The Click Reaction as an Efficient Tool for the Construction of Macrocyclic Structures


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
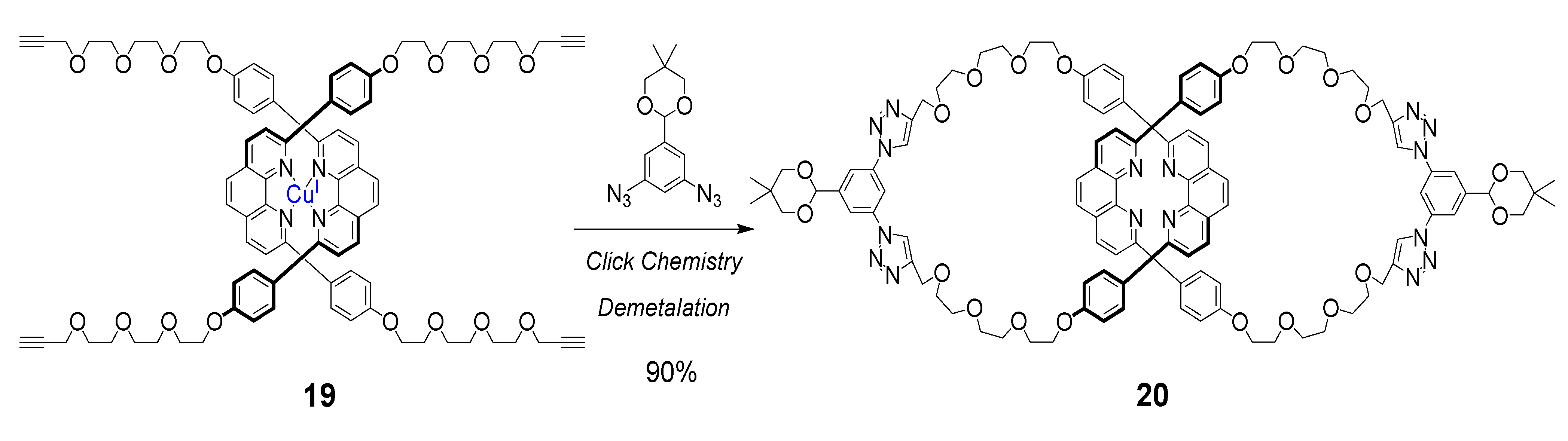{kind=link}
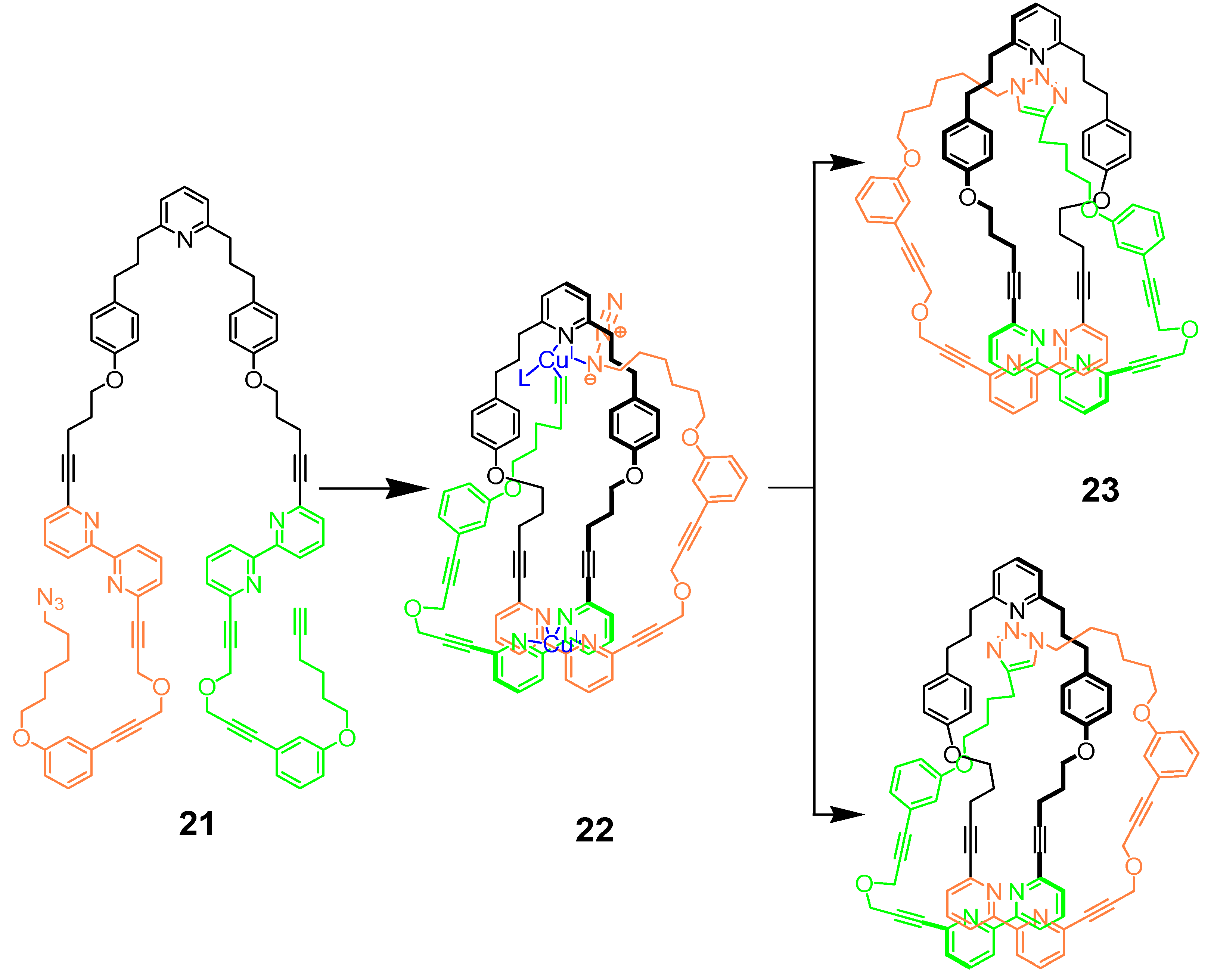{kind=link}
{kind=link}
1. Introduction
- (a)
- the use of high dilution conditions has greatly helped to improve yields in the cyclization process; this tool statistically favors ring closing vs. polymerization reaction, either in single chains containing complementarily reactive chain end functionalities (AB; A reacting with B), or in chains containing complementary reactive functionalities (AA with BB) [5,6,7];
- (b)
- template-directed syntheses: it is the exploitation of coherently designed guest systems, recognized by the forming host, which can effectively act as templates during the reaction, in order to pre-organize the host-guest system for the macrocyclization reaction (forming one or more new covalent bonds), facilitating the desired chemistry [5,6].
- (c)
- Dynamic combinatorial chemistry, as a method for the generation of new molecules by means of reversible reactions between simple building blocks under thermodynamic control. In a dynamic combinatorial library (DCL) all constituents are in equilibrium, and their distribution is determined by their thermodynamic stability within the DCL. This tool has been used with success for the construction, amongst others, of macrocyclic structures [8].
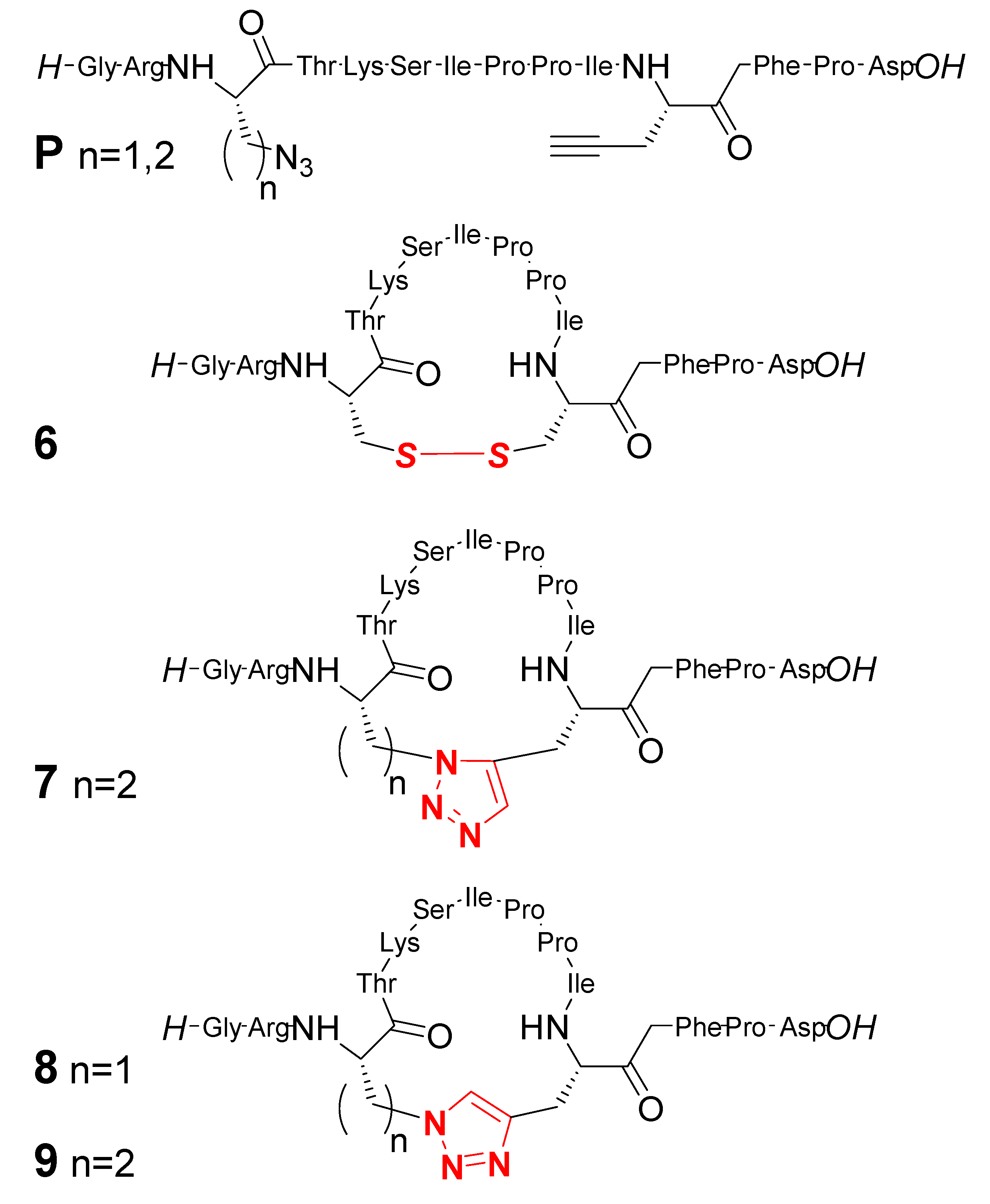
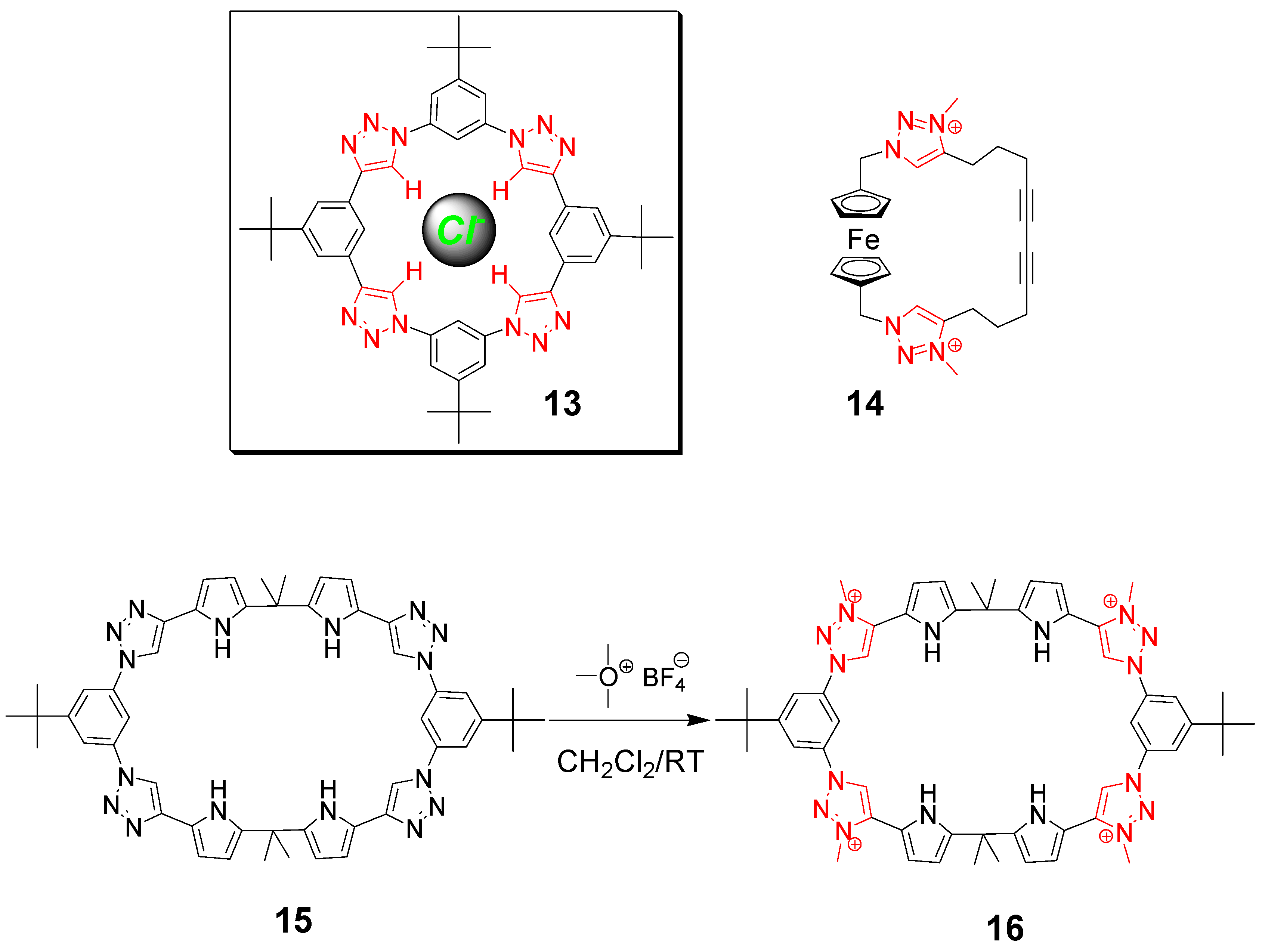
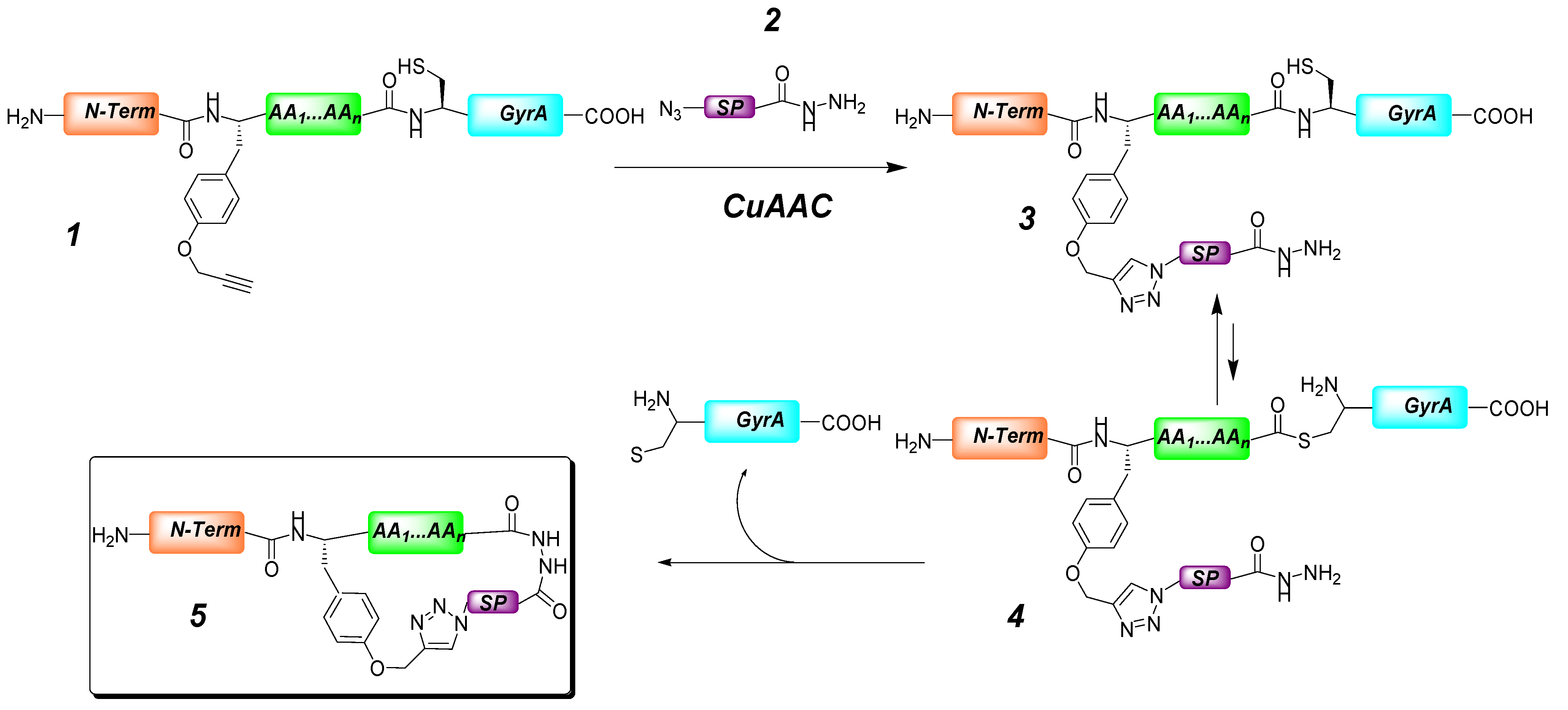
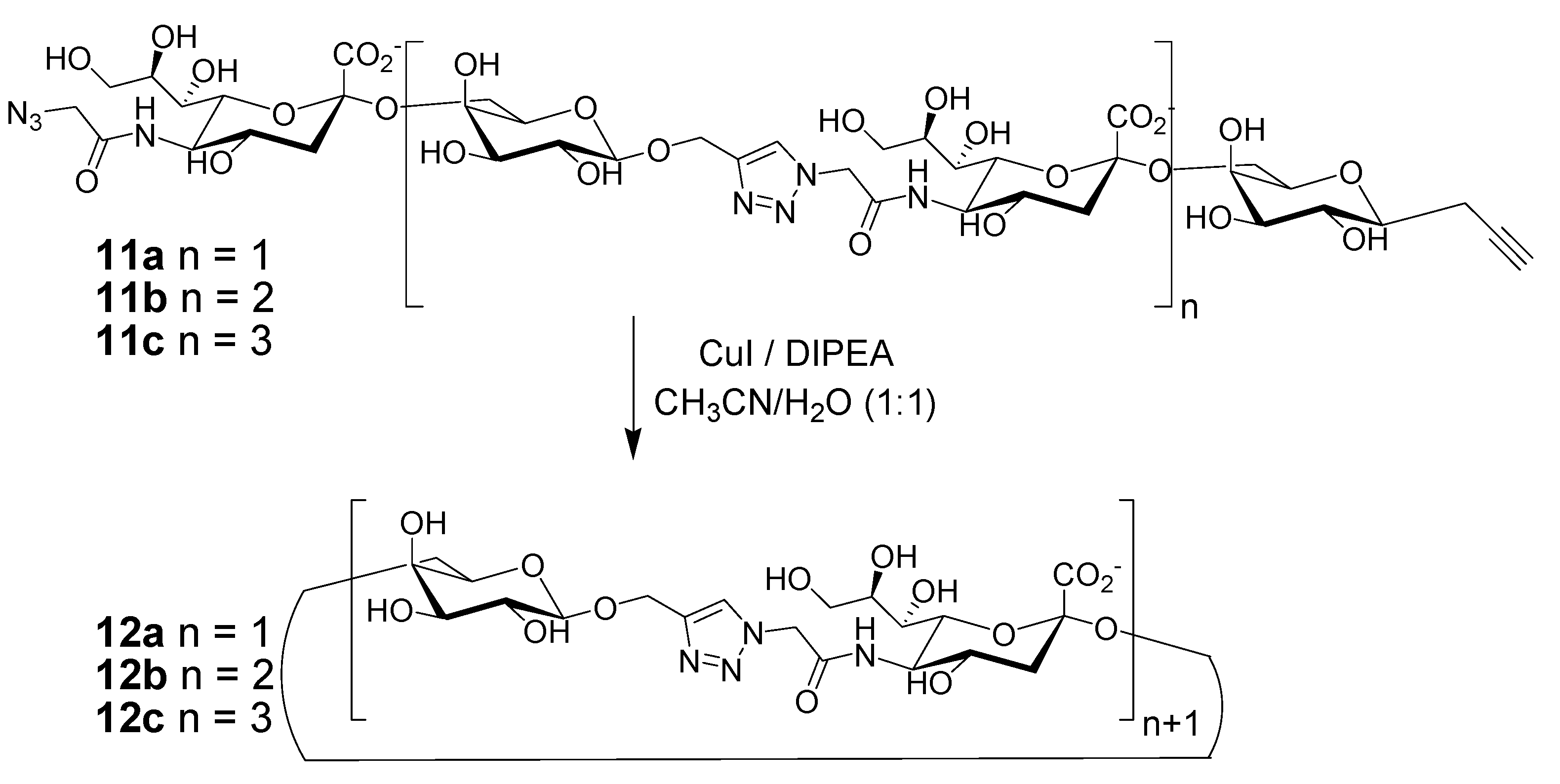
2. Peptide- and Sugar-Containing Click Macrocycles
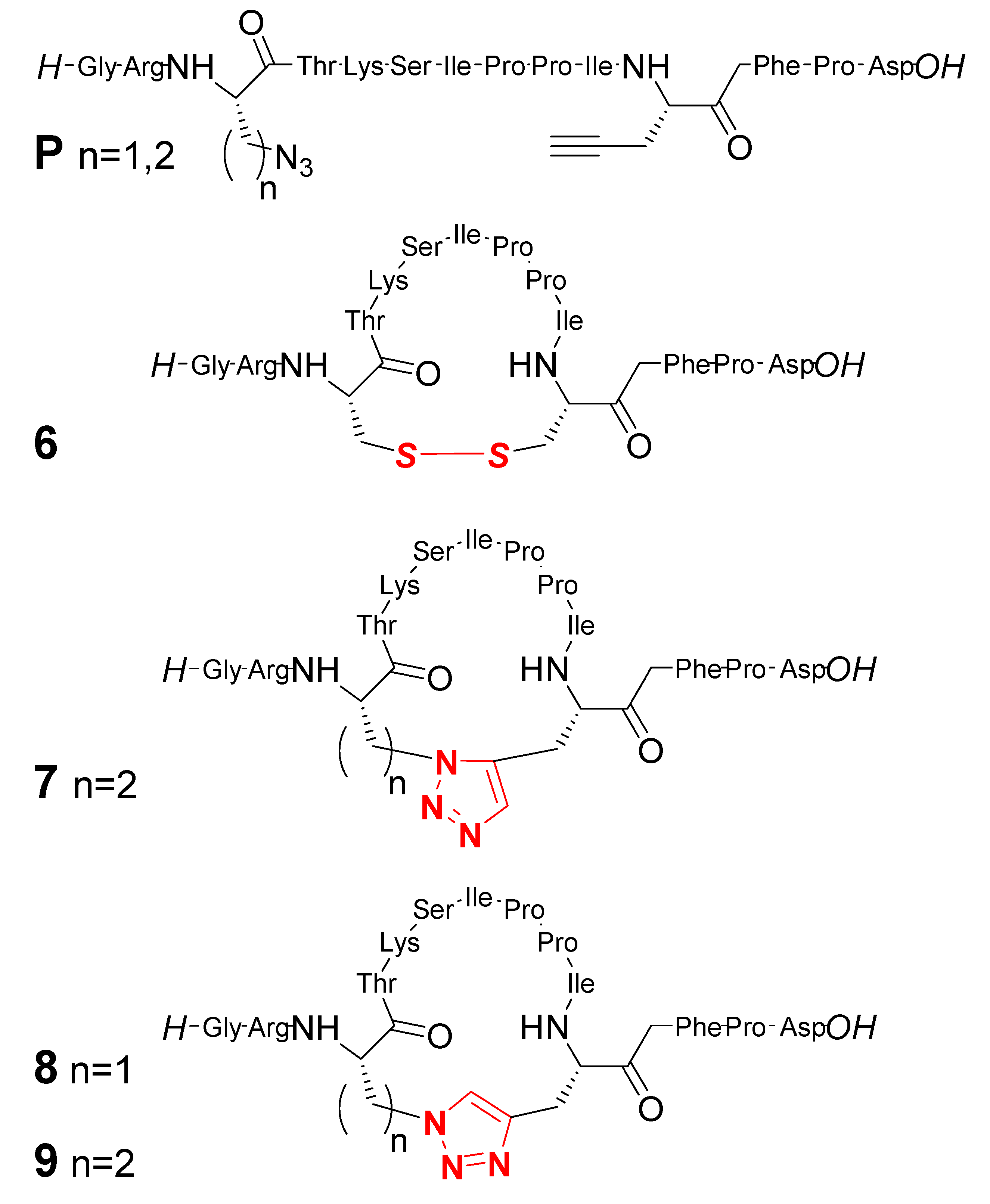
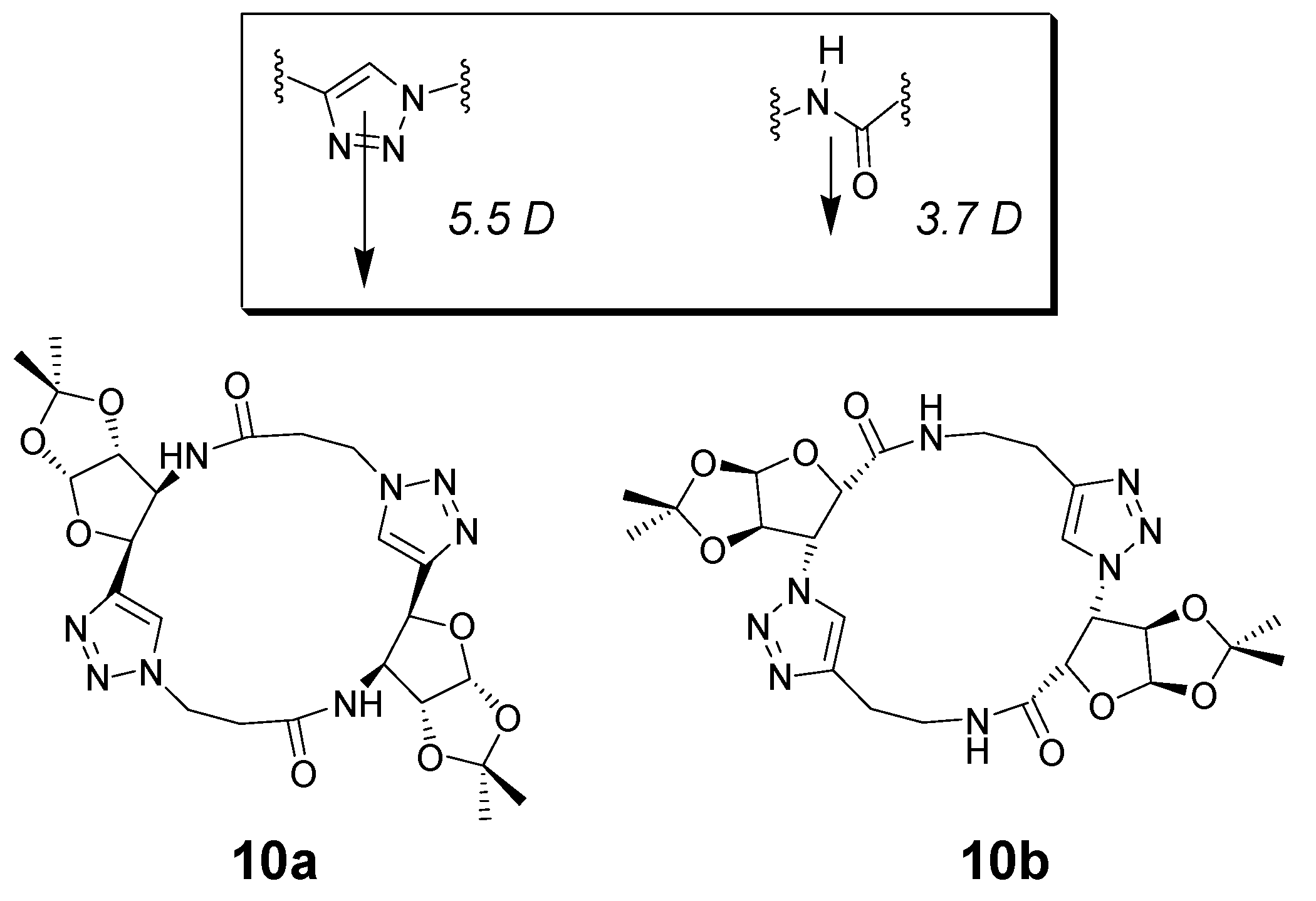
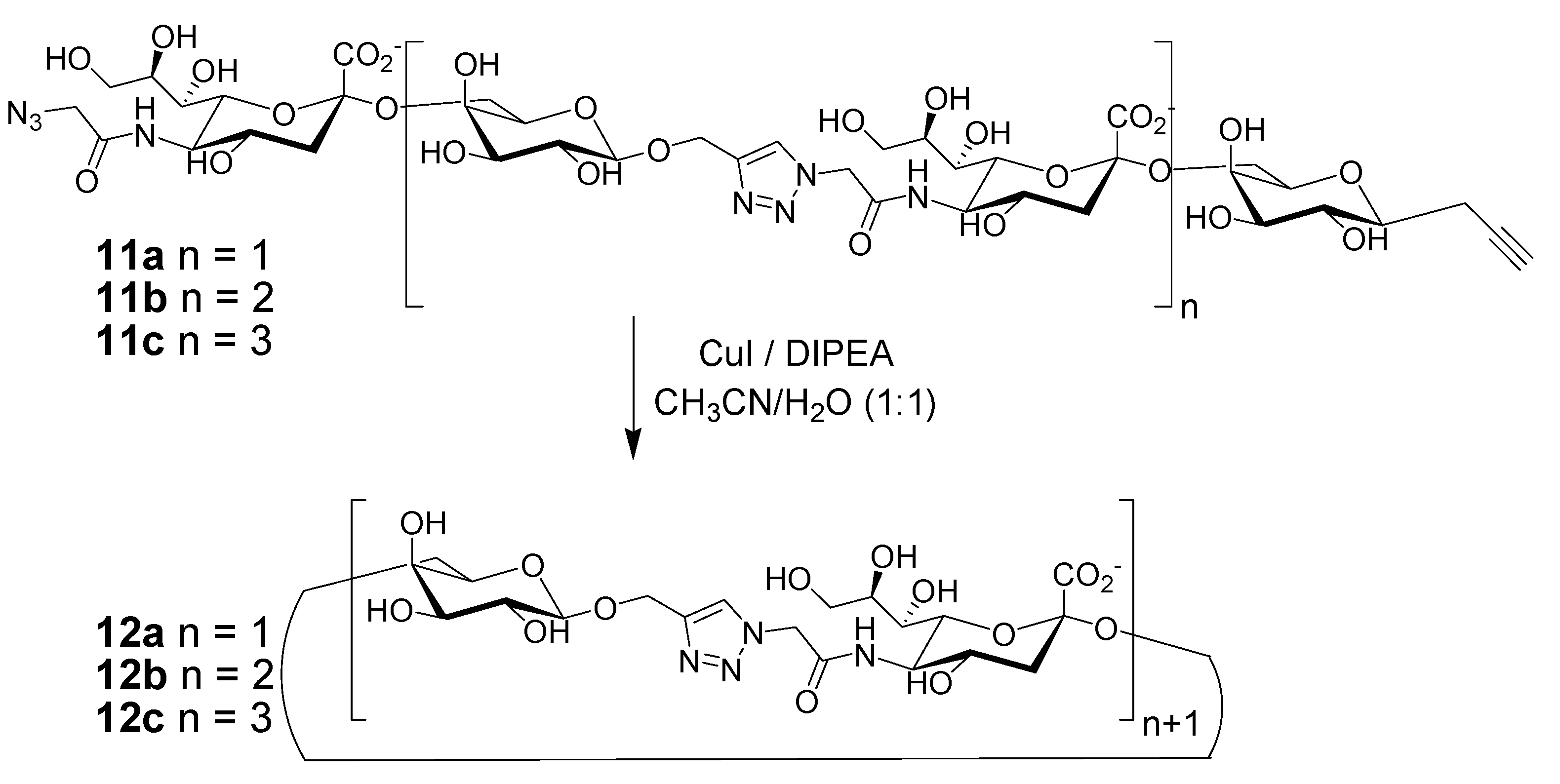
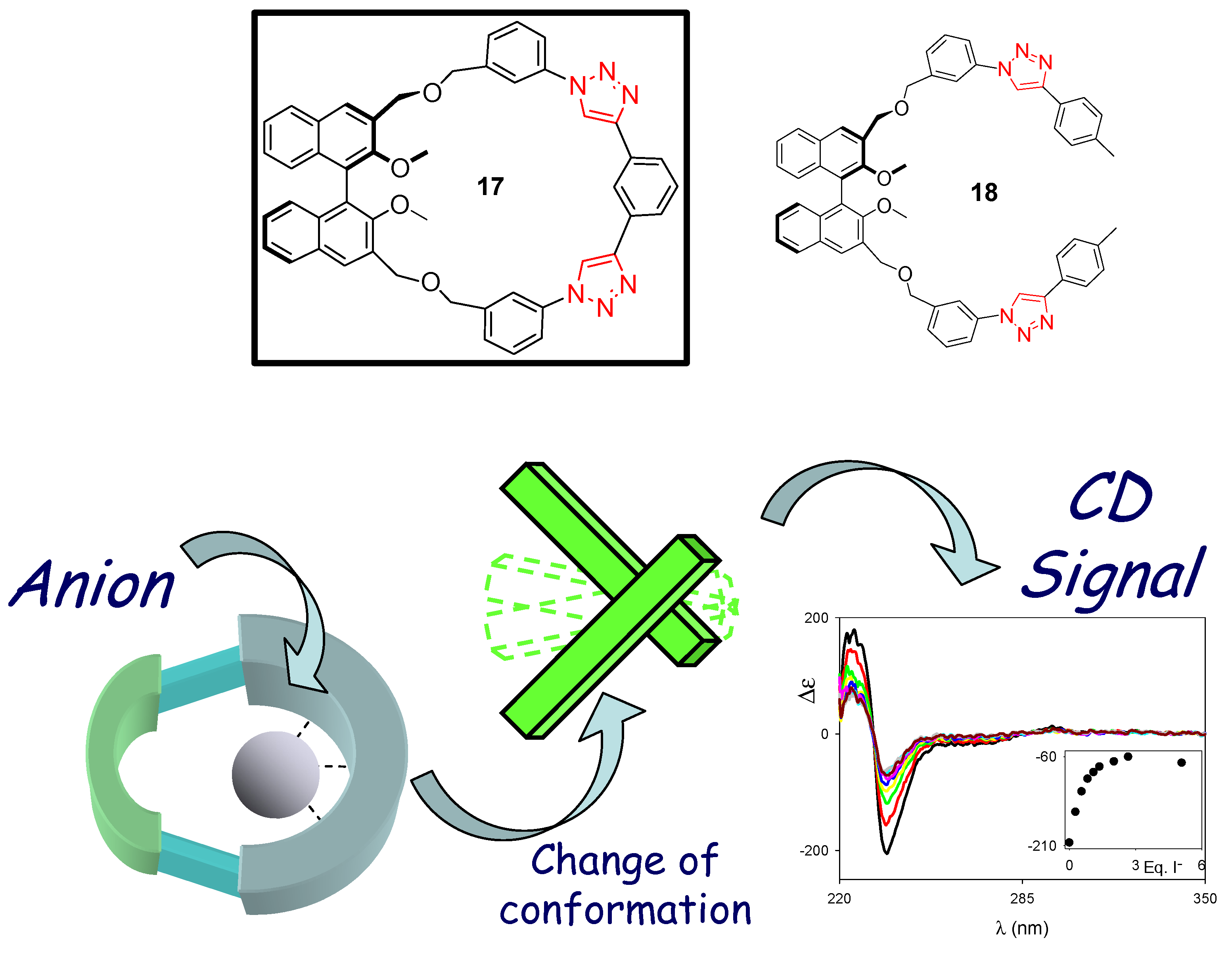
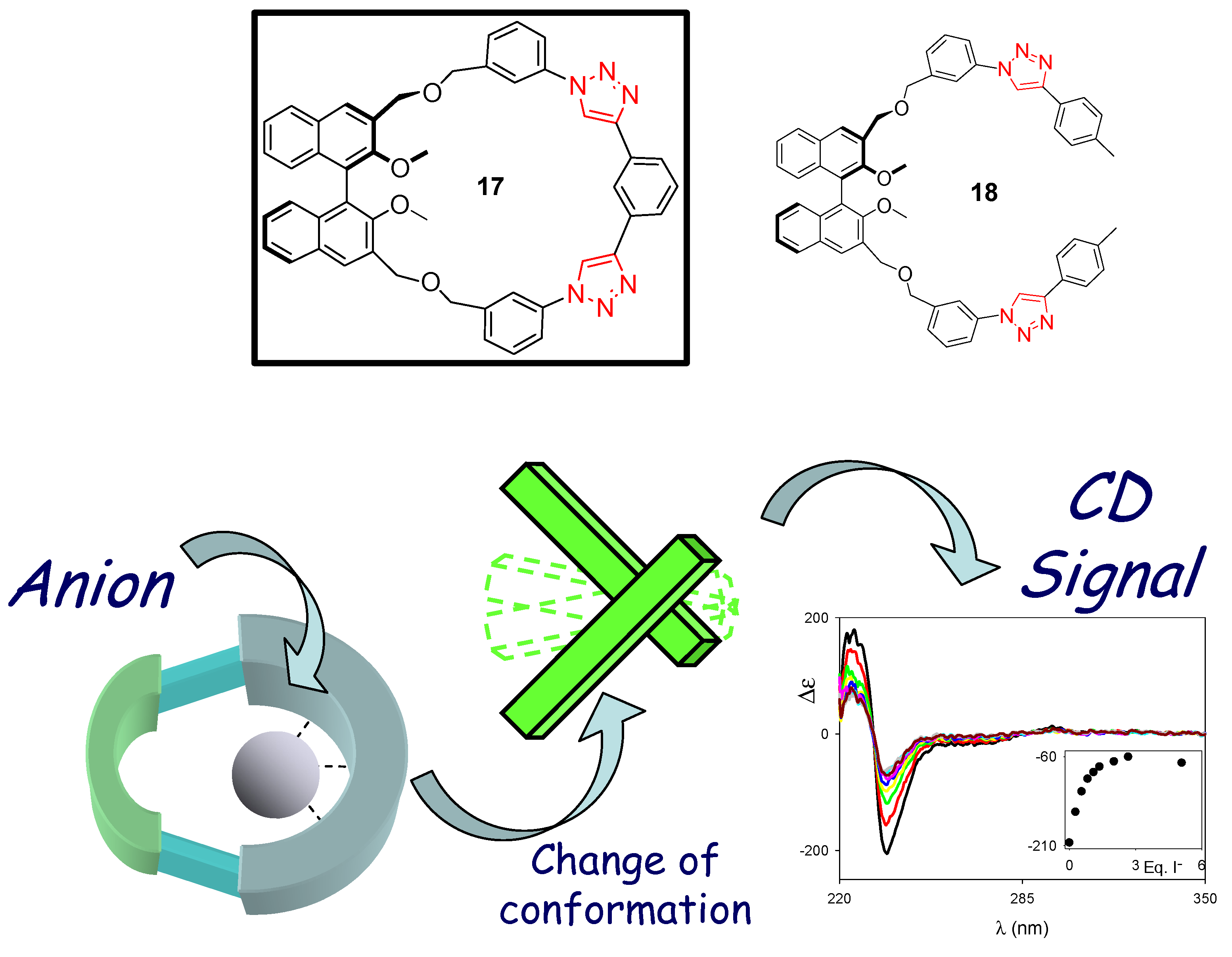
3. Click Macrocycles for Anion Binding and Supramolecular Recognition
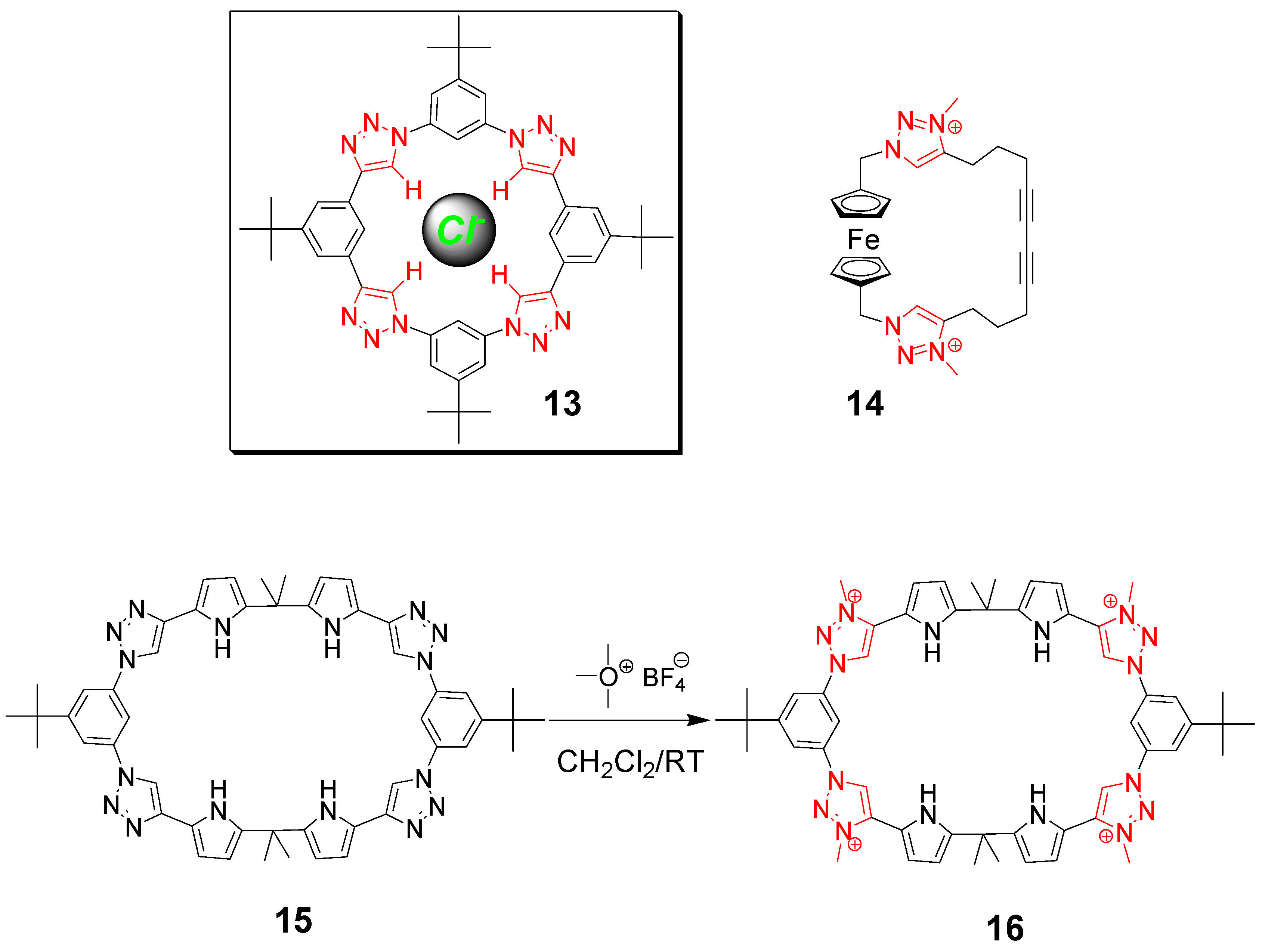
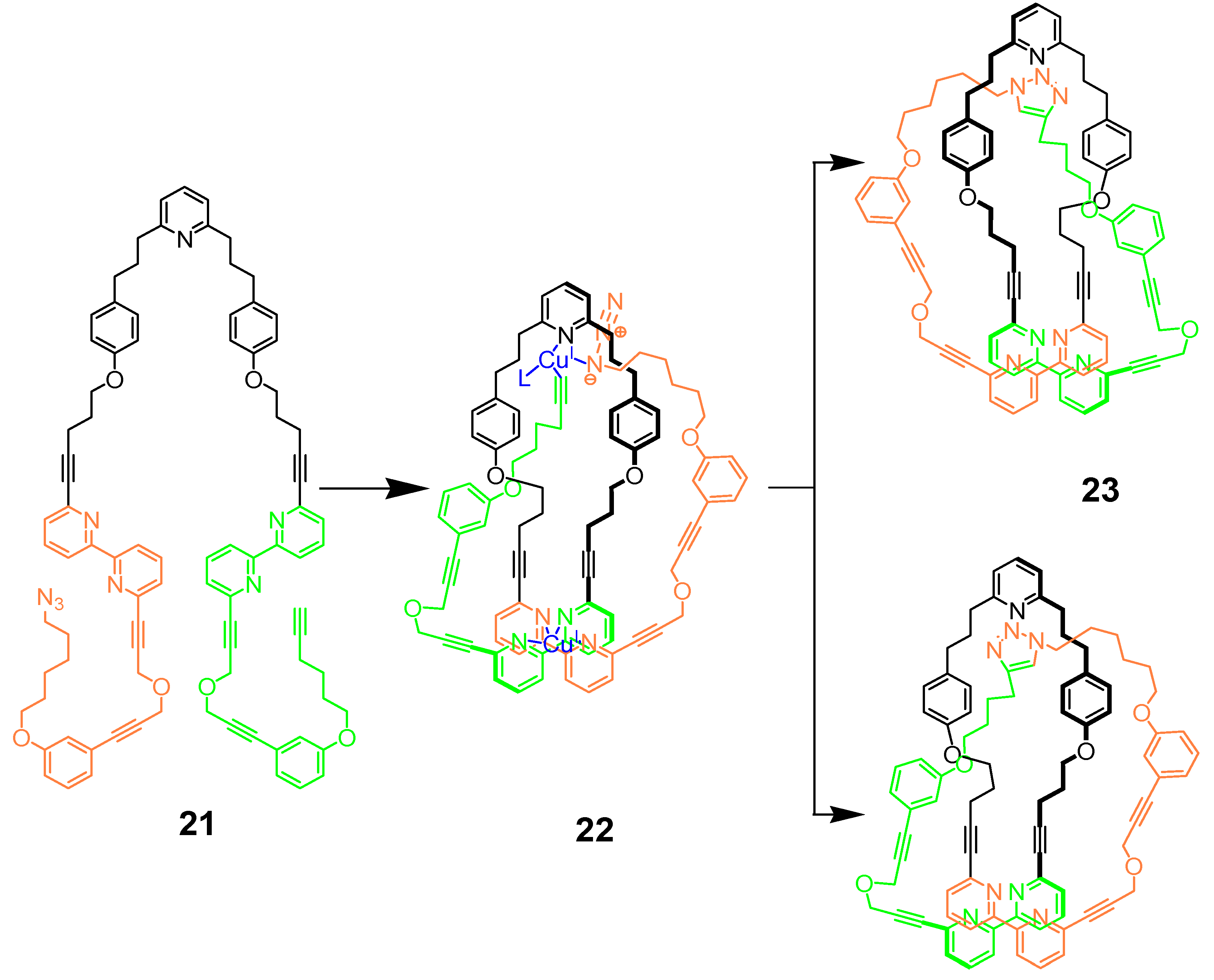
4. Clicking Macrocycles to Form Mechanical Bonds
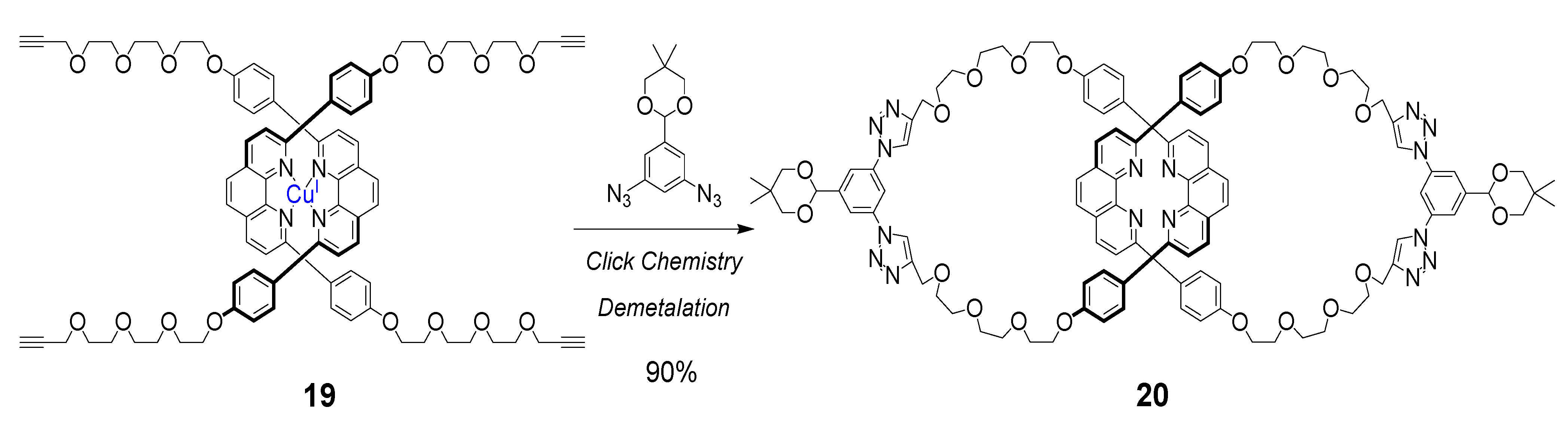
5. Cyclic Polymers Obtained by the CuAAC Click Reaction

6. Summary and Perspectives
Acknowledgments
Conflicts of Interest
References
- IUPAC GOLDBOOK. Available online: http://goldbook.iupac.org/M03662.html (accessed on 18 June 2013).
- Lehn, J.M. Supramolecular chemistry—scope and perspectives molecules, supermolecules, and molecular devices (nobel lecture). Angew. Chem. Int. Ed. 1988, 27, 89–112. [Google Scholar] [CrossRef]
- Cram, D.J. The design of molecular hosts, guests, and their complexes (nobel lecture). Angew. Chem. Int. Ed. 1988, 27, 1009–1020. [Google Scholar] [CrossRef]
- Pedersen, C.J. Cyclic polyethers and their complexes with metal salts. J. Am. Chem. Soc. 1967, 89, 7017–7036. [Google Scholar] [CrossRef]
- Weber, E.; Vögtle, F.; Burrell, A. K. Macrocycles; Springer-Verlag: Berkin, Germany, 1992. [Google Scholar]
- Lehn, J.M. Supramolecular Chemistry: Concepts and Perspectives; Wiley-VCH: Weinheim, Germany, 1995. [Google Scholar]
- Fastrez, J. Macrocyclization versus polymerization in polycondensation reactions under high-dilution conditions: a theoretical study. J. Phys. Chem. 1989, 93, 2635–2642. [Google Scholar] [CrossRef]
- Cougnon, F.B.L.; Sanders, J.K.M. Evolution of dynamic combinatorial chemistry. Acc. Chem. Res. 2012, 45, 2211–2221. [Google Scholar] [CrossRef]
- Wong, C.-H.; Zimmerman, S.C. Orthogonality in organic, polymer, and supramolecular chemistry: from merrifield to click chemistry. Chem. Commun. 2013, 49, 1679–1695. [Google Scholar] [CrossRef]
- Huisgen, R.; Szeimies, G.; Möbius, L. 1.3-Dipolare cycloadditionen, XXXII. Kinetik der Additionen organischer Azide an CC-Mehrfachbindungen. Chem. Ber. 1967, 100, 2494–2507. [Google Scholar] [CrossRef]
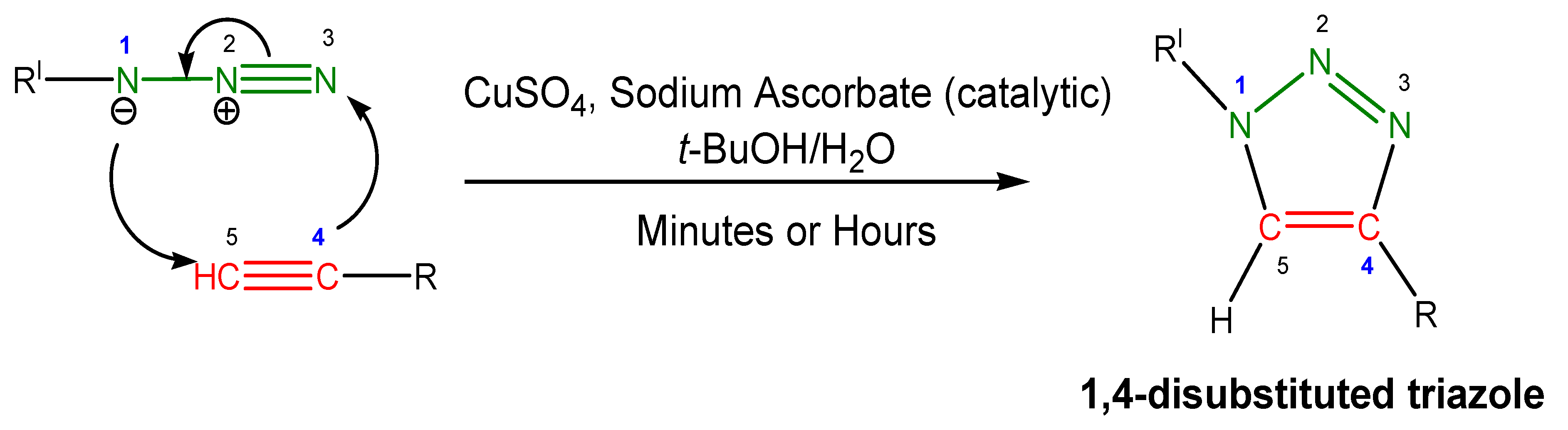
- Meldal, M.; Tornøe, C.W. Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Sen, G.S.; Kuzelka, J.; Singh, P.; Lewis, W.G.; Manchester, M.; Finn, M.G. Accelerated bioorthogonal conjugation: A practical method for the ligation of diverse functional molecules to a polyvalent virus scaffold. Bioconjugate Chem. 2005, 16, 1572–1579. [Google Scholar] [CrossRef]
- Barner-Kowollik, C.; Du Prez, F.E.; Espeel, P.; Hawker, C.J.; Junkers, T.; Schlaad, H.; van Camp, W. Clicking” polymers or just efficient linking: what is the difference? Angew. Chem. Int. Ed. 2011, 50, 60–62. [Google Scholar]
- Touaibia, M.; Shiao, T.C.; Papadopoulos, A.; Vaucher, J.; Wang, Q.; Behamioud, K.; Roy, R. Tri- and hexavalent mannoside clusters as potential inhibitors of type 1 fimbriated bacteria using pentaerythritol and triazole linkages. Chem. Commun 2007. [Google Scholar] [CrossRef]
- Pacini, A.; Caricato, M.; Ferrari, S.; Capsoni, D.; Martínez de Ilarduya, A.; Muñoz-Guerra, S.; Pasini, D. Poly(γ-glutamic acid) esters with reactive functional groups suitable for orthogonal conjugation strategies. J. Polym. Sci. A Polym Chem. 2012, 50, 4790–4799. [Google Scholar] [CrossRef]
- Sharma, A.K.; Caricato, M.; Quartarone, E.; Edizer, S.; Schieroni, A.G.; Mendichi, R.; Pasini, D. Polystyrene-based self-aggregating polymers based on UPy units. Polym. Bull. 2012, 69, 911–923. [Google Scholar] [CrossRef]
- Brennan, J.L.; Hatzakis, N.S.; Tshikhudo, T.R.; Dirvianskyte, N.; Razumas, V.; Patkar, S.; Vind, J.; Svendsen, A.; Nolte, R.J.M.; Rowan, A.E.; et al. Bionanoconjugation via click chemistry: The creation of functional hybrids of lipases and gold nanoparticles. Bioconjugate Chem. 2006, 17, 1373–1375. [Google Scholar] [CrossRef]
- Mynar, J.L.; Choi, T.-L.; Yoshida, M.; Kim, V.; Hawker, C.J.; Fréchet, J.M.J. Doubly-dendronized linear polymers. Chem. Commun. 2005, 5169–5171. [Google Scholar]
- Wu, P.; Feldman, A.K.; Nugent, A.K.; Hawker, C.J.; Scheel, A.; Voit, B.; Pyun, J.; Fréchet, J.M.J.; Sharpless, K.B.; Fokin, V.V. Efficiency and fidelity in a click-chemistry route to triazole dendrimers by the copper(I)-catalyzed ligation of azides and alkynes. Angew. Chem. Int. Ed. 2004, 43, 3928–3932. [Google Scholar] [CrossRef]
- Sletten, E.M.; Bertozzi, C.R. From mechanism to mouse: A tale of two bioorthogonal reactions. Acc. Chem. Res. 2011, 44, 666–676. [Google Scholar] [CrossRef]
- Romuald, C.; Cazals, G.; Enjalbal, C.; Coutrot, F. Straightforward synthesis of a double-lasso macrocycle from a nonsymmetrical [c2]Daisy chain. Org. Lett. 2013, 15, 184–187. [Google Scholar] [CrossRef]
- Lau, Y.H.; Price, J.R.; Todd, M.H.; Rutledge, P.J. A click fluorophore sensor that can distinguish CuII and HgII via selective anion-induced demetallation. Chem. Eur. J. 2011, 17, 2850–2858. [Google Scholar] [CrossRef]
- Nierengarten, I.; Guerra, S.; Holler, M.; Nierengarten, J.-F.; Deschenaux, R. Building liquid crystals from the 5-fold symmetrical pillar[5]arene core. Chem. Commun. 2012, 48, 8072–8074. [Google Scholar]
- Savonnet, M.; Kockrick, E.; Camarata, A.; Bazer-Bachi, D.; Bats, N.; Lecocq, V.; Pinela, C.; Farrusseng, D. Combinatorial synthesis of metal–organic frameworks libraries by click-chemistry. New J. Chem. 2011, 35, 1892–1897. [Google Scholar] [CrossRef]
- Driggers, E.M.; Hale, S.P.; Lee, J.; Terrett, N.K. The exploration of macrocycles for drug discovery-an underexploited structural class. Nat. Rev. Drug Discovery 2008, 7, 608–624. [Google Scholar] [CrossRef]
- Walensky, L.D.; Kung, A.L.; Escher, I.; Malia, T.J.; Barbuto, S.; Wright, R.D.; Wagner, G.; Verdine, G.L.; Korsmeyer, S.J. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 2004, 305, 1466–1470. [Google Scholar] [CrossRef]
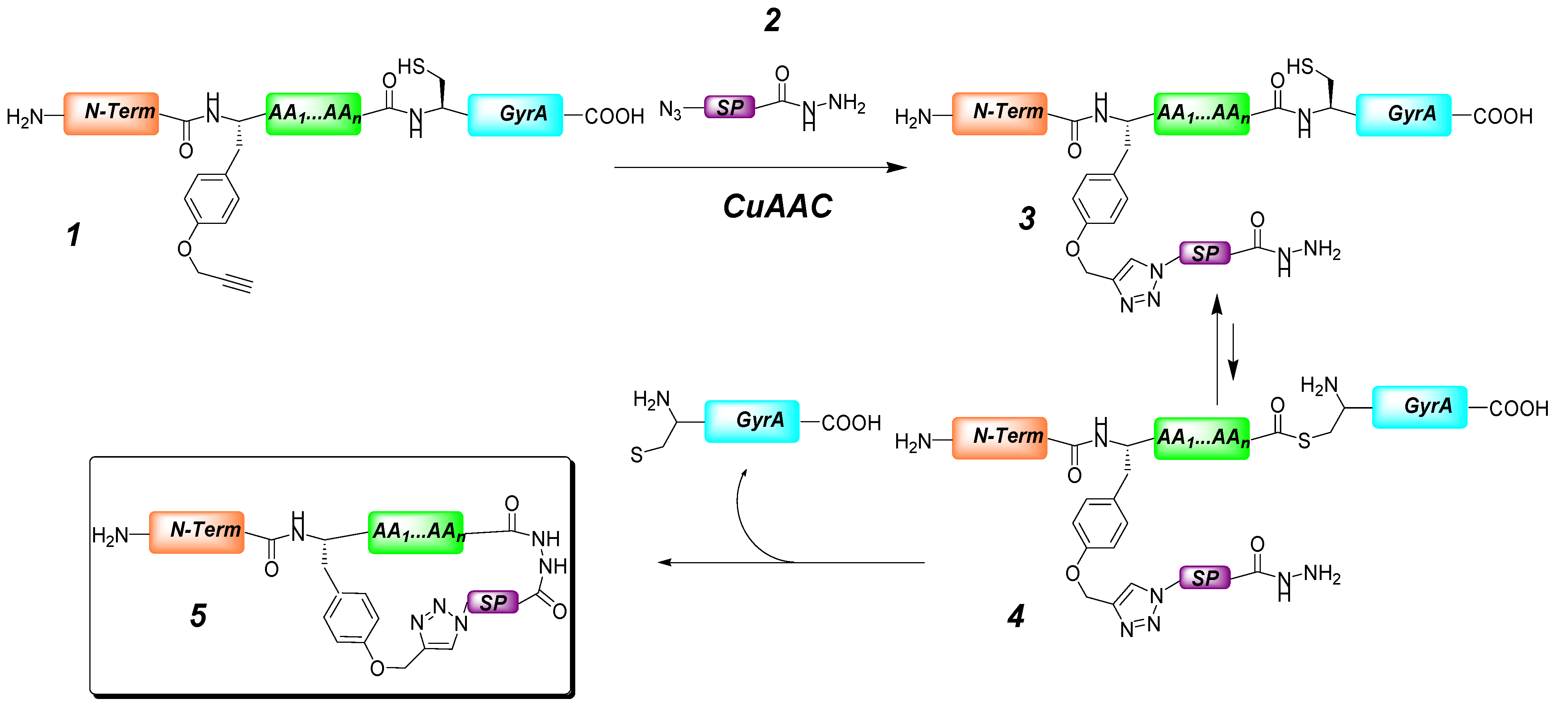
- Holub, J.M.; Kirshenbaum, K. Tricks with clicks: modification of peptidomimetic oligomers via copper-catalyzed azide-alkyne [3 + 2] cycloaddition. Chem. Soc. Rev. 2010, 39, 1325–1337. [Google Scholar] [CrossRef]
- Smith, J.M.; Vitali, F.; Archer, S.A.; Fasan, R. Modular assembly of macrocyclic organo-peptide hybrids using synthetic and genetically encoded precursors. Angew. Chem. Int. Ed. 2011, 50, 5075–5080. [Google Scholar] [CrossRef]
- Niu, J.; Hili, R.; Liu, D.R. Enzyme-free translation of DNA into sequence defined synthetic polymers structurally unrelated to nucleic acids. Nat. Chem. 2013, 5, 282–292. [Google Scholar] [CrossRef]
- Turner, R.A.; Oliver, A.G.; Lokey, R.S. Click chemistry as a macrocyclization tool in the solid-phase synthesis of small cyclic peptides. Org. Lett. 2007, 9, 5011–5014. [Google Scholar] [CrossRef]
- Ingale, S.; Dawson, P.E. On resin side-chain cyclization of complex peptides using CuAAC. Org. Lett. 2011, 13, 2822–2825. [Google Scholar] [CrossRef]
- Saludes, J.P.; Morton, L.A.; Ghosh, N.; Beninson, L.A.; Chapman, E.R.; Fleshner, M.; Yin, H. Detection of highly curved membrane surfaces using a cyclic peptide derived from synaptotagmin-I. ACS Chem. Biol. 2012, 7, 1629–1635. [Google Scholar]
- Empting, M.; Avrutina, O.; Meusinger, R.; Fabritz, S.; Reinwarth, M.; Biesalski, M.; Voigt, S.; Buntkowsky, G.; Kolmar, H. Triazole Bridge: Disulfide-bond replacement by ruthenium-catalyzed formation of 1,5-disubstituted 1,2,3-triazoles. Angew. Chem. Int. Ed. 2011, 50, 5207–5211. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, X.; Xue, P.; Sun, H.H.Y.; Williams, I.D.; Sharpless, K.B.; Fokin, V.V.; Jia, G. Ruthenium-catalyzed cycloaddition of alkynes and organic azides. J. Am. Chem. Soc. 2005, 127, 15998–15999. [Google Scholar]
- Boz, E.; Tüzün, N.Ş. Reaction mechanism of ruthenium-catalyzed azide-alkyne cycloaddition reaction: A DFT study. J. Organomet. Chem. 2013, 724, 167–176. [Google Scholar]
- Boren, B.C.; Narayan, S.; Rasmussen, L.K.; Zhang, L.; Zhao, H.T.; Lin, Z.Y.; Jia, G.C.; Fokin, V.V. Ruthenium-catalyzed azide-alkyne cycloaddition: scope and mechanism. J. Am. Chem. Soc. 2008, 130, 8923–8930. [Google Scholar]
- Majireck, M.M.; Weinreb, S.M. A study of the scope and regioselectivity of the ruthenium-catalyzed [3 + 2]-cycloaddition of azides with internal alkynes. J. Org. Chem. 2006, 71, 8680–8683. [Google Scholar] [CrossRef]
- Ruan, Y.-B.; Yu, Y.; Li, C.; Bogliotti, N.; Tang, J.; Xie, J. Triazolyl benzothiadiazole fluorescent chemosensors: A systematic investigation of 1,4- or 1,5-disubstituted mono- and bis-triazole derivatives. Tetrahedron 2013, 69, 4603–4609. [Google Scholar] [CrossRef]
- Zhang, J.; Kemmink, J.; Rijkers, D.T.S.; Liskamp, R.M.J. Synthesis of 1,5-triazole bridged vancomycin CDE-ring bicyclic mimics using RuAAC macrocyclization. Chem. Commun. 2013, 49, 4498–4500. [Google Scholar] [CrossRef]
- Arisawa, M.; Fujii, Y.; Kato, H.; Fukuda, H.; Matsumoto, T.; Ito, M.; Abe, H.; Ito, Y.; Shuto, S. One-pot ring-closing metathesis/1,3-dipolar cycloaddition through assisted tandem ruthenium catalysis: Synthesis of a dye with isoindolo[2,1-a]quinoline structure. Angew. Chem. Int. Ed. 2013, 52, 1003–1007. [Google Scholar] [CrossRef]
- Pasini, D.; Ricci, M. Macrocycles as precursors for organic nanotubes. Curr. Org. Synth. 2007, 4, 59–80. [Google Scholar] [CrossRef]
- Baudry, Y.; Bollot, G.; Gorteau, V.; Litvinchuk, S.; Mareda, J.; Nishihara, M.; Pasini, D.; Perret, F.; Ronan, D.; Sakai, N.; et al. Molecular recognition by synthetic multifunctional pores in practice: Are structural studies really helpful? Adv. Funct. Mat. 2006, 16, 169–179. [Google Scholar] [CrossRef]
- Shimizu, L.S. Perspectives on main-chain hydrogen bonded supramolecular polymers. Polym. Int. 2007, 56, 444–452. [Google Scholar] [CrossRef]
- Bong, D.T.; Clark, T.D.; Granja, J.R.; Ghadiri, M.R. Self-assembling organic nanotubes. Angew. Chem. Int. Ed. 2001, 40, 988–1011. [Google Scholar] [CrossRef]
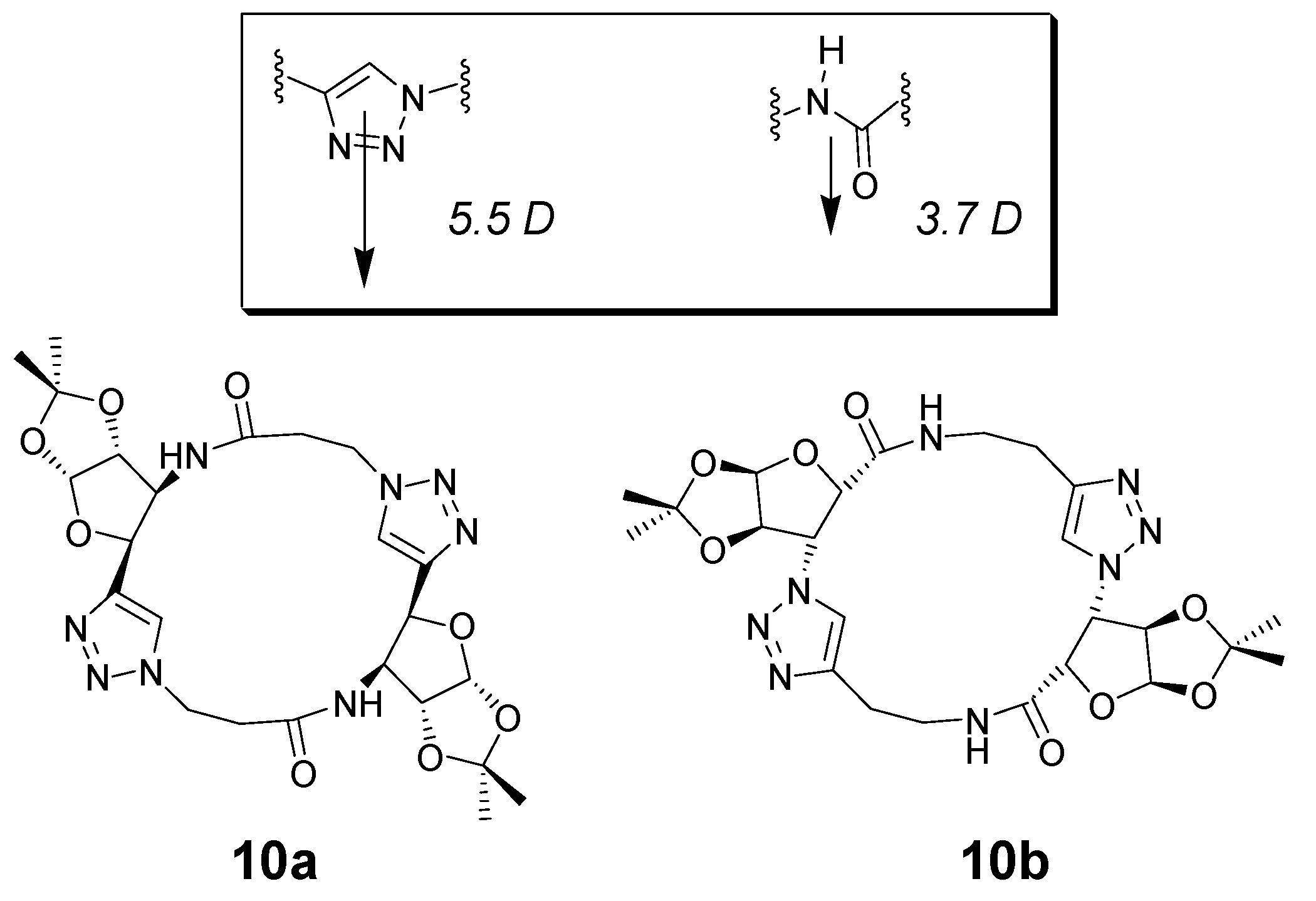
- Ghorai, A.; Padmanaban, E.; Mukhopadhyay, C.; Acharia, B.; Chattopadhyay, P. Design and synthesis of regioisomeric triazole based peptidomimetic macrocycles and their dipole moment controlled self-assembly. Chem. Commun. 2012, 48, 11975–11977. [Google Scholar] [CrossRef]
- Ghorai, A.; Gayen, A.; Kulsi, G.; Padmanaban, E.; Laskar, A.; Achari, B.; Mukhopadhyay, C.; Chattopadhyay, P. Simultaneous parallel and antiparallel self-assembly in a triazole/amide macrocycle conformationally homologous to D-,L-α-amino acid based cyclic peptides: NMR and molecular modeling study. Org. Lett. 2011, 13, 5512–5515. [Google Scholar] [CrossRef]
- Pehere, A.D.; Sumby, C.J.; Abell, A.D. New cylindrical peptide assemblies defined by extended parallel β-sheets. Org. Biomol. Chem. 2013, 11, 425–429. [Google Scholar] [CrossRef]
- Chouhan, G.; James, K. Efficient construction of proline-containing β-Turn mimetic cyclic tetrapeptides via CuAAC macrocyclization. Org. Lett. 2013, 15, 1206–1209. [Google Scholar] [CrossRef]
- Ajay, A.; Sharma, S.; Gupt, M.P.; Bajpai, V.; Kumar, H.B.; Kaushik, M.P.; Konwar, R.; Ampapathi, R.S.; Tripathi, R.P. Diversity oriented synthesis of pyran based polyfunctional stereogenic macrocyles and their conformational studies. Org. Lett. 2012, 14, 4306–4309. [Google Scholar] [CrossRef]
- Allam, A.; Dupont, L.; Behr, J.-B.; Plantier-Royon, R. Convenient synthesis of a galacturonic acid based macrocycle with potential copper-complexation ability. Eur. J. Org. Chem. 2012, 817–823. [Google Scholar]
- Kumar, A.; Geng, Y.; Schmidt, R.R. Intramolecular glycosidation by click reaction mediated spacer generation followed by spacer cleavage. Eur. J. Org. Chem. 2012, 6846–6851. [Google Scholar] [CrossRef]
- Lewandowski, B.; Jarosz, S. Amino-acid templated assembly of sucrose-derived macrocycles. Org. Lett. 2010, 12, 2532–2535. [Google Scholar] [CrossRef]
- Muthana, S.; Yu, H.; Cao, H.; Cheng, J.; Chen, X. Chemoenzymatic synthesis of a new class of macrocyclic oligosaccharides. J. Org. Chem. 2009, 74, 2928–2936. [Google Scholar] [CrossRef]
- Schulz, M.; Christoffers, J. New macrocyclic bistriazolophanes with thioindigo chromophore. Tetrahedron 2013, 69, 802–809. [Google Scholar] [CrossRef]
- Hradilová, L.; Grepl, M.; Dvoráková, B.; Hradil, P. Study of direct macrocycle formation via the cyclisation of propargyl 2-azidobenzoate. Tetrahedron Lett. 2013, 54, 1218–1221. [Google Scholar] [CrossRef]
- Li, Y.; Flood, A.H. Pure CH hydrogen bonding to chloride ions: A pre-organized and rigid macrocyclic receptor. Angew. Chem. Int. Ed. 2008, 47, 2649–2652. [Google Scholar] [CrossRef]
- Hua, Y.; Flood, A.H. Click chemistry generates privileged CH hydrogen-bonding triazoles: the latest addition to anion supramolecular chemistry. Chem. Soc. Rev. 2010, 39, 1262–1271. [Google Scholar]
- White, N.G.; Beer, P.D. A ferrocene redox-active triazolium macrocycle that binds and senses chloride. Beilstein J. Org. Chem. 2012, 8, 246–252. [Google Scholar] [CrossRef]
- Cai, J.; Hay, B.P.; Young, N.J.; Yang, X.; Sessler, J.L. A pyrrole-based triazolium-phane with NH and cationic CH donor groups as a receptor for tetrahedral oxyanions that functions in polar media. Chem. Sci. 2013, 4, 1560–1567. [Google Scholar]
- Caricato, M.; Olmo, A.; Gargiulli, C.; Gattuso, G.; Pasini, D. A “clicked” macrocyclic probe incorporating Binol as the signalling unit for the chiroptical sensing of anions. Tetrahedron 2012, 68, 7861–7866. [Google Scholar] [CrossRef]
- Moletti, A.; Coluccini, C.; Pasini, D.; Taglietti, A. A chiral probe for the detection of Cu(II) by UV, CD and emission spectroscopies. Dalton Trans. 2007, 16, 1588–1592. [Google Scholar]
- Boiocchi, M.; Bonizzoni, M.; Moletti, A.; Pasini, D.; Taglietti, A. Linear recognition of dicarboxylates by ditopic macrocyclic complexes. New J. Chem. 2007, 31, 352–356. [Google Scholar]
- Bencini, A.; Coluccini, C.; Garau, A.; Giorgi, C.; Lippolis, V.; Messori, L.; Pasini, D.; Puccioni, S. A BINOL-based chiral polyammonium receptor for highly enantioselective recognition and fluorescence sensing of (S,S)-tartaric acid in aqueous solution. Chem. Commun. 2012, 48, 10428–10430. [Google Scholar] [CrossRef]
- Coluccini, C.; Mazzanti, A.; Pasini, D. Locked chromophores as CD and NMR probes for the helical conformation of tetraamidic macrocycles. Org. Biomol. Chem. 2010, 8, 1807–1815. [Google Scholar] [CrossRef]
- Colombo, S.; Coluccini, C.; Caricato, M.; Gargiulli, C.; Gattuso, G.; Pasini, D. Shape selectivity in the synthesis of chiral macrocyclic amides. Tetrahedron 2010, 66, 4206–4211. [Google Scholar] [CrossRef]
- Coluccini, C.; Dondi, D.; Caricato, M.; Taglietti, A.; Boiocchi, M.; Pasini, D. Structurally-variable, rigid and optically-active D2 and D3 macrocycles possessing recognition properties towards C60. Org. Biomol. Chem. 2010, 8, 1640–1649. [Google Scholar]
- Caricato, M.; Leza, N.J.; Gargiulli, C.; Gattuso, G.; Dondi, D.; Pasini, D. Synthesis and anion recognition properties of shape-persistent binaphthyl-containing chiral macrocyclic amides. Beilstein J. Org. Chem. 2012, 8, 967–976. [Google Scholar]
- Caricato, M.; Coluccini, C.; Dondi, D.; Vander Griend, D.A.; Pasini, D. Nesting complexation of C60 with large, rigid D2 symmetrical macrocycles. Org. Biomol. Chem. 2010, 8, 3272–3280. [Google Scholar]
- Ricci, M.; Pasini, D. Rigid optically-active D2 and D3 macrocycles. Org. Biomol. Chem. 2003, 1, 3261–3262. [Google Scholar] [CrossRef]
- Coluccini, C.; Castelluccio, A.; Pasini, D. Chemoselective functionalization of 3,3'-substituted BINOL derivatives. J. Org. Chem. 2008, 73, 4237–4240. [Google Scholar] [CrossRef]
- Caricato, M.; Coluccini, C.; Vander Griend, D.A.; Forni, A.; Pasini, D. From red to blue shift: switching the binding affinity from the acceptor to the donor end by increasing the π-bridge in push-pull chromophores with coordinative ends. New J. Chem. 2013. [Google Scholar] [CrossRef]
- Caricato, M.; Leza, N.J.; Roy, K.; Dondi, D.; Gattuso, G.; Shimizu, L.S.; Vander Griend, D.A.; Pasini, D. A Chiroptical probe for sensing metal Ions in water. Eur. J. Org. Chem. 2013. [Google Scholar] [CrossRef]
- Asakawa, M.; Ashton, P.R.; Hayes, W.; Janssen, H.M.; Meijer, E.W.; Menzer, S.; Pasini, D.; Stoddart, J.F.; White, A.J.P.; Williams, D.J. Constitutionally-asymmetric and chiral [2]Pseudorotaxanes. J. Am. Chem. Soc. 1998, 120, 920–931. [Google Scholar] [CrossRef]
- Asakawa, M.; Ashton, P.R.; Boyd, S.E.; Brown, C.L.; Menzer, S.; Pasini, D.; Stoddart, J.F.; Tolley, M.S.; White, A.J.P.; Williams, D.J.; et al. Axially-chiral catenanes and π-electron deficient receptors. Chem. Eur. J. 1997, 3, 463–481. [Google Scholar]
- Asakawa, M.; Brown, C.L.; Pasini, D.; Stoddart, J.F.; Wyatt, P.G. Enantioselective recognition of amino acids by axially-chiral π-electron deficient receptors. J. Org. Chem. 1996, 61, 7234–7235. [Google Scholar] [CrossRef]
- Asakawa, M.; Janssen, H.M.; Meijer, E.W.; Pasini, D.; Stoddart, J.F. Enantioselective differentiation in the self-assembly of [2]Pseudorotaxanes. Eur. J. Org. Chem. 1998, 1998, 983–986. [Google Scholar]
- Ashton, P.R.; Heiss, A.; Pasini, D.; Raymo, F.M.; Shipway, A.N.; Stoddart, J.F.; Spencer, N. Diastereoselective self-assembly of [2]Catenanes. Eur. J. Org. Chem. 1999, 1999, 995–1004. [Google Scholar] [CrossRef]
- Miljanić, O.Š.; Dichtel, W.R.; Aprahamian, I.; Rohde, R.D.; Agnew, H.D.; Heath, J.R.; Stoddart, J.F. Rotaxanes and catenanes by click chemistry. QSAR Comb. Sci. 2007, 26, 1165–1174. [Google Scholar] [CrossRef]
- Megiatto, J.D., Jr.; Schuster, D.I. Introduction of useful peripheral functional groups on [2]Catenanes by combining Cu(I) template synthesis with “click” chemistry. New J. Chem. 2010, 34, 276–286. [Google Scholar] [CrossRef]
- Megiatto, J.D., Jr.; Schuster, D.I.; Abwandner, S.; de Miguel, G.; Guldi, D.M. [2]Catenanes decorated with porphyrin and [60]Fullerene groups: design, convergent synthesis, and photoinduced processes. J. Am. Chem. Soc. 2010, 132, 3847–3861. [Google Scholar]
- Romuald, C.; Coutrot, F. Combining coordination chemistry and catalysis to tie a knot by an active-metal template strategy. Angew. Chem. Int. Ed. 2012, 51, 2544–2545. [Google Scholar] [CrossRef]
- Barran, P.E.; Cole, H.L.; Goldup, S.M.; Leigh, D.A.; McGonigal, P.R.; Symes, M.D.; Wu, J.; Zengerle, M. Active-metal template synthesis of a molecular trefoil knot. Angew. Chem. Int. Ed. 2011, 50, 12280–12284. [Google Scholar] [CrossRef]
- Fox, M.E.; Szoka, F.C.; Frechet, J.M.J. Soluble polymer carriers for the treatment of cancer: The importance of molecular architecture. Acc. Chem. Res. 2009, 42, 1141–1151. [Google Scholar] [CrossRef]
- Binauld, S.; Hawker, C.J.; Fleury, E.; Drockenmuller, E. A modular approach to functionalized and expanded crown ether based macrocycles using click chemistry. Angew. Chem. Int. Ed. 2009, 48, 6654–6658. [Google Scholar] [CrossRef]
- Schulz, M.; Tanner, S.; Barqawi, H.; Binder, W.H. Macrocyclization of polymers via ring-closing metathesis and azide/alkyne-“click”-reactions: An approach to cyclic polyisobutylenes. J. Polym. Sci. A. Polym Chem. 2010, 48, 671–680. [Google Scholar] [CrossRef]
- Narumi, A.; Zeidler, H.S.; Barqawi, H.; Enders, C.; Binder, W.H. Cyclic alkoxyamine-initiator tethered by azide/alkyne-“click”-chemistry enabling ring-expansion vinyl polymerization providing macrocyclic polymers. J. Polym. Sci. A. Polym Chem. 2010, 48, 3402–3416. [Google Scholar] [CrossRef]
- Wan, X.; Liu, T.; Liu, S. Synthesis of amphiphilic tadpole-shaped linear-cyclic diblock copolymers via ring-opening polymerization directly initiating from cyclic precursors and their application as drug nanocarriers. Biomacromolecules 2011, 12, 1146–1154. [Google Scholar] [CrossRef]
- Sharma, A.K.; Cornaggia, C.; Pasini, D. Controlled RAFT Cyclopolymerization of oriented styrenic difunctional monomers. Macromol. Chem. Phys. 2010, 211, 2254–2259. [Google Scholar] [CrossRef]
- Edizer, S.; Veronesi, B.; Karahan, O.; Aviyente, V.; Değirmenci, I.; Galbiati, A.; Pasini, D. Efficient Free-Radical Cyclopolymerization of oriented styrenic difunctional monomers. Macromolecules 2009, 49, 1860–1866. [Google Scholar]
- Coluccini, C.; Metrangolo, P.; Parachini, M.; Pasini, D.; Resnati, G.; Righetti, P. Push-pull supramolecular chromophores supported on cyclopolymers. J. Polym. Sci. A Polym Chem. 2008, 46, 5202–5213. [Google Scholar] [CrossRef]
- Cagnoni, E.; Pasini, D.; Galbiati, A.; Ricci, M.; Righetti, P.P. Cyclopolymers as liquid membrane carriers. Macromolecules 2003, 36, 8894–8897. [Google Scholar] [CrossRef]
- Blazquez, E.; Mustarelli, P.; Pasini, D.; Righetti, P.P.; Tomasi, C. Thermal and conductivity properties of polyethylene glycol-based cyclopolymers. J. Mater. Chem. 2004, 14, 2524–2529. [Google Scholar] [CrossRef]
- Laurent, B.A.; Grayson, S.M. An efficient route to well-defined macrocyclic polymers via "click" cyclization. J. Am. Chem. Soc. 2006, 128, 4238–4239. [Google Scholar] [CrossRef]
- Laurent, B.A.; Grayson, S.M. Synthesis of cyclic dendronized polymers via divergent “graft-from” and convergent click “graft-to” routes: Preparation of modular toroidal macromolecules. J. Am. Chem. Soc. 2011, 133, 13421–13429. [Google Scholar] [CrossRef]
- Lonsdale, D.E.; Bell, C.A.; Monteiro, M.J. Strategy for rapid and high-purity monocyclic polymers by CuAAC “click” reactions. Macromolecules 2010, 43, 3331–3339. [Google Scholar] [CrossRef]
- Ulloora, S.; Shabaraya, R.; Adhikari, A.V. Facile synthesis of new imidazo[1,2-a]pyridines carrying 1,2,3-triazoles via click chemistry and their antiepileptic studies. Bioorg. Med. Chem. Lett. 2013, 23, 3368–3372. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pasini, D. The Click Reaction as an Efficient Tool for the Construction of Macrocyclic Structures. Molecules 2013, 18, 9512-9530. https://doi.org/10.3390/molecules18089512
Pasini D. The Click Reaction as an Efficient Tool for the Construction of Macrocyclic Structures. Molecules. 2013; 18(8):9512-9530. https://doi.org/10.3390/molecules18089512
Chicago/Turabian StylePasini, Dario. 2013. "The Click Reaction as an Efficient Tool for the Construction of Macrocyclic Structures" Molecules 18, no. 8: 9512-9530. https://doi.org/10.3390/molecules18089512