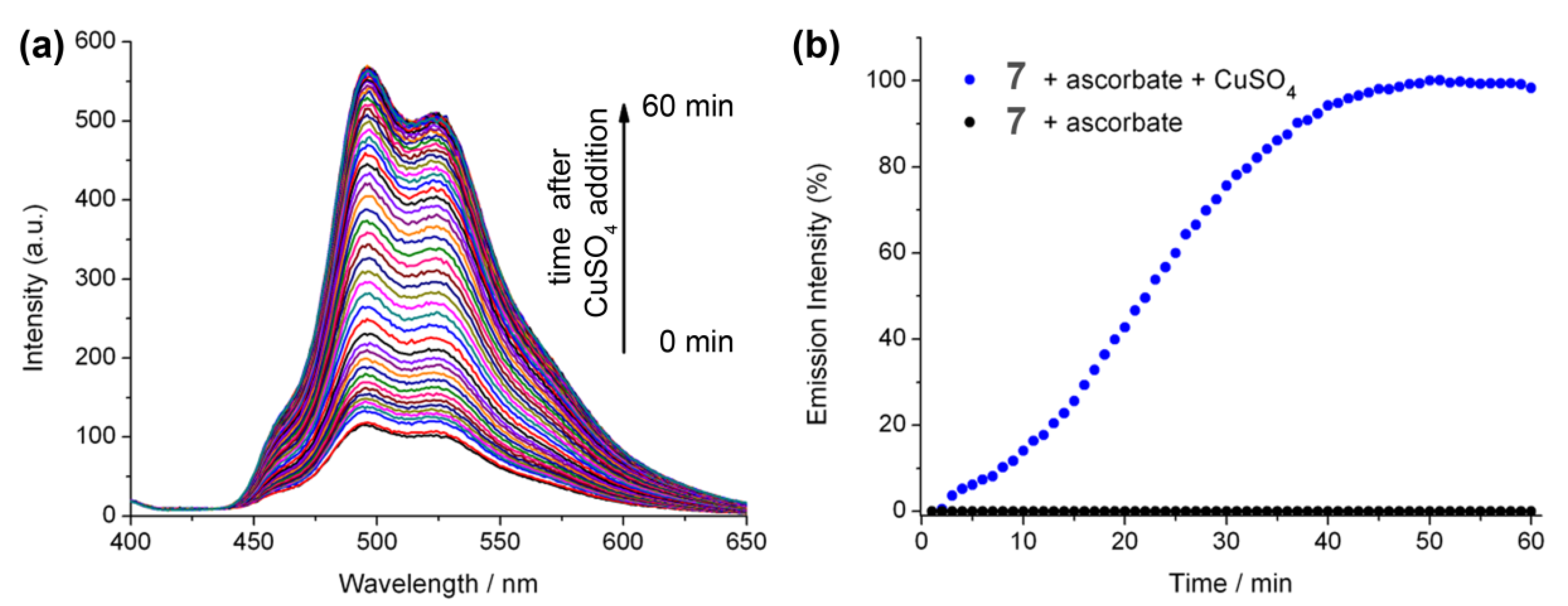
2.5. The Effect of Ascorbate on Using Luminescence to Monitor the “Click” Reaction

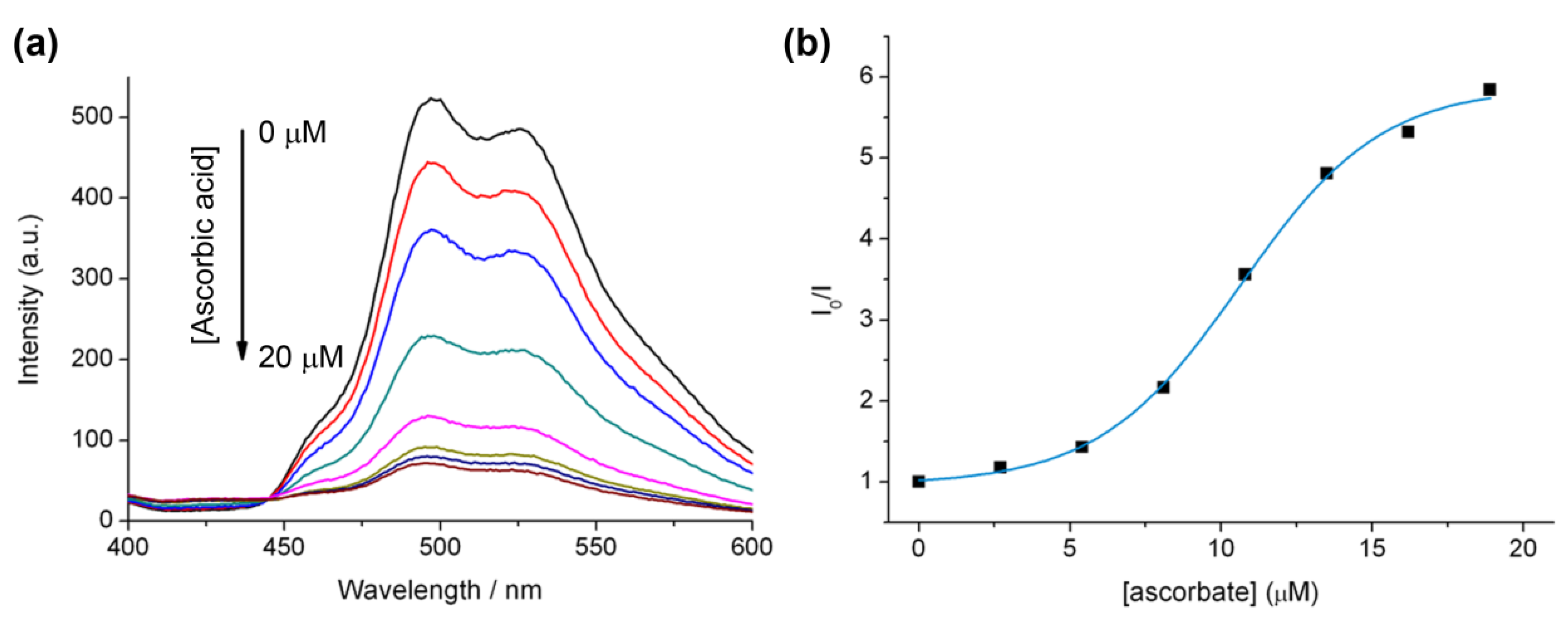

2.6. Luminescent Lifetimes of the Iridium(III) Azide and Triazole Bisterpyridine Complexes
| CH3CN | H2O | |||||
| λa (nm) | τb (μs) | 102 x Φec | λa (nm) | τb (μs) | 102 × Φec | |
| 5 | 498 | 1.5 (5.7) | 0.7 | 497 | 3.8 (3.9) | 0.9 |
| 6 | 498 | 1.3 (8.9) | 0.5 | 497 | 4.0 (4.0) | 0.3 |
| 7 | 493 | 1.3 (2.9) | 1.4 | 494 | 3.6 (4.5) | 2.5 |
| 8 | 495 | 2.3 (5.2) | 2.1 | 493 | 2.3 (2.7) | 2.2 |
3. Experimental
3.1. Chemicals
3.2. Spectroscopy
3.3. Steady-State and Lifetime Fluorescence
3.4. Syntheses
3.4.1. General Method A: Preparation of Ir(terpy-R)Cl3
3.4.2. General Method B: Preparation of [Ir(terpy-R1)(terpy-R2)](PF6)3
3.4.3. General Method C: Direct Conversion of Alcohols to Azides Using Diphenyl Phosphorazidate (SN2)
3.4.4. General Method D: Copper Catalysed Huisgen 1,3-Dipolar Cycloaddition
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Flamigni, L.; Barbieri, A.; Sabatini, C.; Ventura, B.; Barigelletti, F. Photochemistry and photophysics of coordination compounds: Iridium. Top. Curr. Chem. 2007, 281, 143–203. [Google Scholar] [CrossRef]
- Williams, J.A.G.; Wilkinson, A.J.; Whittle, V.L. Light-emitting iridium complexes with tridentate ligands. Dalton Trans. 2008, 2081–2099. [Google Scholar]
- Chi, Y.; Chou, P.-T. Transition-metal phosphors with cyclometalatingligands: Fundamentals and applications. Chem. Soc. Rev. 2010, 39, 638–655. [Google Scholar] [CrossRef]
- You, Y.; Park, S.Y. Phosphorescent iridium(iii) complexes: Toward high phosphorescence quantum efficiency through ligand control. Dalton Trans. 2009, 1267–1282. [Google Scholar] [CrossRef]
- Zhao, Q,; Huang, C.; Li, F. Phosphorescent heavy-metal complexes for bioimaging. Chem. Soc. Rev. 2011, 40, 2508–2524. [Google Scholar] [CrossRef]
- Sauvage, J.P.; Collin, J.P.; Chambron, J.C.; Guillerez, S.; Coudret, C.; Balzani, V.; Barigelletti, F.; De Cola, L.; Flamigni, L. Ruthenium(ii) and osmium(ii) bis(terpyridine) complexes in covalently-linked multicomponent systems: Synthesis, electrochemical behavior, absorption spectra, and photochemical and photophysical properties. Chem. Rev. 1994, 94, 993–1019. [Google Scholar] [CrossRef]
- Medlycott, E.A.; Hanan, G.S. Synthesis and properties of mono- and oligo-nuclear ru(ii) complexes of tridentate ligands: The quest for long-lived excited states at room temperature. Coord. Chem. Rev. 2006, 250, 1763–1782. [Google Scholar] [CrossRef]
- Flamigni, L.; Collin, J.P.; Sauvage, J.P. Iridium terpyridine complexes as functional assembling units in arrays for the conversion of light energy. Acc. Chem. Res. 2008, 41, 857–871. [Google Scholar] [CrossRef]
- Sánchez-Barragán, I.; Costa-Fernández, J.M.; Sanz-Medel, A.; Valledor, M.; Campo, J.C. Room-temperature phosphorescence (RTP) for optical sensing. Trends Anal. Chem. 2006, 25, 958–967. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 2nd ed.; Kluwer Academic/Plenum Press: New York, NY, USA, 1999. [Google Scholar]
- Lo, K.K.-W.; Chung, C.-K.; Ng, D.C.-M.; Zhu, N. Syntheses, characterisation and photophysical studies of novel biological labelling reagents derived from luminescent iridium (iii) terpyridine complexes. New J. Chem. 2002, 26, 81–88. [Google Scholar] [CrossRef]
- Aihara, M.; Kishita, H.; Misumi, S. Polargraphic studies of bis(2,2',2"-terpyridine) complexes of manganese(ii), cobalt(ii), nickel(ii) and europium(iii) in acetonitrile. Bull. Chem. Soc. Jpn. 1975, 48, 680–683. [Google Scholar] [CrossRef]
- Eryazici, I.; Moorefield, C.N.; Newkome, G.R. Square-planar Pd(ii), Pt(ii), and Au(iii) terpyridine complexes: Their syntheses, physical properties, supramolecular constructs, and biomedical activities. Chem. Rev. 2008, 108, 1834–1895. [Google Scholar] [CrossRef]
- Cotton, S.A.; Noy, O.E.; Liesener, F.; Raithby, P.R. Unequivocal characterisation of a [Ln(terpy)(NO3)3·(H2O)] complex.: The synthesis and structure of [M(terpy)(NO3)3·(H2O)] (M=Eu, Tb); a comparison with the structure of [Eu(bipy)2(NO3)3] and with other europium nitrate complexes {terpy=2,2':6',2''-terpyridyl; bipy=2,2'-bipyridyl}. Inorg. Chim. Acta 2003, 344, 37–42. [Google Scholar]
- Morari, C. Bond energy and electronic structure in M-bis-terpyridine complexes (M=Os, Co and Ru). Phys. Lett. A 2008, 372, 1885–1889. [Google Scholar]
- Collin, J.-P.; Dixon, I.M.; Sauvage, J.-P.; Williams, J.A.G.; Barigelletti, F.; Flamigni, L. Synthesis and photophysical properties of iridium(iii) bisterpyridine and its homologues: A family of complexes with a long-lived excited state. J. Am. Chem. Soc 1999, 121, 5009–5016. [Google Scholar] [CrossRef]
- Goldstein, D.C.; Cheng, Y.Y.; Schmidt, T.W.; Bhadbhade, M.; Thordarson, P. Photophysical properties of a new series of water soluble iridium bisterpyridine complexes functionalised at the 4[prime or minute] position. Dalton Trans. 2011, 40, 2053–2061. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgencycloaddition process: Copper(i)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Huisgen, R. Centenary Lecture – 1,3-Dipolar Cycloadditions. Proc. Chem. Soc. 1961, 357–396. [Google Scholar]
- Baranoff, E.; Dixon, I.M.; Collin, J.-P.; Sauvage, J.-P.; Ventura, B.; Flamigni, L. Dyads containing iridium(iii) bis-terpyridine as photoactive center: Synthesis and electron transfer study. Inorg. Chem. 2004, 43, 3057–3066. [Google Scholar] [CrossRef]
- Ayala, N.P.; Flynn, C.M.; Sacksteder, L.; Demas, J.N.; DeGraff, B.A. Synthesis, luminescence, and excited-state complexes of the tris(1,10-phenanthroline)- and bis(terpyridine)iridium(iii) cations. J. Am. Chem. Soc. 1990, 112, 3837–3844. [Google Scholar] [CrossRef]
- Seok, W.K.; Yim, S.B.; Klapötke, T.M.; White, P.S. Synthesis, structural characterization and semiempirical calculations of the ruthenium azide complex [Ru(tpy)(PPh3)2(N3)][ClO4]. J. Organomet. Chem. 1998, 559, 165–171. [Google Scholar] [CrossRef]
- Hofmeier, H.; Andres, P.R.; Hoogenboom, R.; Herdtweck, E.; Schubert, U.S. Terpyridine-ruthenium complexes as building blocks for new metallo-supramolecular architectures. Aust. J. Chem. 2004, 57, 419–426. [Google Scholar]
- Thompson, A.S.; Humphrey, G.R.; DeMarco, A.M.; Mathre, D.J.; Grabowski, E.J.J. Direct conversion of activated alcohols to azides using diphenyl phosporazidate. A practical alternative to mitsunobu conditions. J. Org. Chem. 1993, 58, 5886–5888. [Google Scholar] [CrossRef]
- Beusker, P.H.; Groot, F.H.D. Triazole-containing releasable linkers, conjugates thereof, and methods of preparation. WO2007018431, 15 February 2007. [Google Scholar]
- Thordarson, P.; Droumaguet, B.L.; Velonia, K. Well-defined protein–polymer conjugates—Synthesis and potential applications. Appl. Microbiol. Biotechnol. 2006, 73, 243–254. [Google Scholar] [CrossRef]
- Peterson, J.R.; Smith, T.A.; Thordarson, P. Photoinduced reduction of catalytically and biologically active Ru(ii)bisterpyridine-cytochrome c bioconjugates. Chem. Commun. 2007, 2007, 1899–1901. [Google Scholar] [CrossRef]
- Peterson, J.R.; Smith, T.A.; Thordarson, P. Synthesis and room temperature photo-induced electron transfer in biologically active bis(terpyridine)ruthenium(ii)-cytochrome c bioconjugates and the effect of solvents on the bioconjugation of cytochrome c. Org. Biomol. Chem. 2010, 8, 151–162. [Google Scholar] [CrossRef]
- Hvasanov, D.; Mason, A.F.; Goldstein, D.C.; Bhadbhade, B.; Thordarson, P. Optimising the synthesis, polymer membrane encapsulation and photoreduction performance of Ru(ii)- and Ir(iii) bis(terpyridine) cytochrome c bioconjugates. Org. Biomol. Chem. 2013, 11, 4602–4612. [Google Scholar] [CrossRef]
- Clevenger, R.C.; Turnbull, K.D. Synthesis on n-alkylatedmaleimides. Synth. Commun. 2000, 30, 1379–1388. [Google Scholar] [CrossRef]
- Walker, M.A. A high yielding synthesis of n-alkyl maleimides using a novel modification of the mitsunobu reaction. J. Org. Chem. 1995, 60, 5352–5355. [Google Scholar] [CrossRef]
- Mehta, N.B.; Phillips, A.P.; Lui, F.F.; Brooks, R.E. Maleamic and citraconamic acids, methyl esters, and imides. J. Org. Chem. 1960, 25, 1012–1015. [Google Scholar] [CrossRef]
- Dirks, A.J.; Cornelissen, J.J.L.M.; Nolte, R.J.M. Monitoring protein-polymer conjugation by a fluorogenicCu(i)-catalyzed azide-alkyne 1,3-dipolar cycloaddition. Bioconjugate Chem. 2009, 20, 1129–1138. [Google Scholar] [CrossRef]
- Bisby, R.H.; Morgan, C.G.; Hamblett, I.; Gorman, A.A. Quenching of singlet oxygen by trolox c, ascorbate, and amino acids: Effects of Ph and temperature. J. Phys. Chem. A 1999, 103, 7454–7459. [Google Scholar] [CrossRef]
- Yoshikawa, N.; Yamabe, S.; Kanehisa, N.; Kai, Y.; Takashima, H.; Tsukahara, K. Synthesis, characterization, and dft investigation of iriiitolylterpyridine complexes. Eur. J. Inorg. Chem. 2007, 2007, 1911–1919. [Google Scholar] [CrossRef]
- Holmes, D.T. Quenching of the triplet state of chlorophyll a by ascorbic acid and ascorbate in ethanolic solution. Photochem. Photobiol. 1965, 4, 631–632. [Google Scholar] [CrossRef]
- Meucci, E.; Mordente, A.; Donato Miggiano, G.A.; Martorana, G.E. Fluorescence studies of ascorbate-melittin interaction. Biochim. Biophys. Acta 1989, 999, 58–63. [Google Scholar] [CrossRef]
- Sasso, M.G.; Quina, F.H.; Bechara, E.J.H. Ruthenium(ii) tris(bipyridyl) ion as a luminescent probe for oxygen uptake. Anal. Biochem. 1986, 156, 239–243. [Google Scholar]
- Crosby, G.A.; Demas, J.N. Measurement of photoluminescence quantum yields. Review. J. Phys. Chem. 1971, 75, 991–1024. [Google Scholar] [CrossRef]
- Winter, A.; Ulbricht, C.; Holder, E.; Risch, N.; Schubert, U.S. Unusual terpyridines as ligands for novel light-emitting iridium(iii) complexes: Synthesis and characterization. Aust. J. Chem. 2006, 59, 773–782. [Google Scholar] [CrossRef]
- Dirks, A.J.; van Berkel, S.S.; Hatzakis, N.S.; Opsteen, J.A.; van Delft, F.L.; Cornelissen, J.J.L.M.; Rowan, A.E.; van Hest, J.C.M.; Rutjes, F.P.J.T.; Nolte, R.J.M. Preparation of biohybrid amphiphiles via the copper catalysed huisgen [3+2] dipolar cycloaddition reaction. Chem. Commun. 2005, 2005, 4172–4174. [Google Scholar]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Goldstein, D.C.; Peterson, J.R.; Cheng, Y.Y.; Clady, R.G.C.; Schmidt, T.W.; Thordarson, P. Synthesis and Luminescence Properties of Iridium(III) Azide- and Triazole-Bisterpyridine Complexes. Molecules 2013, 18, 8959-8975. https://doi.org/10.3390/molecules18088959
Goldstein DC, Peterson JR, Cheng YY, Clady RGC, Schmidt TW, Thordarson P. Synthesis and Luminescence Properties of Iridium(III) Azide- and Triazole-Bisterpyridine Complexes. Molecules. 2013; 18(8):8959-8975. https://doi.org/10.3390/molecules18088959
Chicago/Turabian StyleGoldstein, Daniel C., Joshua R. Peterson, Yuen Yap Cheng, Raphael G. C. Clady, Timothy W. Schmidt, and Pall Thordarson. 2013. "Synthesis and Luminescence Properties of Iridium(III) Azide- and Triazole-Bisterpyridine Complexes" Molecules 18, no. 8: 8959-8975. https://doi.org/10.3390/molecules18088959