Breathing Some New Life into an Old Topic: Chalcogen-Nitrogen π-Heterocycles as Electron Acceptors †



Abstract
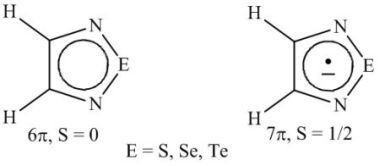
:
1. Introduction
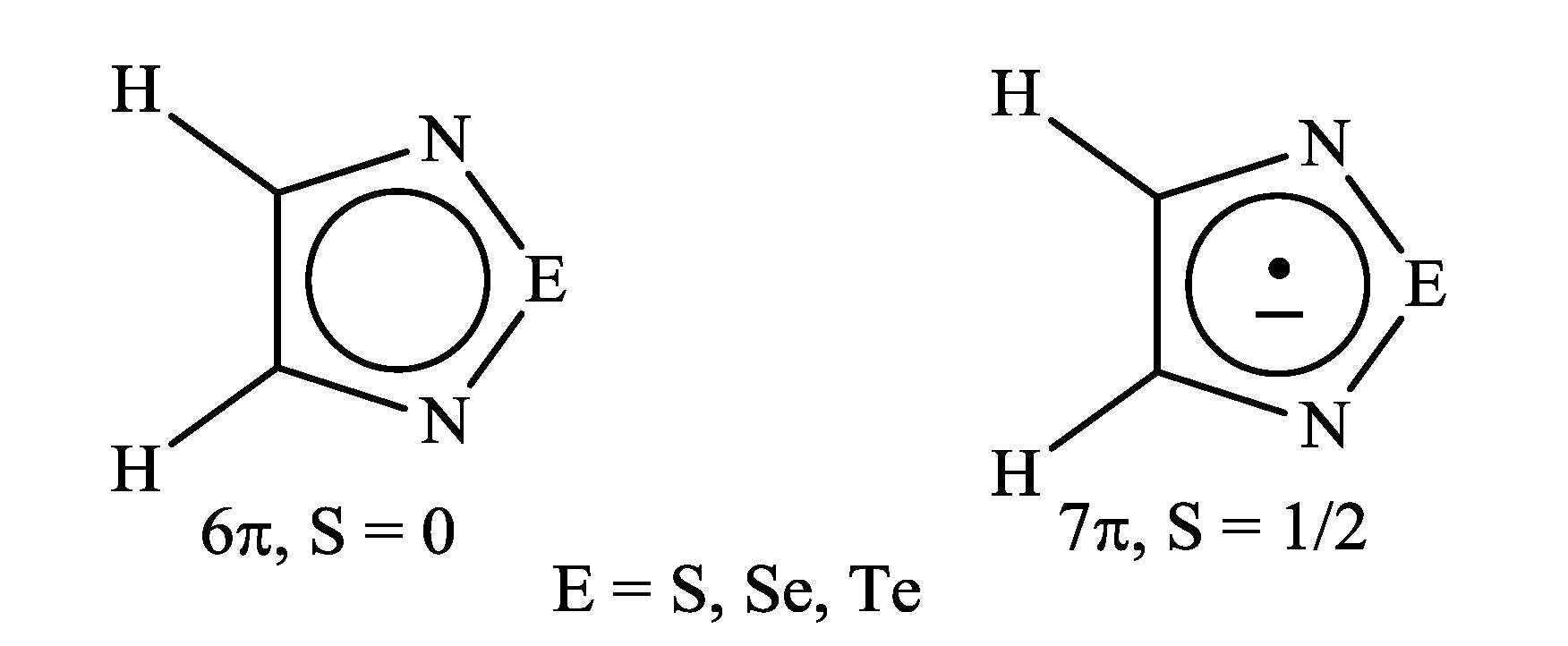
2. Electron Affinity and Redox Properties
2.1. Electron Affinity

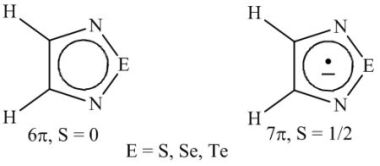{kind=link}
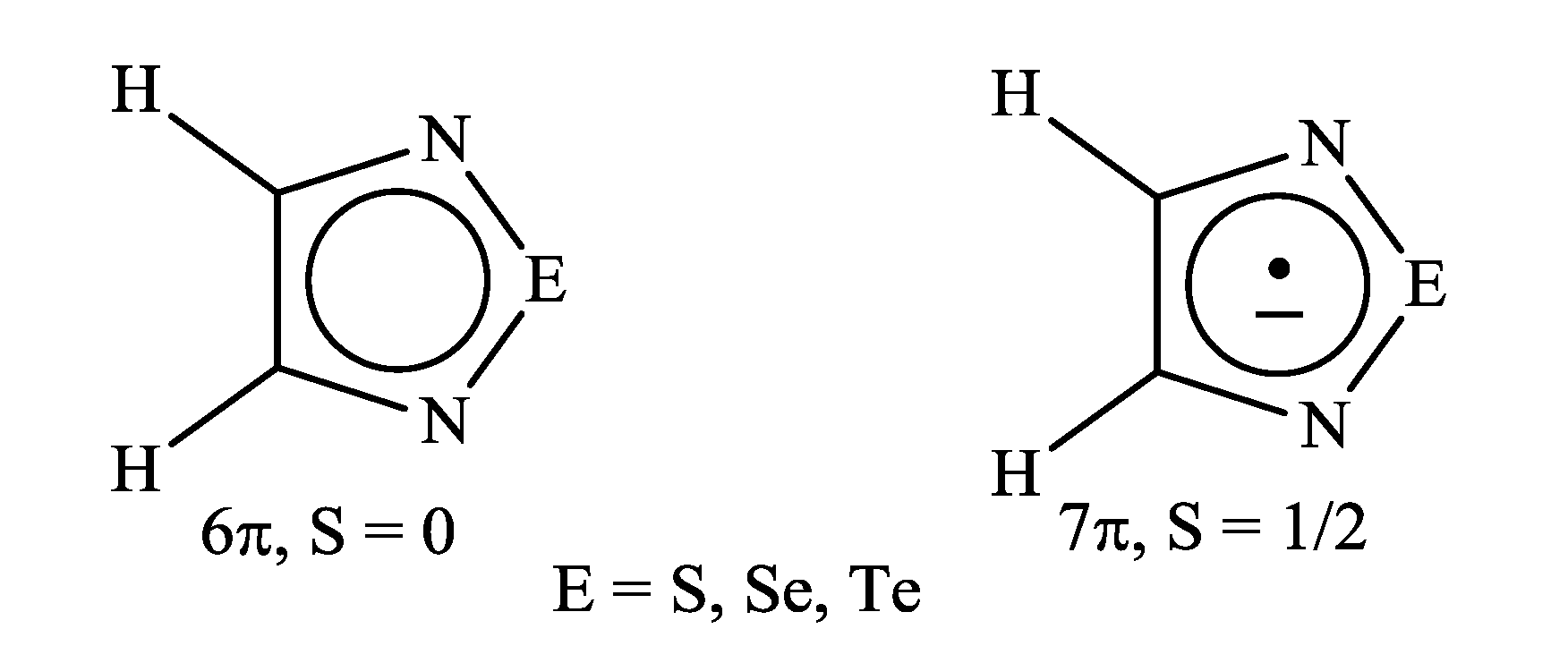{kind=link}
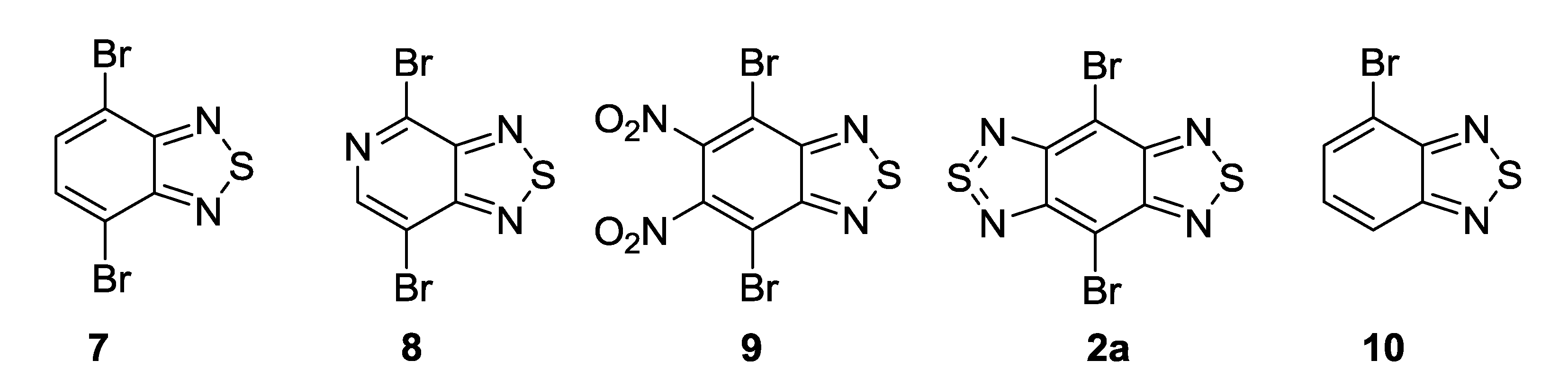{kind=link}
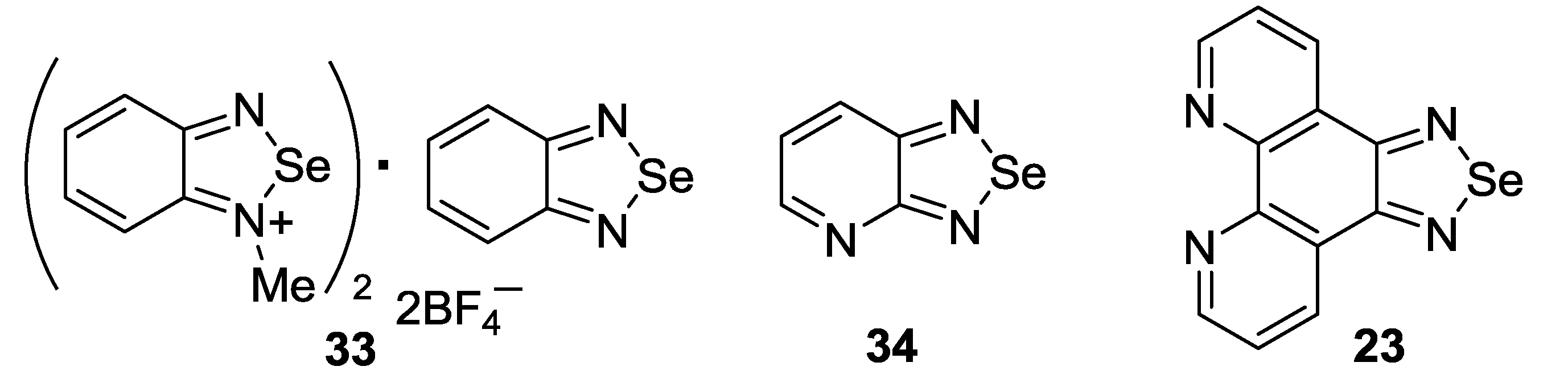{kind=link}
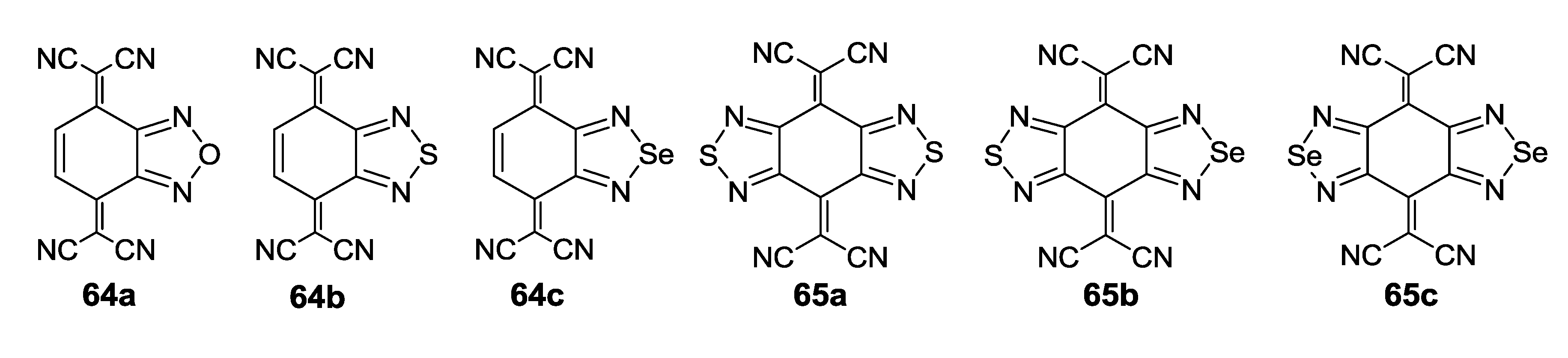{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
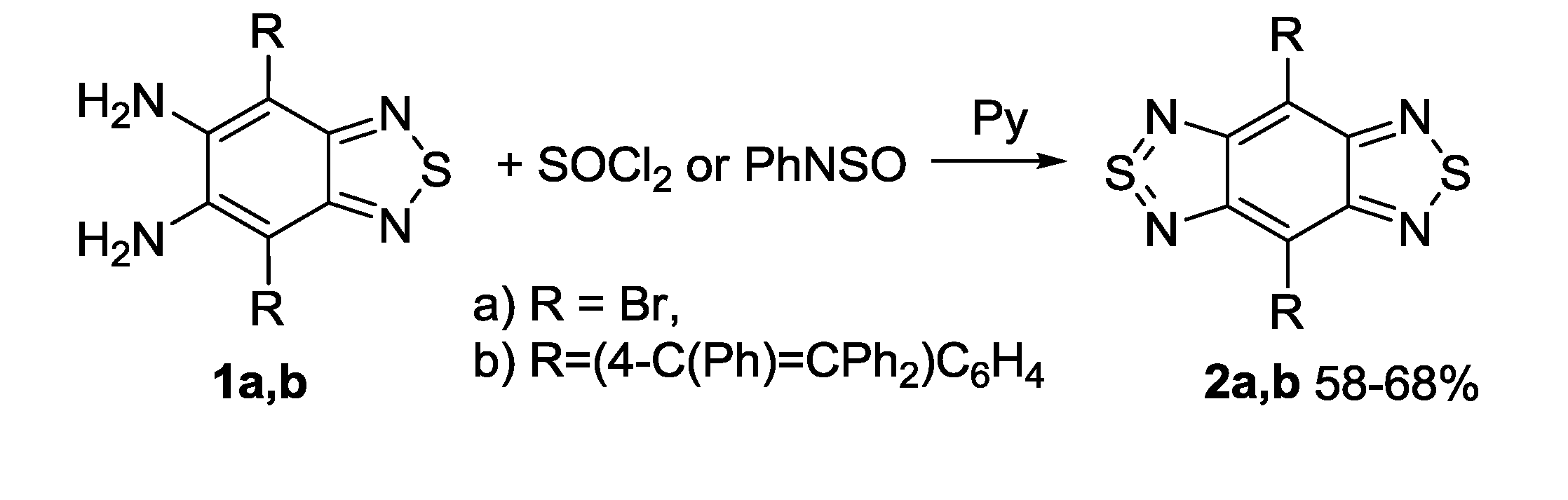{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
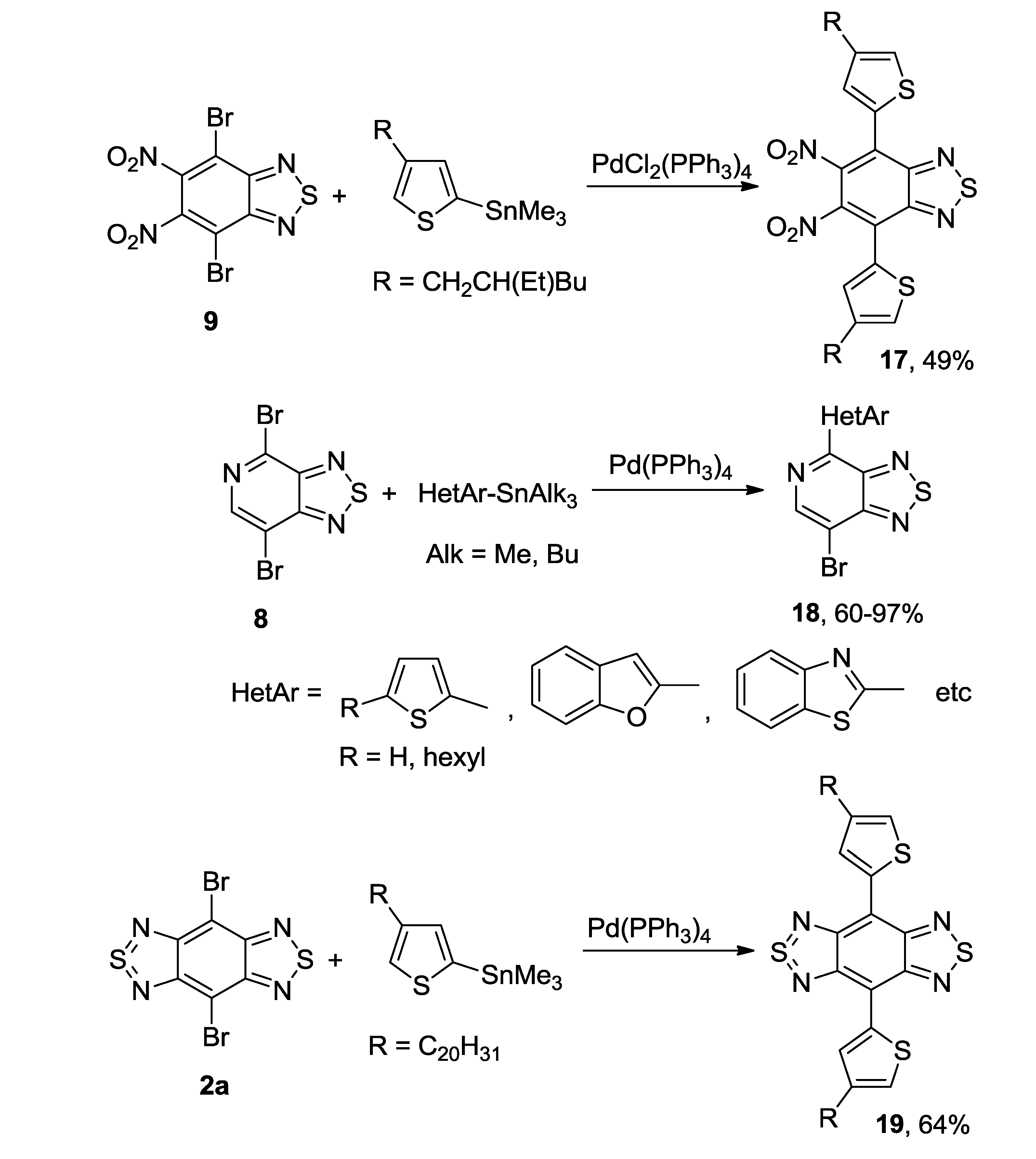{kind=link}
{kind=link}
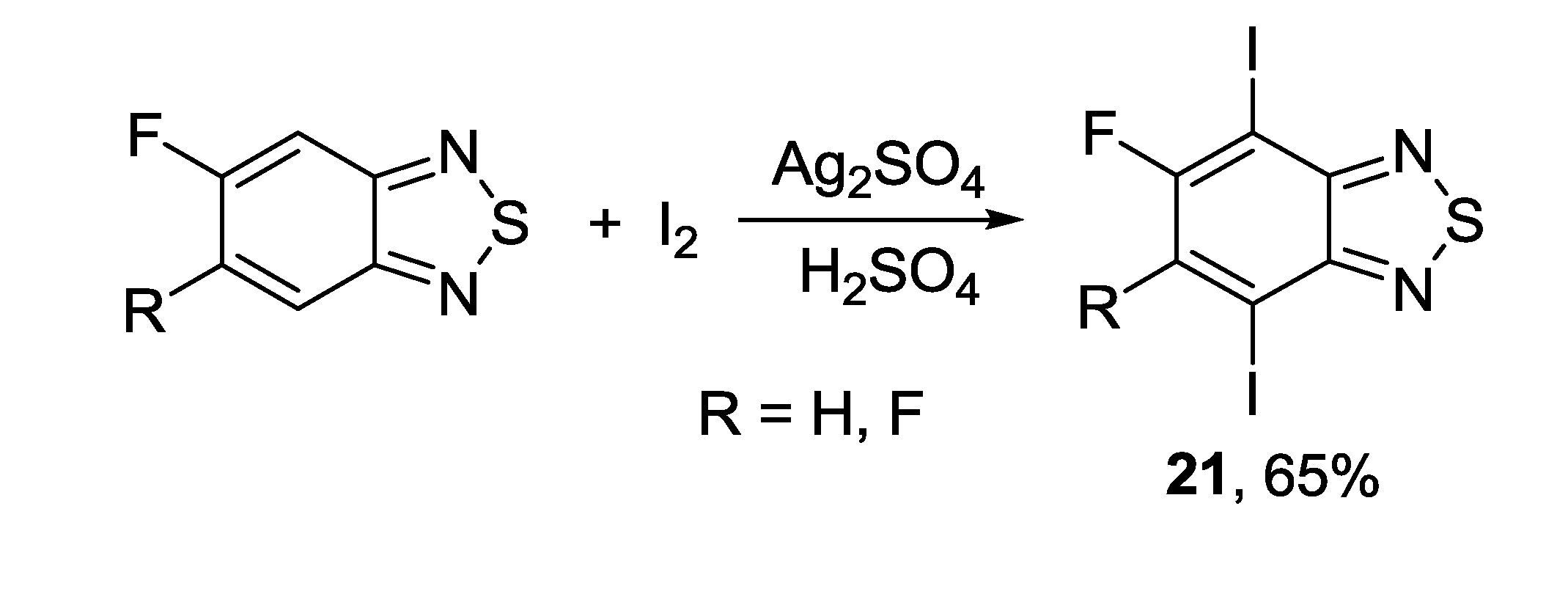{kind=link}
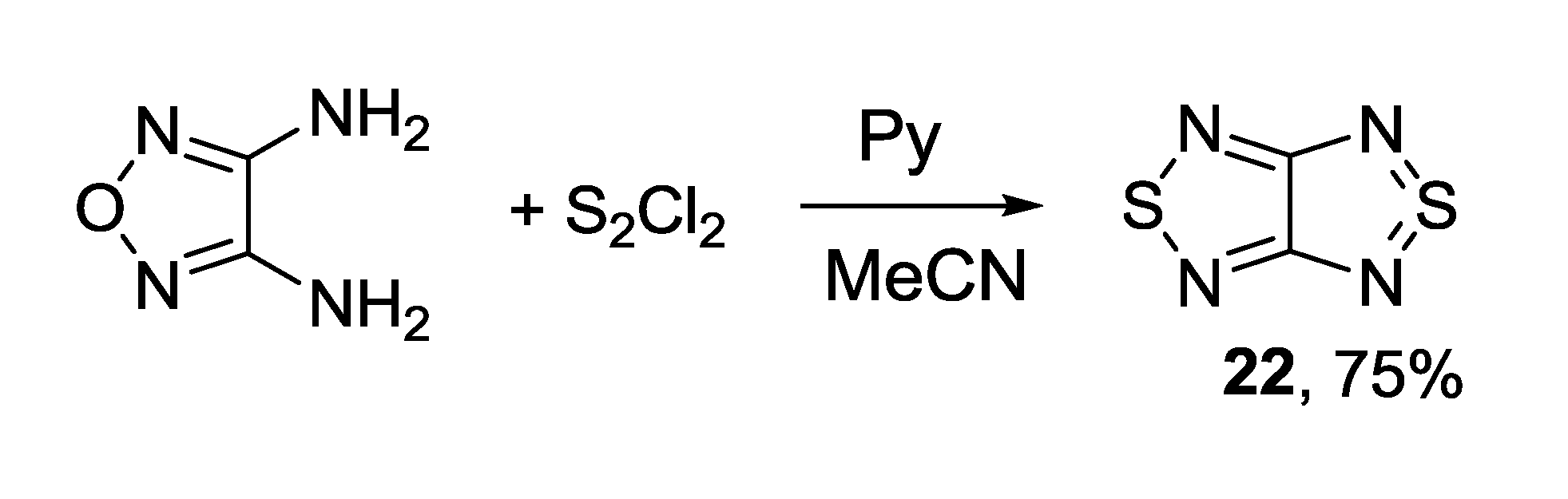{kind=link}
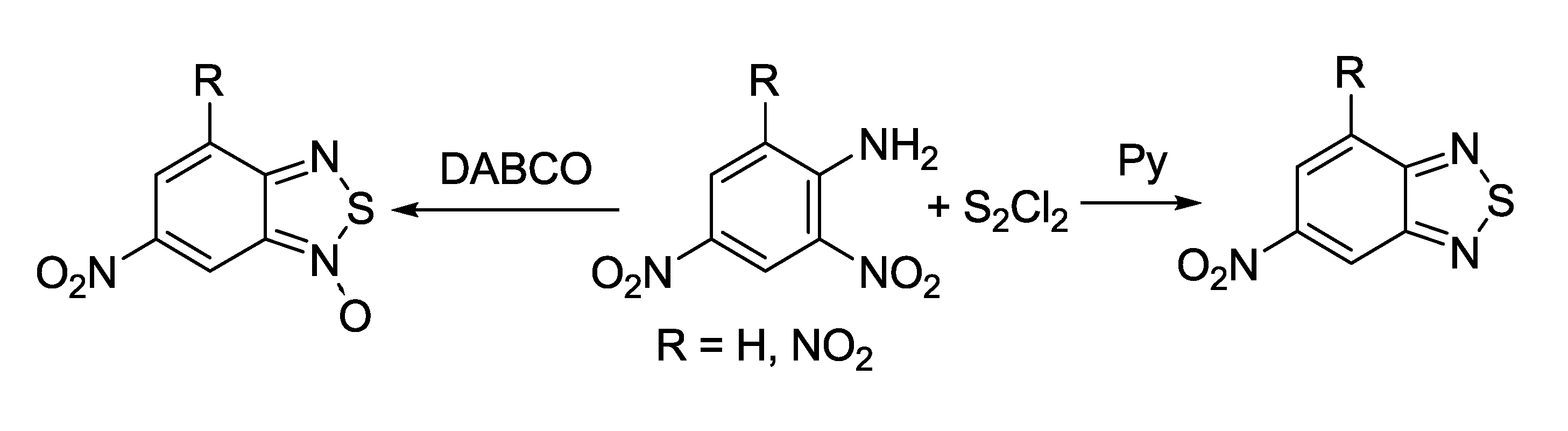{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
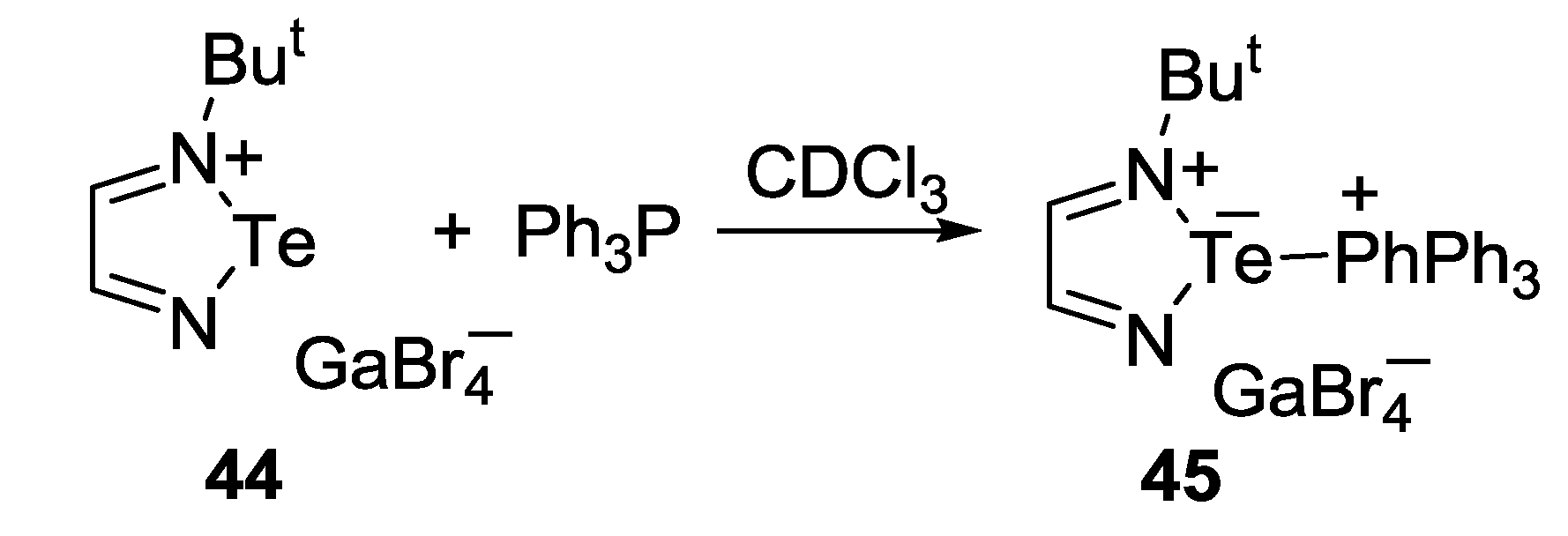{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
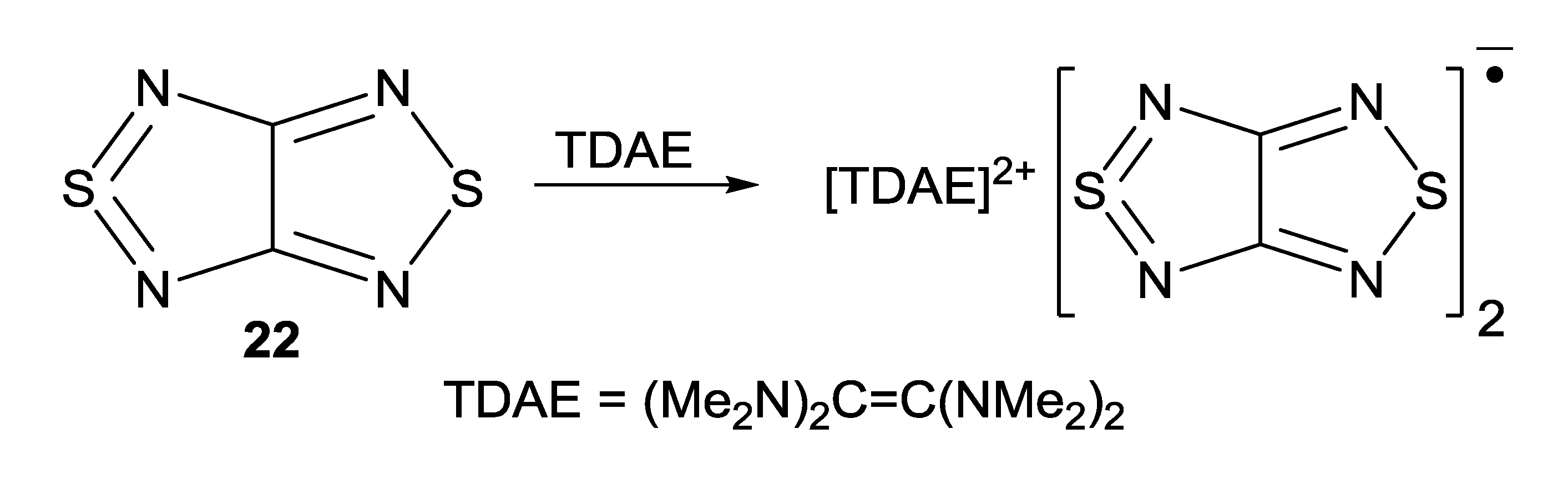{kind=link}
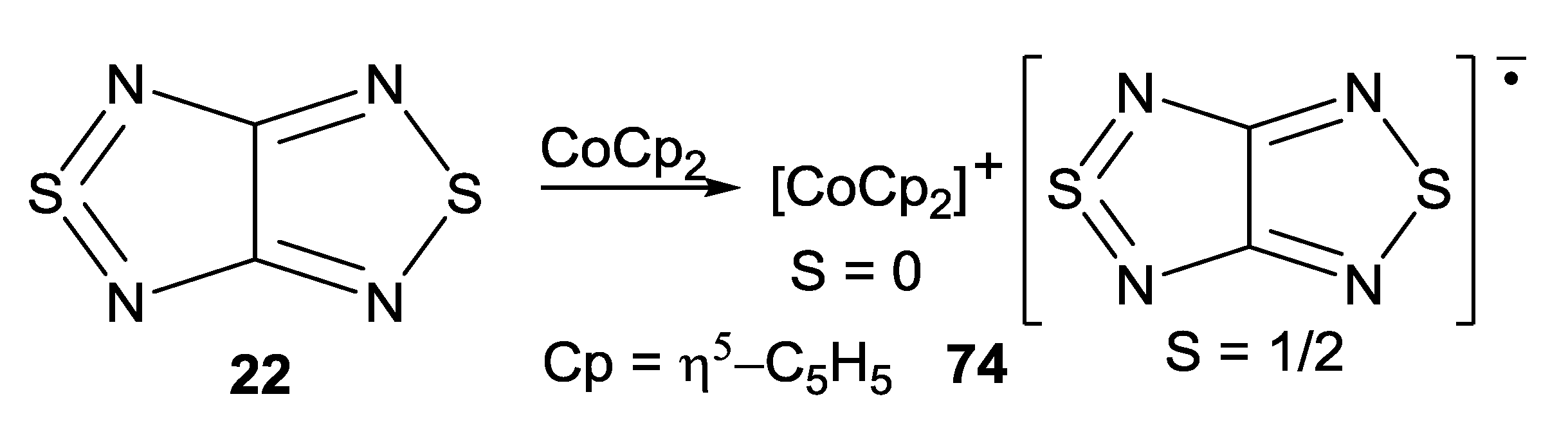{kind=link}
{kind=link}
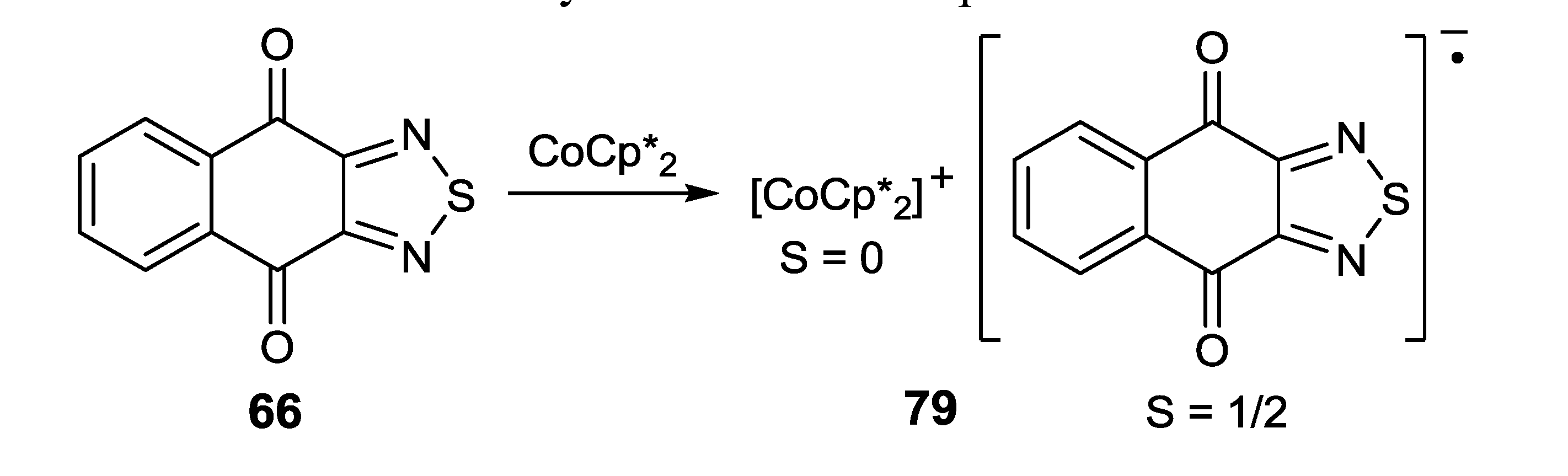{kind=link}
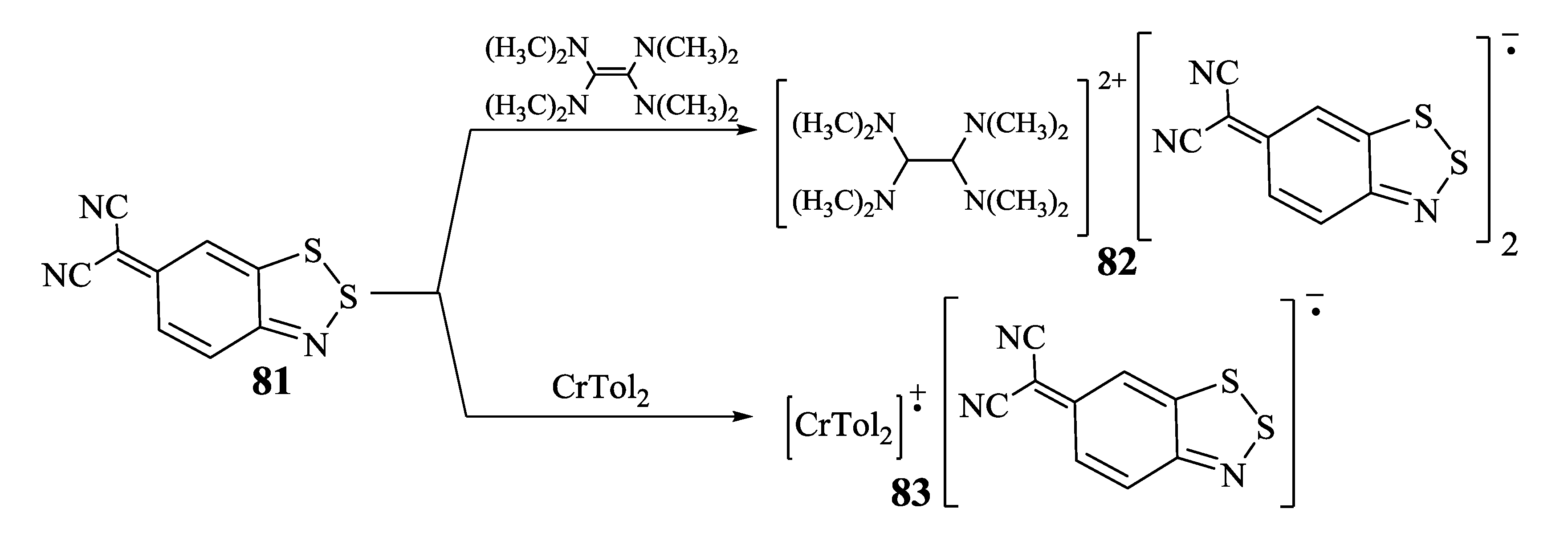{kind=link}
| Method/Compound |  |  |  |  |  |
|---|---|---|---|---|---|
| MP2/6-311+G(3df,2p) a | 0.94 | 1.13 | 1.25 b | 1.16 | 2.38 |
| PMP2/6-311+G(3df,2p) a | 1.33 | 1.54 | 1.67 b | 1.48 | 2.93 |
| (U)B3LYP/6-31+G(d) | 1.91/1.84 | 2.01/1.94 | 2.16/2.10 c | 2.19/2.14 | 3.51/3.48 |
| (U)B3LYP/6-311+G(3df,2p) a | 1.86 | 2.04 | 2.05 b | 2.07 | 3.50 |
| G3B3 | 1.74 | ( d) | ( d) | 1.96 | 3.17 e |
 |  |  |  |  |  |
| 0.05 | 1.32 | 1.84 | 0.22 | 1.42 | 1.94 |
 |  |  |  |  |  |
| −0.35 | 1.47 | 2.20 | −0.15 | 1.55 | 2.25 |
 |  |  |  |  |  |
| 0.08 | 1.63 | 2.23 | ( b) | 1.74 | 2.31 |
 |  |  |  |  |  |
| −0.13 | 1.44 | 2.05 | 0.01 | 1.54 | 2.11 |
 |  |  | |||
| 0.75 | 1.58 | 2.10 |
 |  |  |  | |
| 0.95 | 1.06 | 1.15 | 2.11 | |
 |  |  |  | |
| 1.57 | 1.67 | 1.83 | 2.86 | |
 |  |  |  | |
| 1.91 | 1.24 | 1.42 | 1.50 | |
 |  |  | ||
| 1.67 | 0.95 | 1.29 | ||
 |  |  |  |  |
| 0.95 | 1.75 | 1.75 | 1.79 | 1.88 |
 |  |  |  |  |
| 0.58 | 1.77 | 2.45 | 2.99 | 2.21 |
 |  |  |  |  |
| 2.14 | 2.19 | 2.44 | 1.24 | 2.26 |
 |  |  |  | |
| 3.04 | 3.39 | 1.24 | 2.32 |
 |  |  |  |
| 2.69 | 2.55 | 2.85 | 2.66 |
 |  |  |  |
| 2.94 | 2.94 | 2.82 | 3.07 |
 |  |  |  |
| 3.31 | 3.10 | 3.46 | 2.69 |
 |  |  | |
| 2.81 | 2.88 | 2.81 |
2.2. Redox Properties
 |  |  |  |  |
| −2.17 | (b) | (b) | −1.07 | −1.03 |
 |  |  | |  |
| (c) | −1.48 | (−1.51)–(−1.53) d | (−1.30)–(−1.38) d | (b) |
 |  |  |  |  |
| −1.64 | 0.22 | 0.12 | 0.04 | −0.02 |
 |  |  |  |  |
| −0.12 | −0.23 | −1.73 | −1.72 | −1.57 |
 |  |  |  | |
| −1.21 | −1.08 | (b) | −1.29 | |
 |  |  |  |  |
| −0.59 | −0.53 | 0.10 | (b) | −1.45 |
 |  |  |  |  |
| −0.96 | −1.71 | −0.46 | −1.64 | −0.87 |
 |  |  |  |  |
| −1.16 | −0.46 | −0.70, e −0.80 f | −0.80 e | −0.58 e |
 |  |  | TCNQ | |
| −0.67e | −0.43 | 0.15 | 0.18 |
3. Synthesis
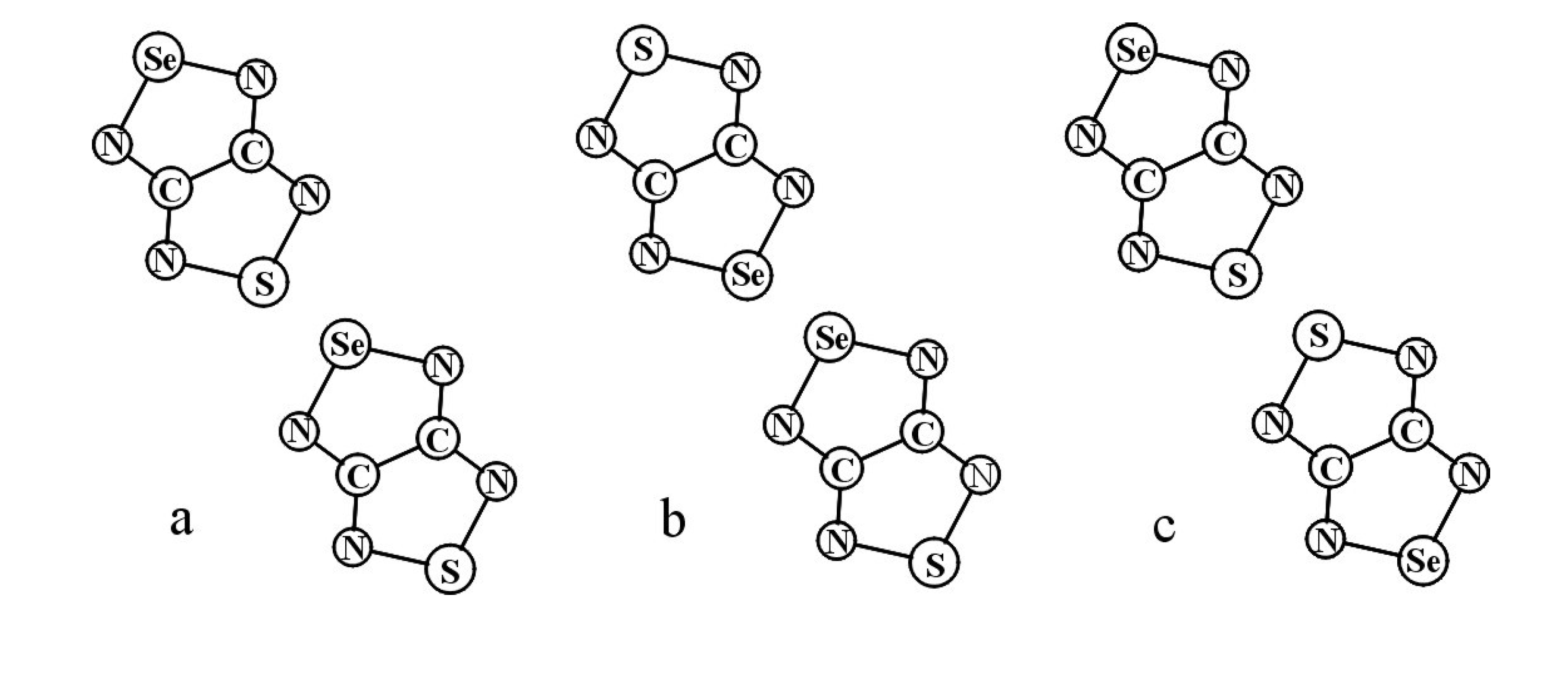
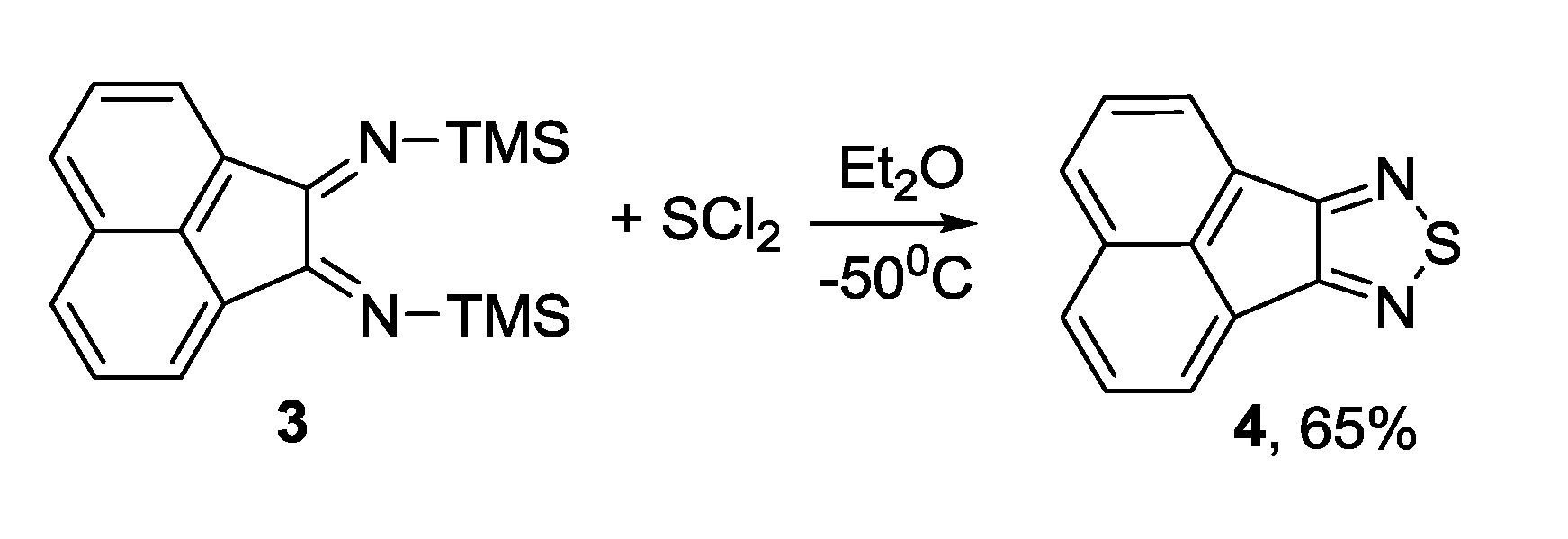
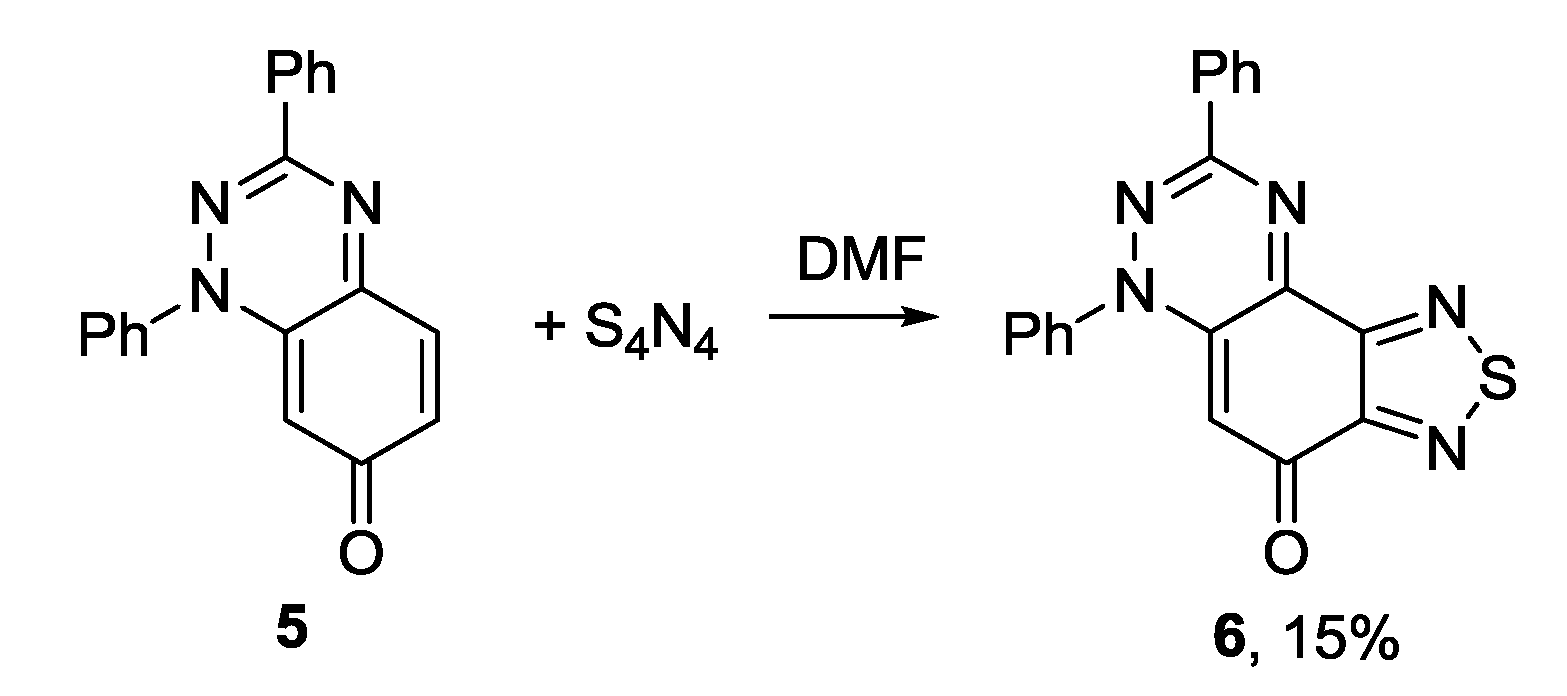
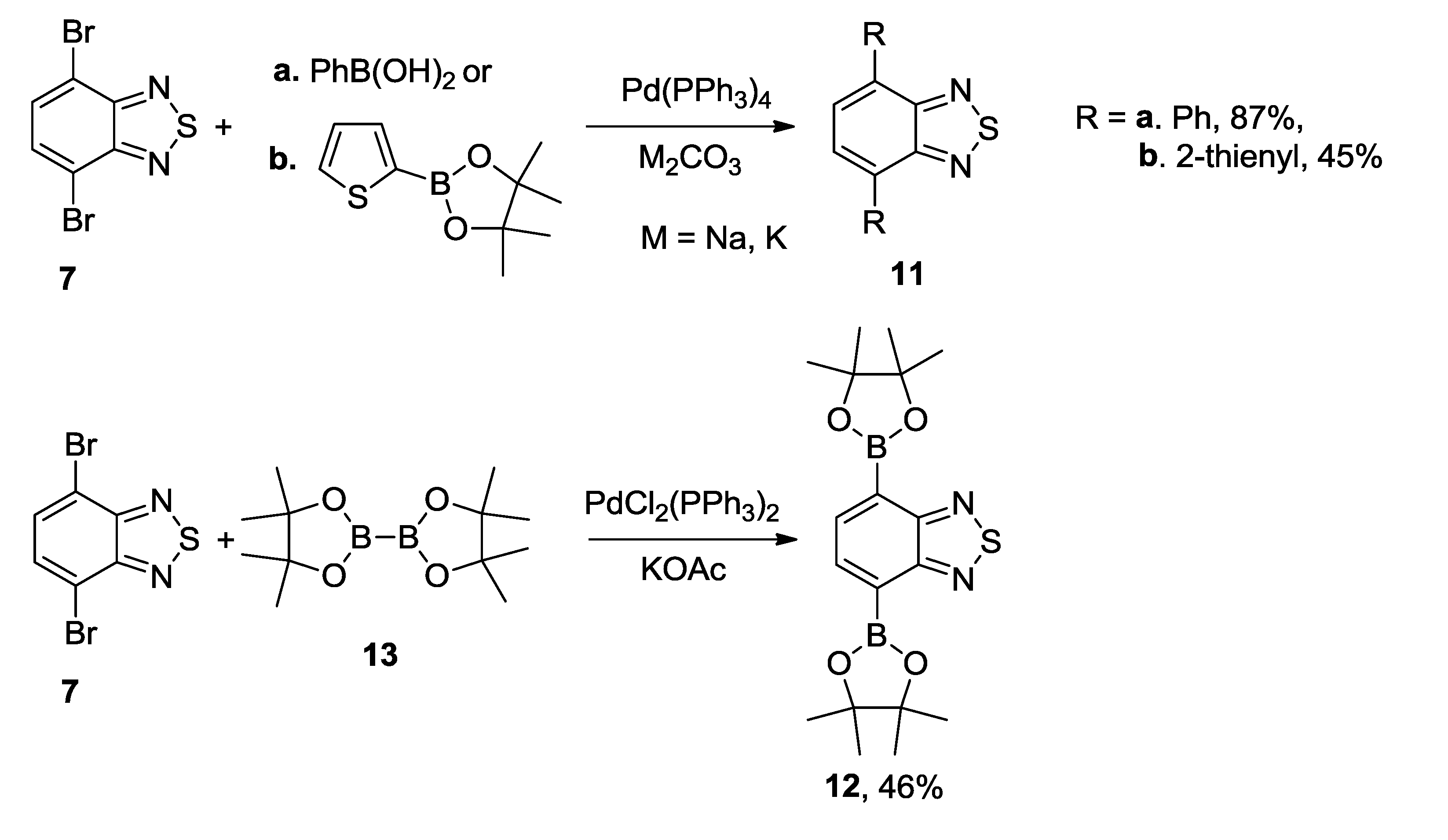
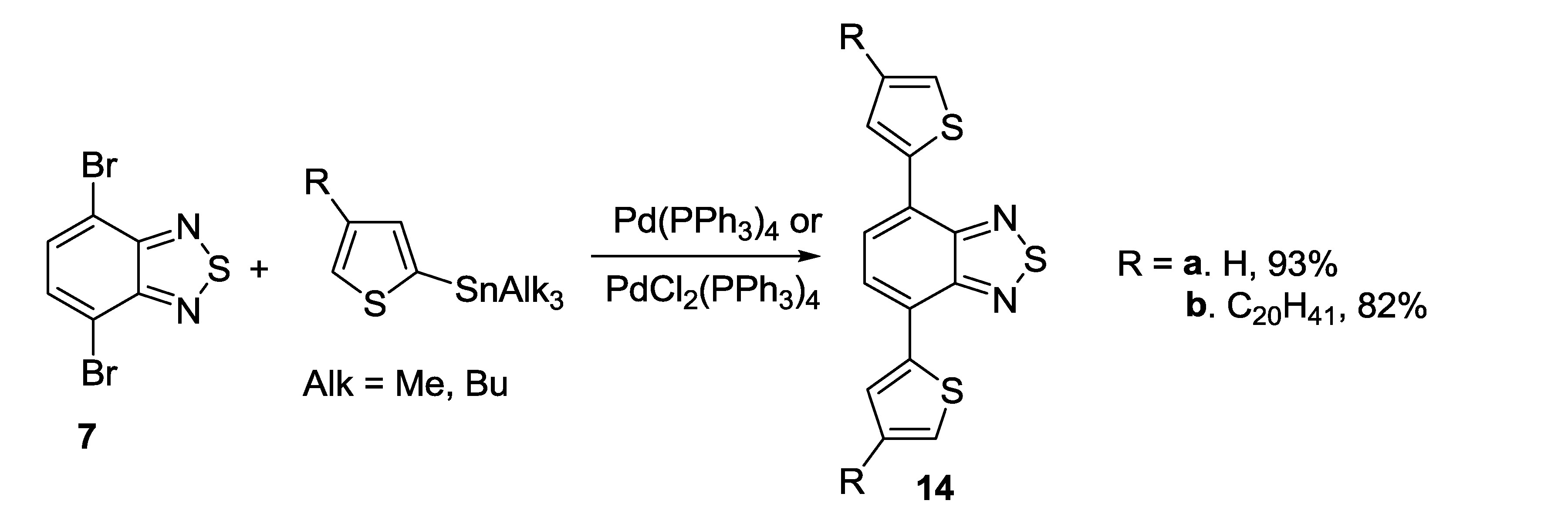
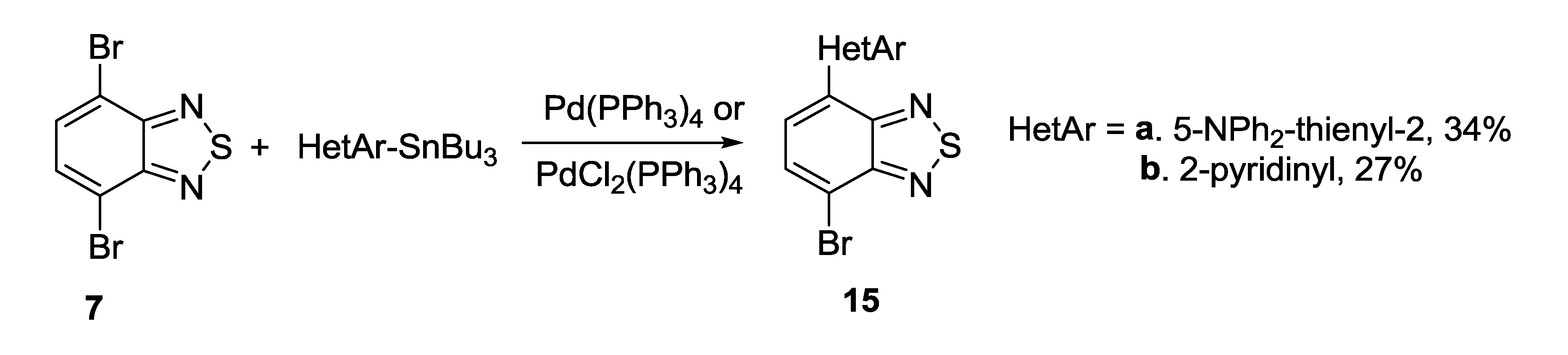
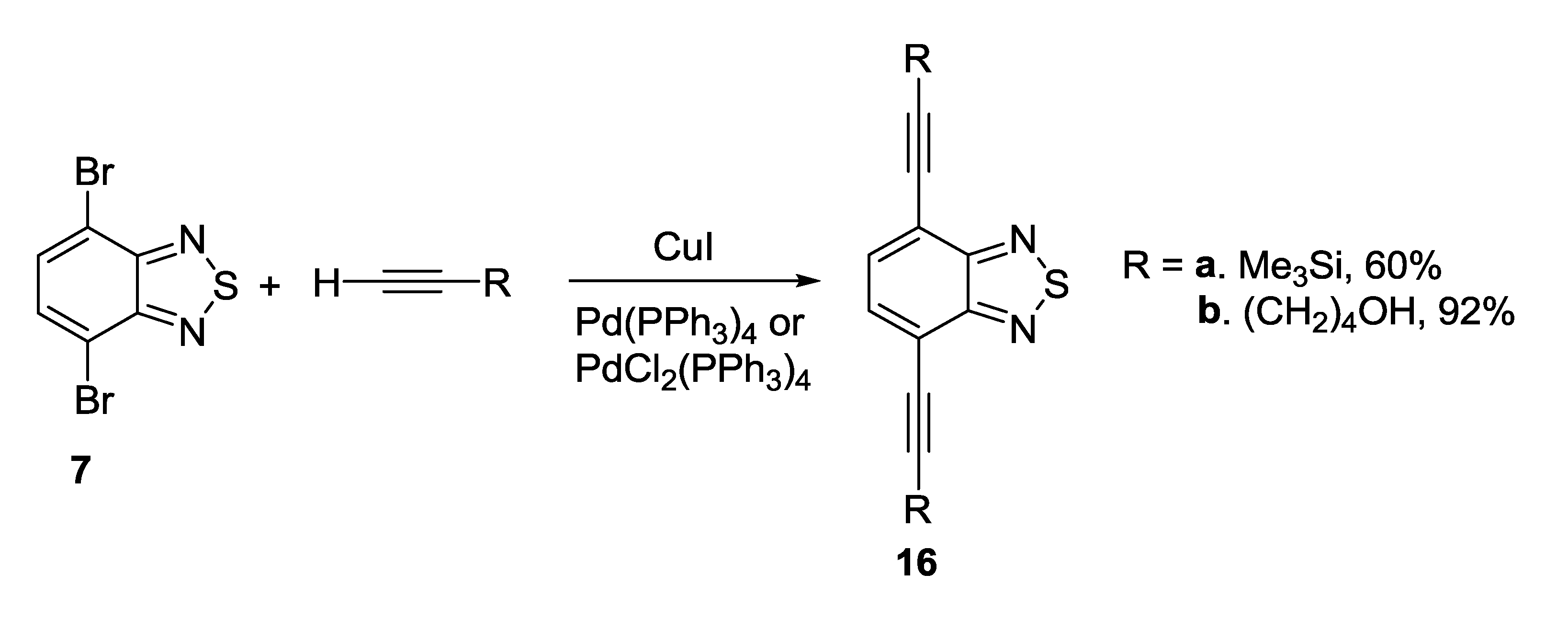
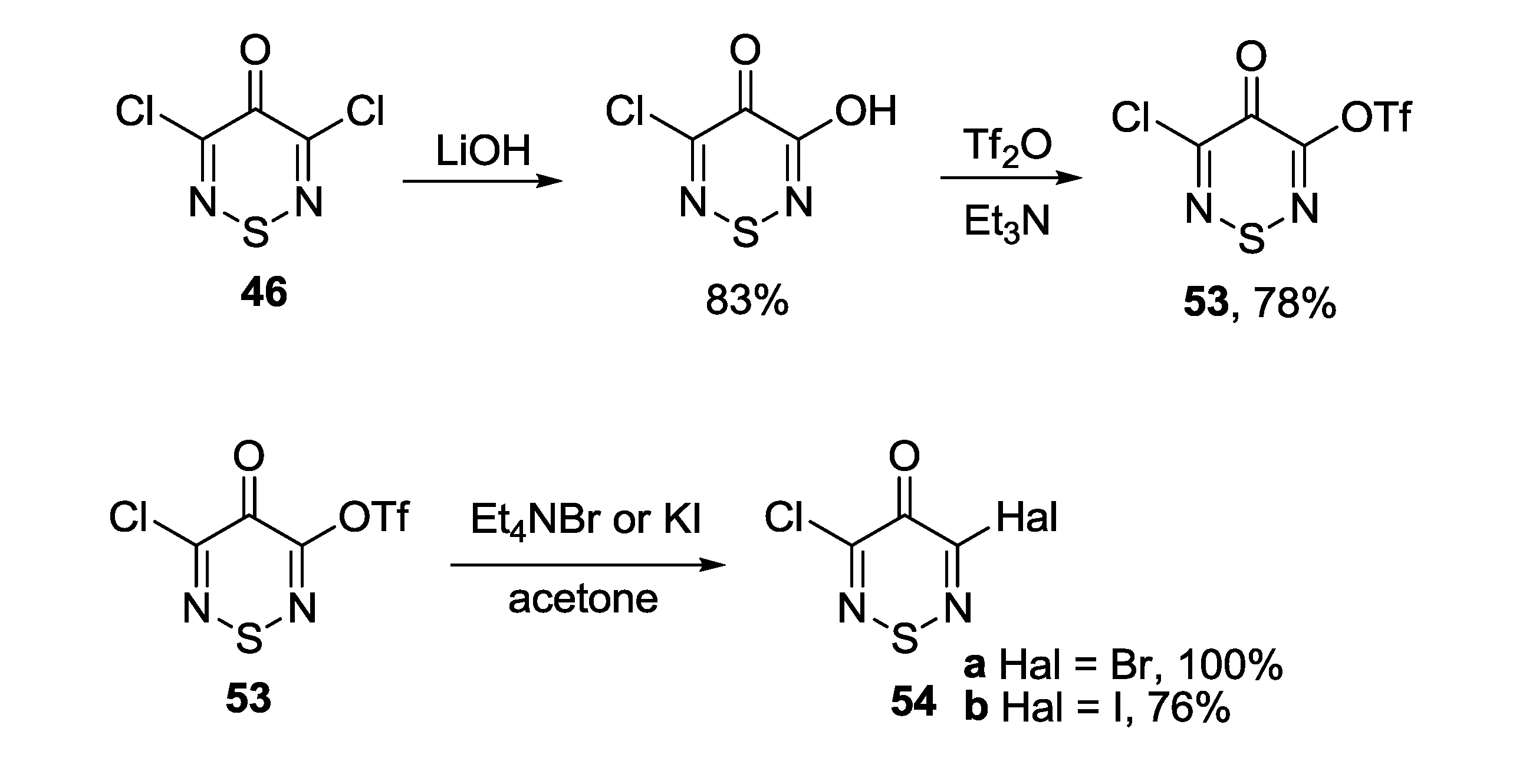
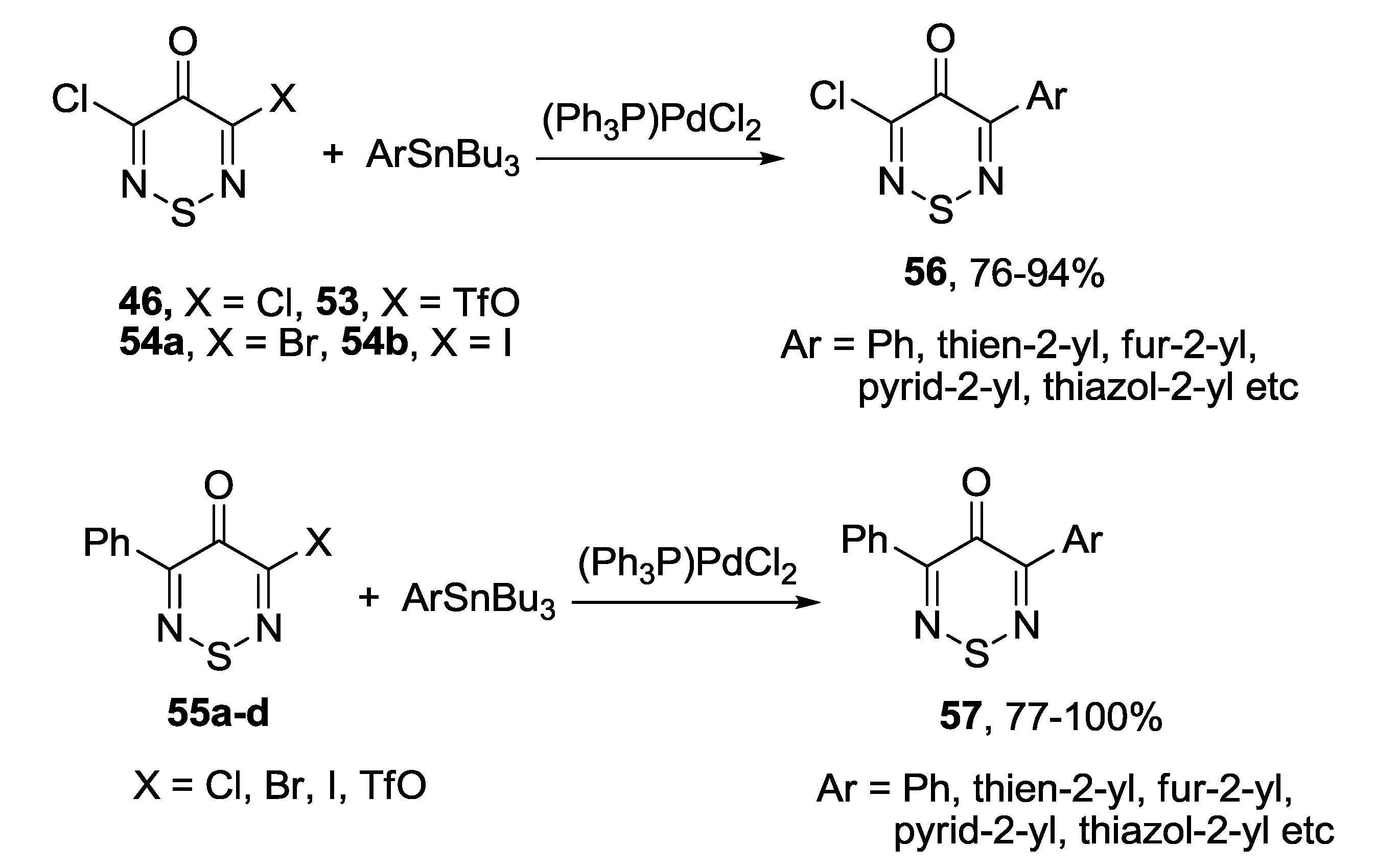
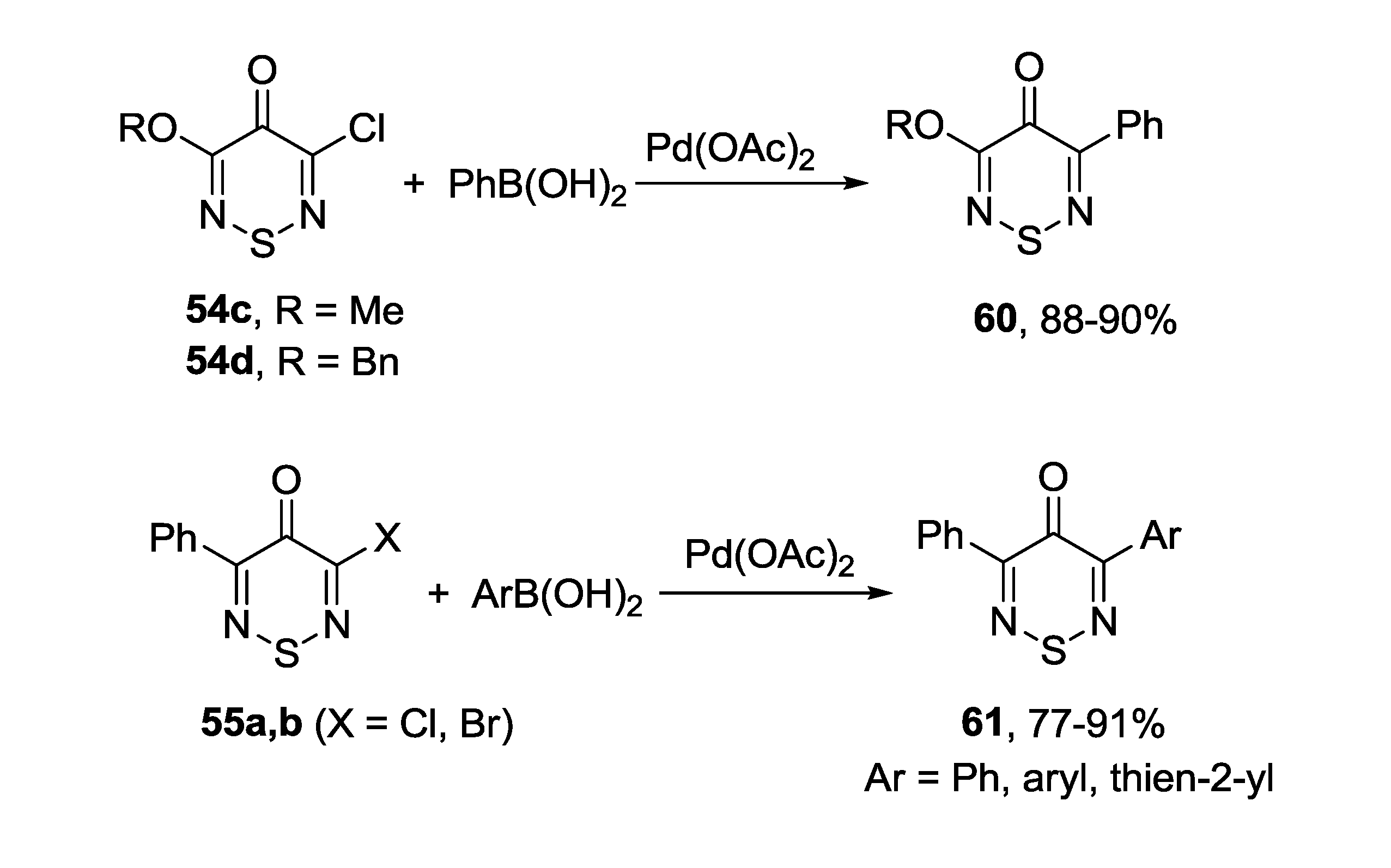
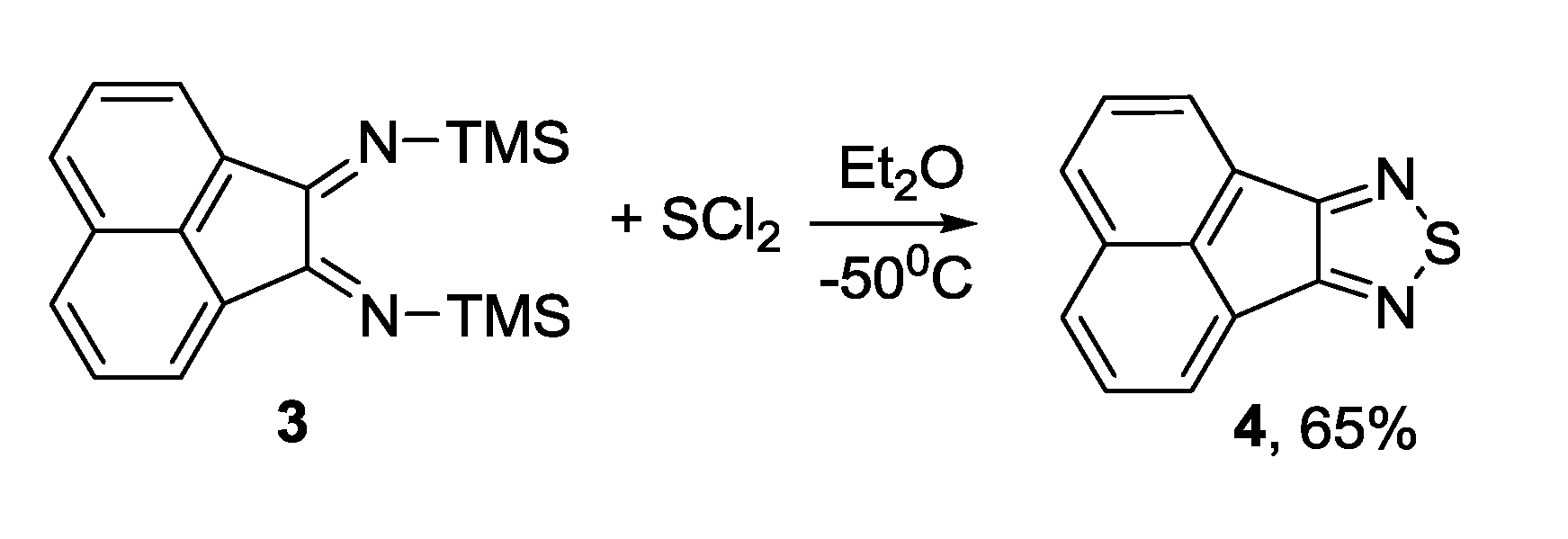
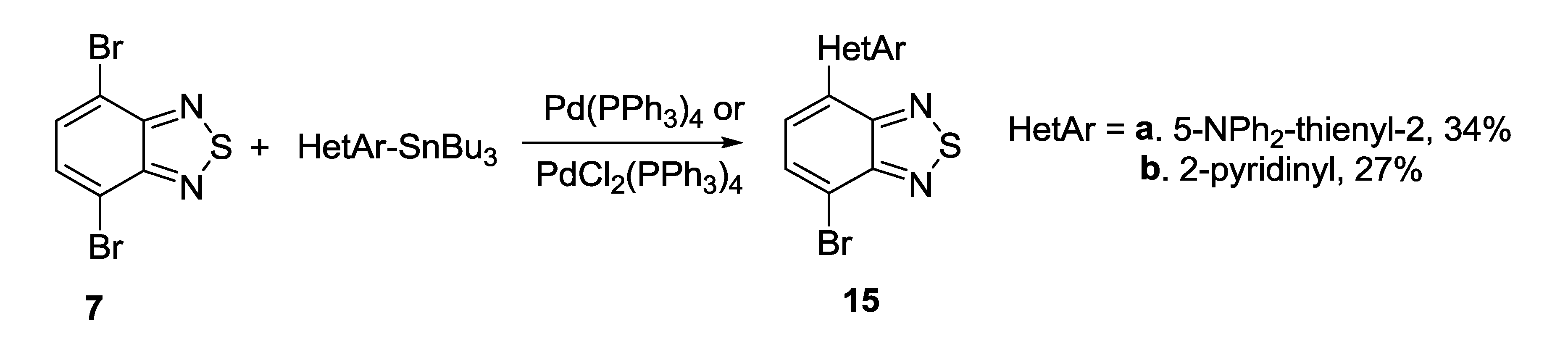
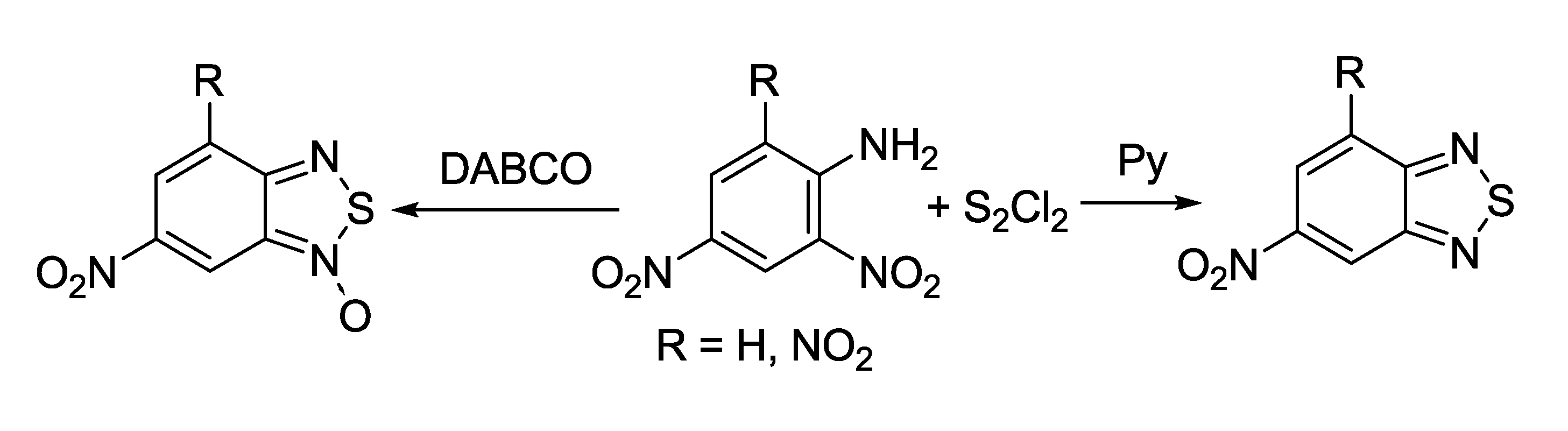
3.1. 1,2,5-Thiadiazoles
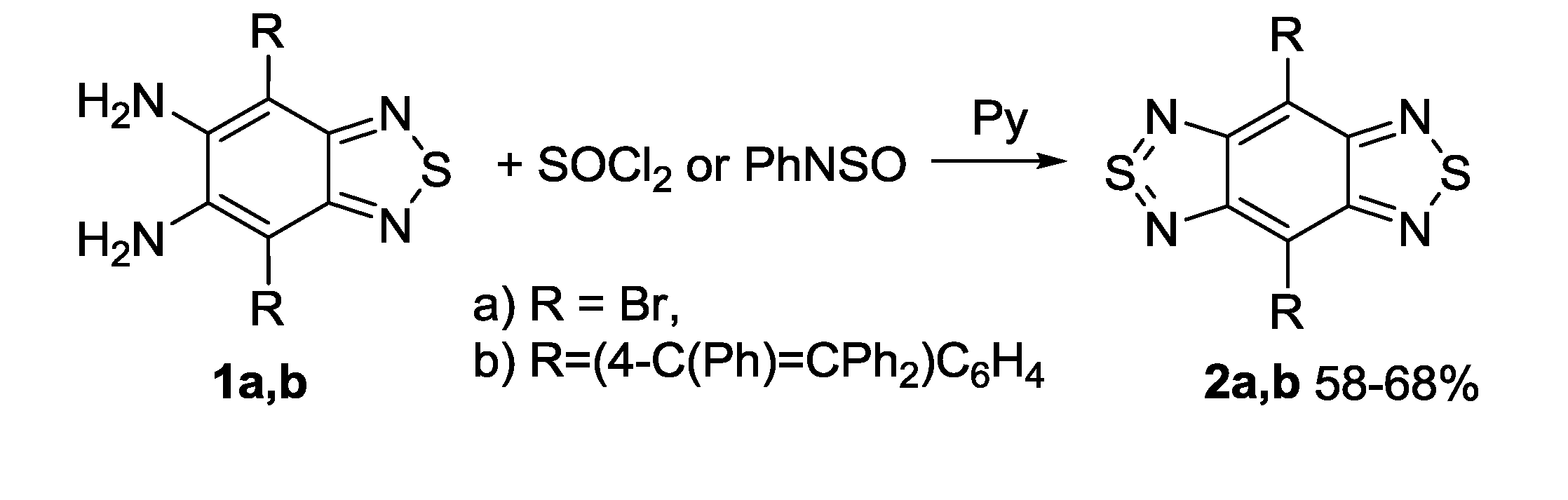




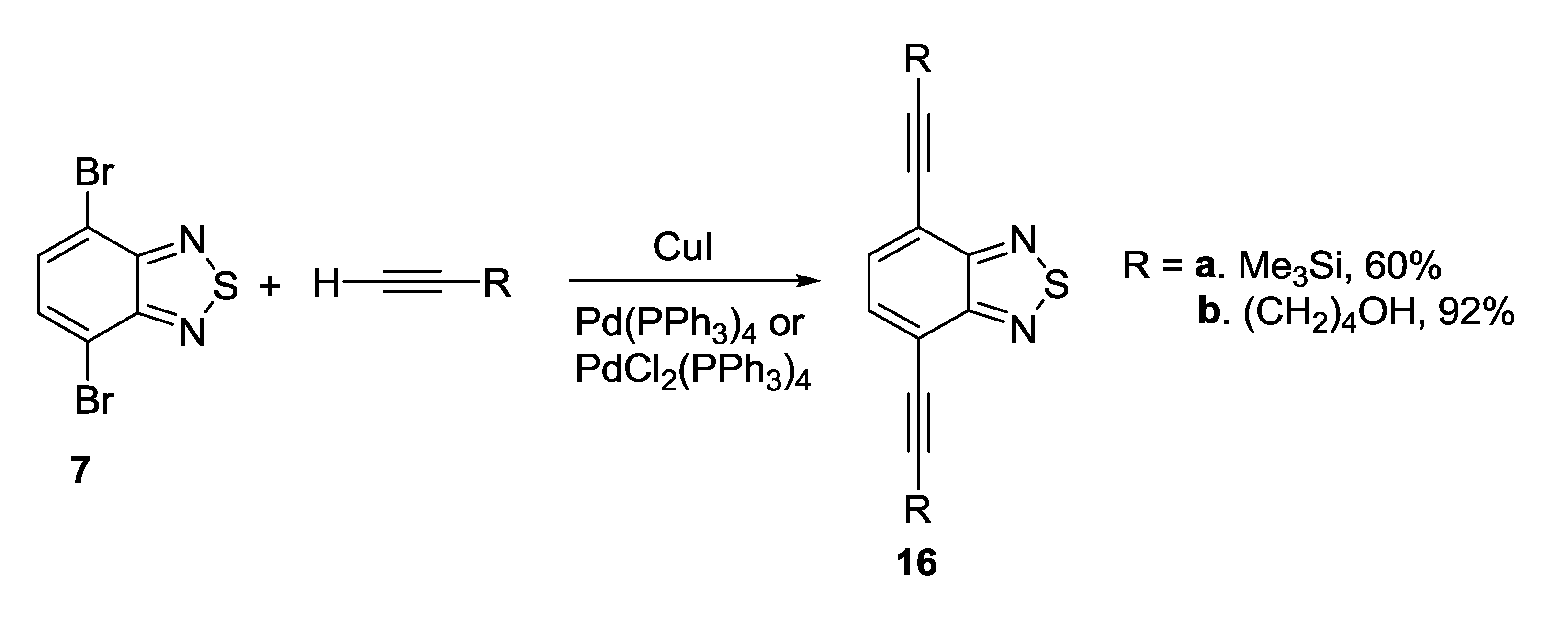

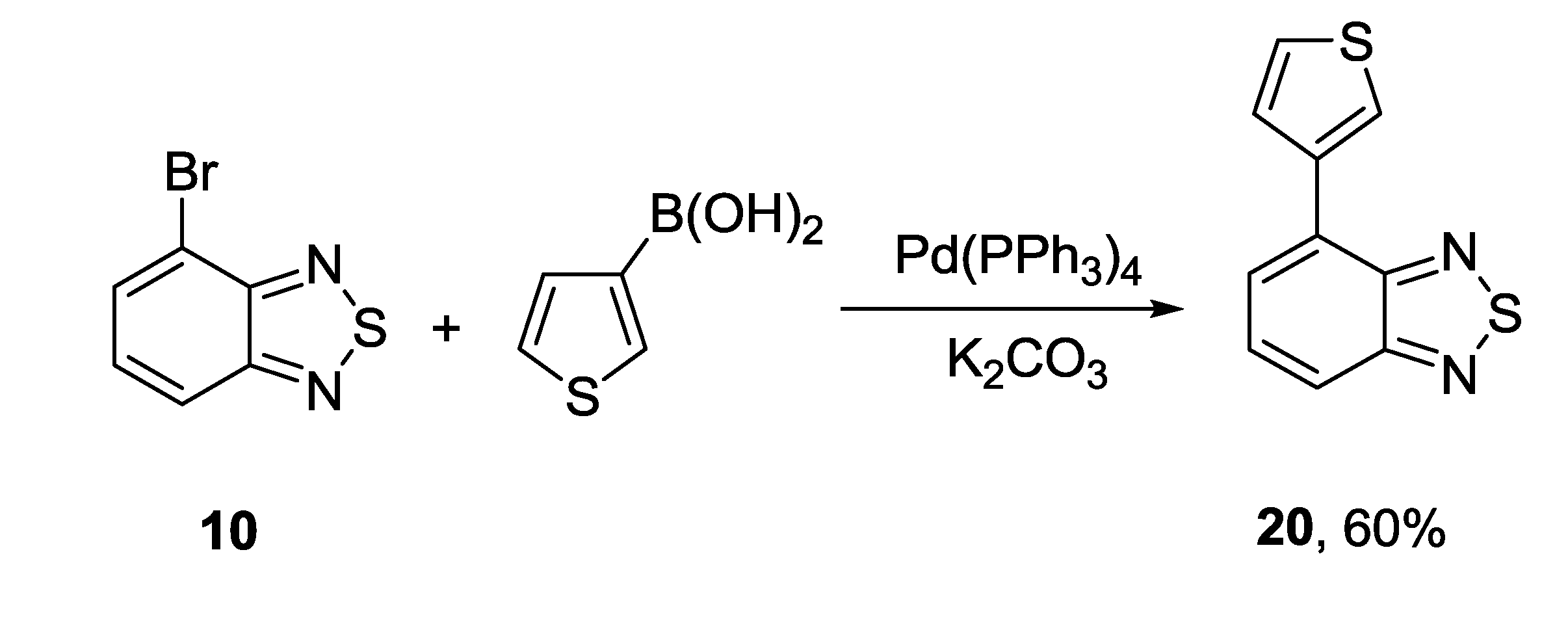
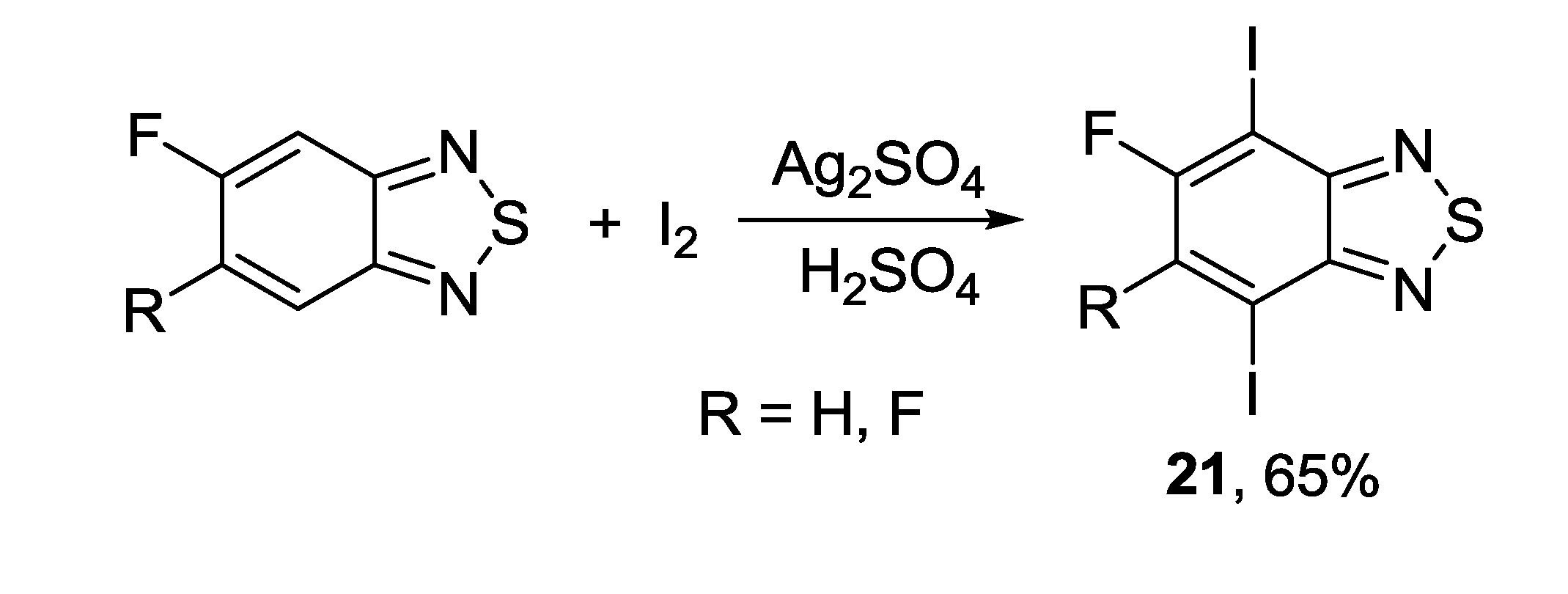
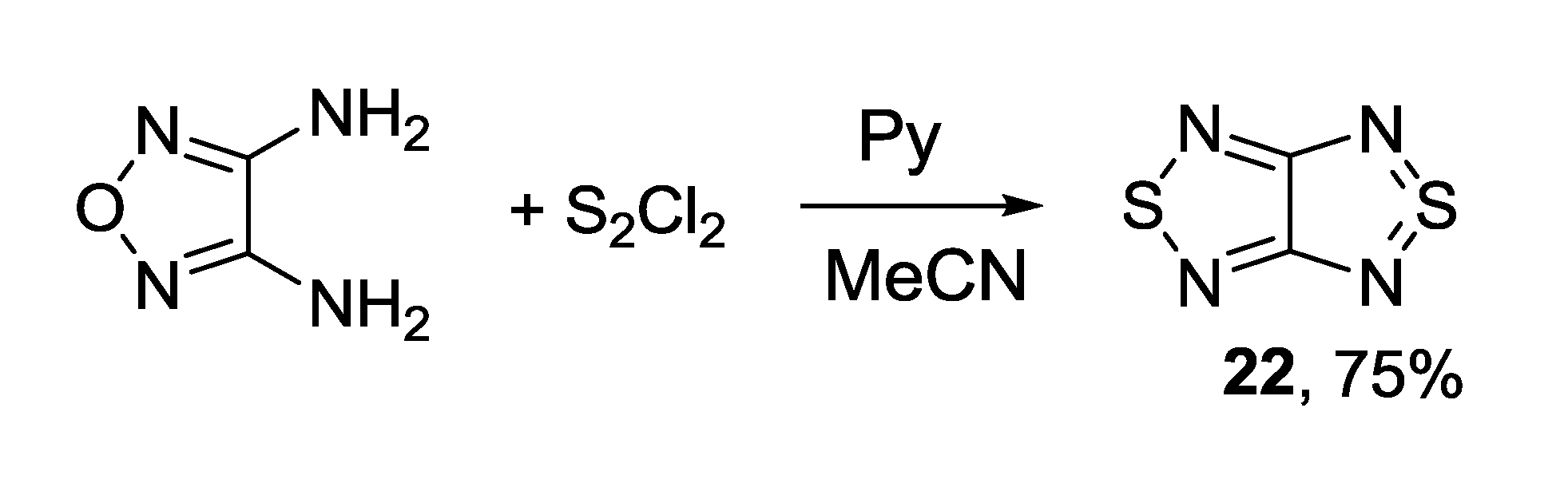
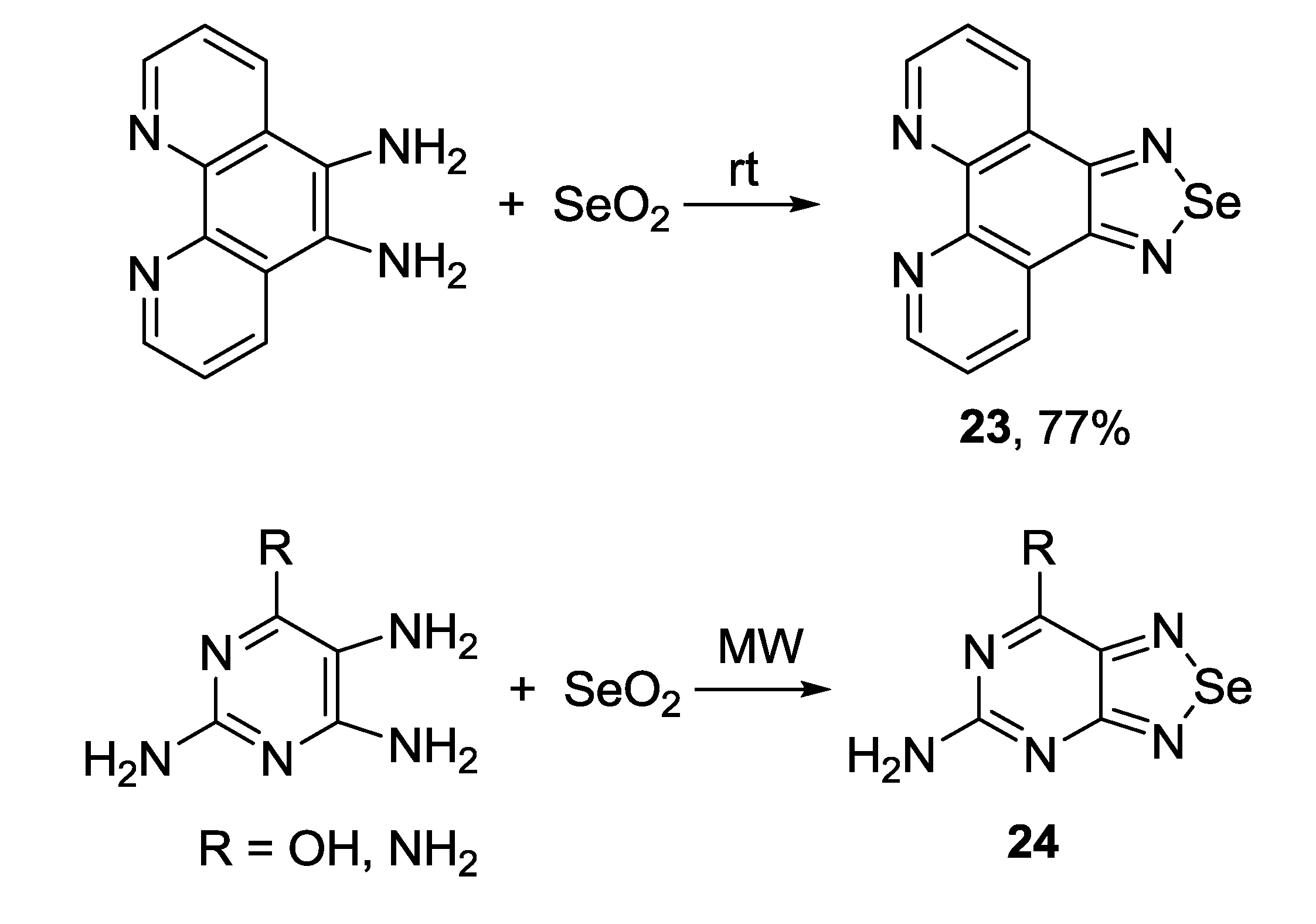
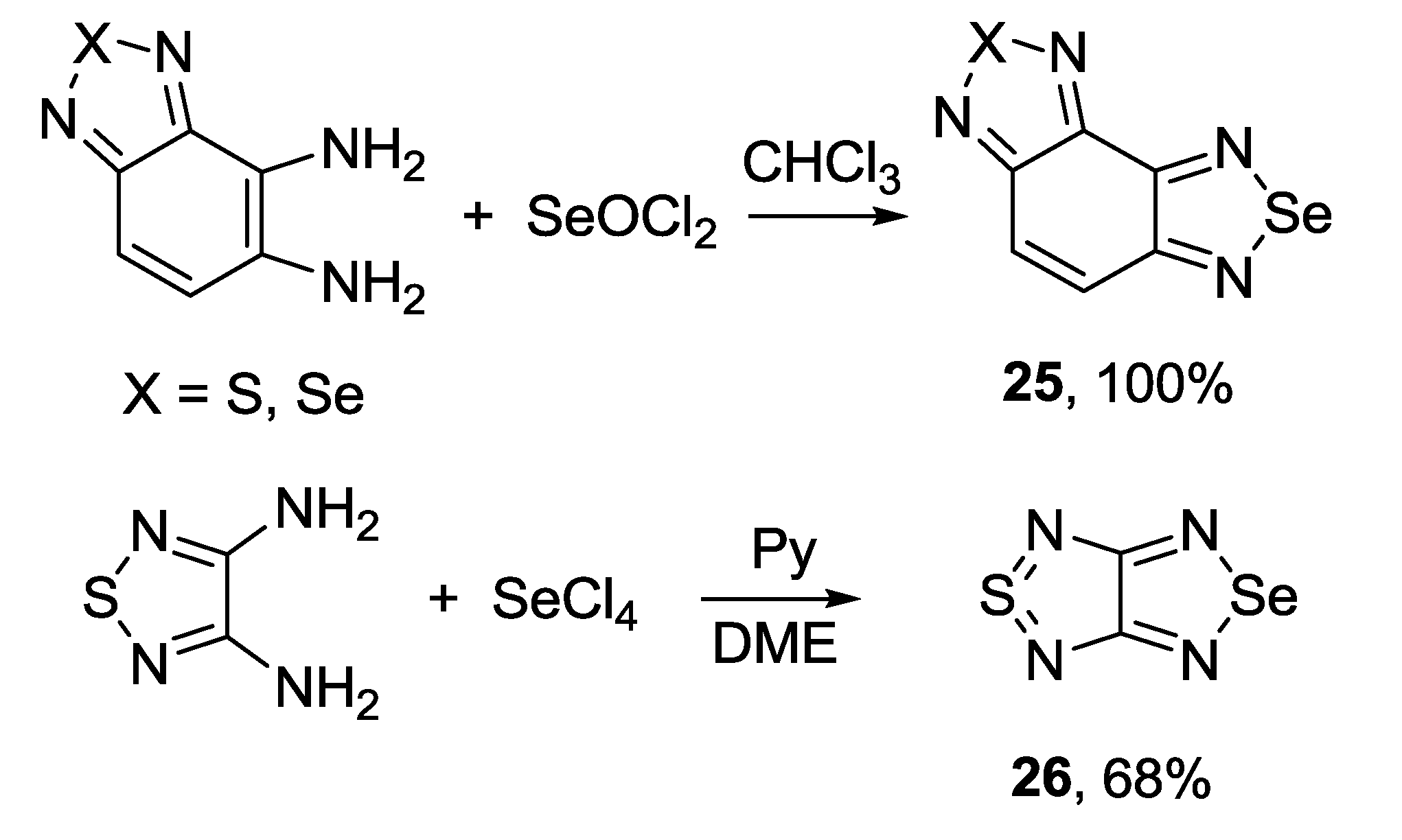
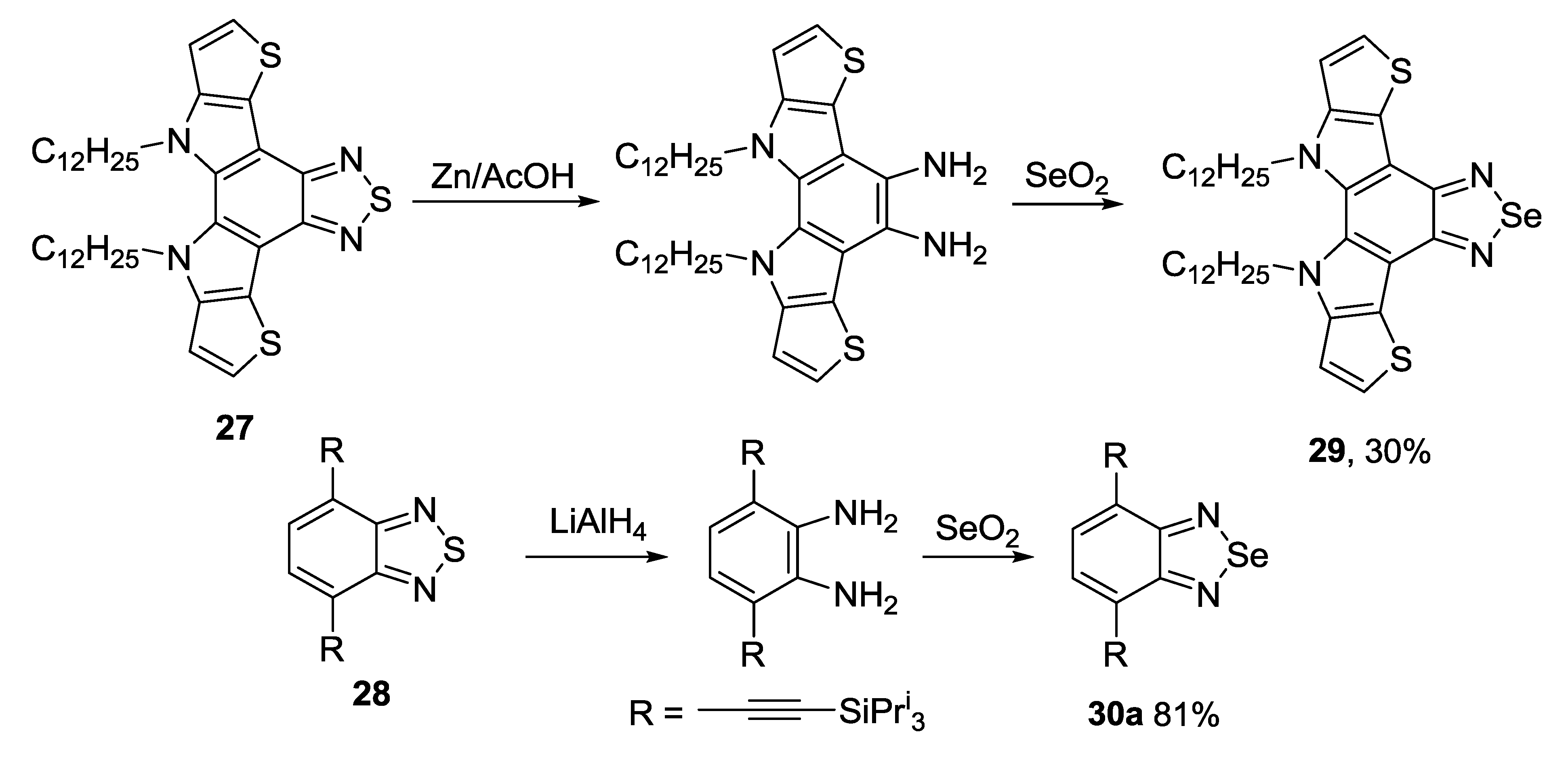
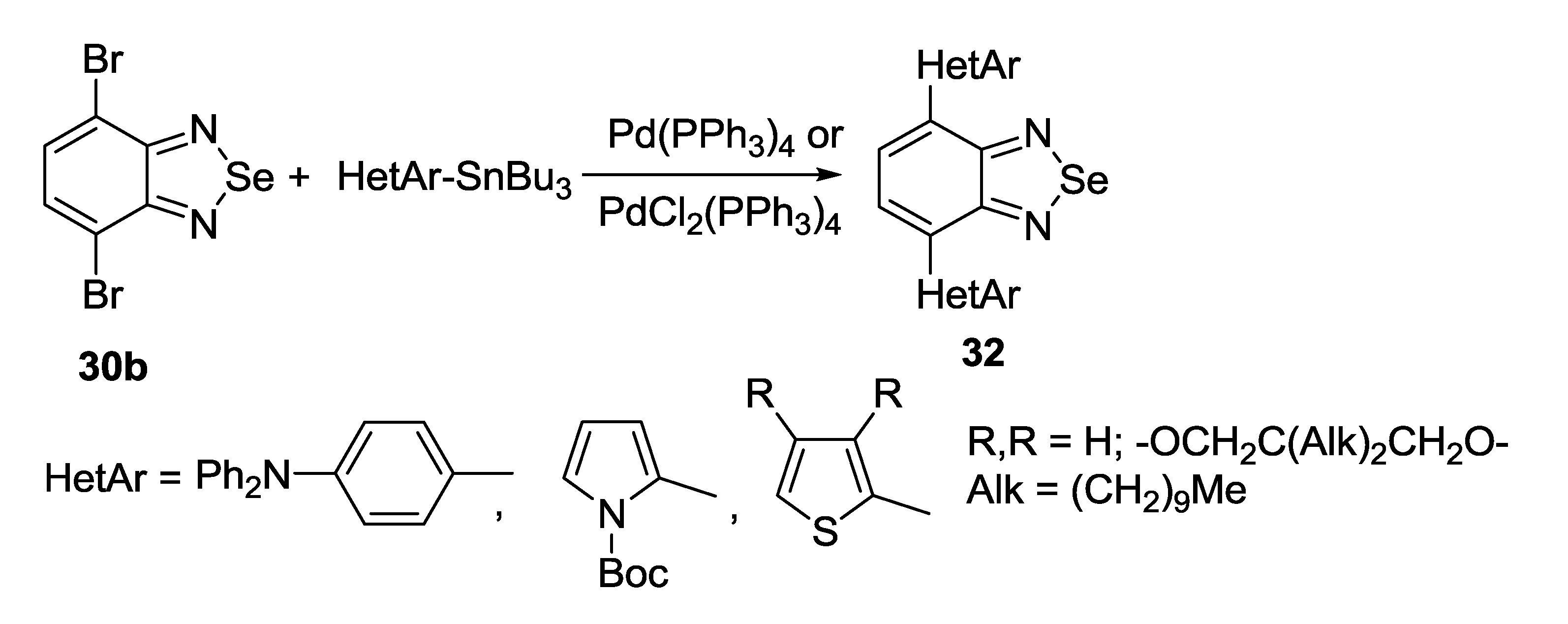
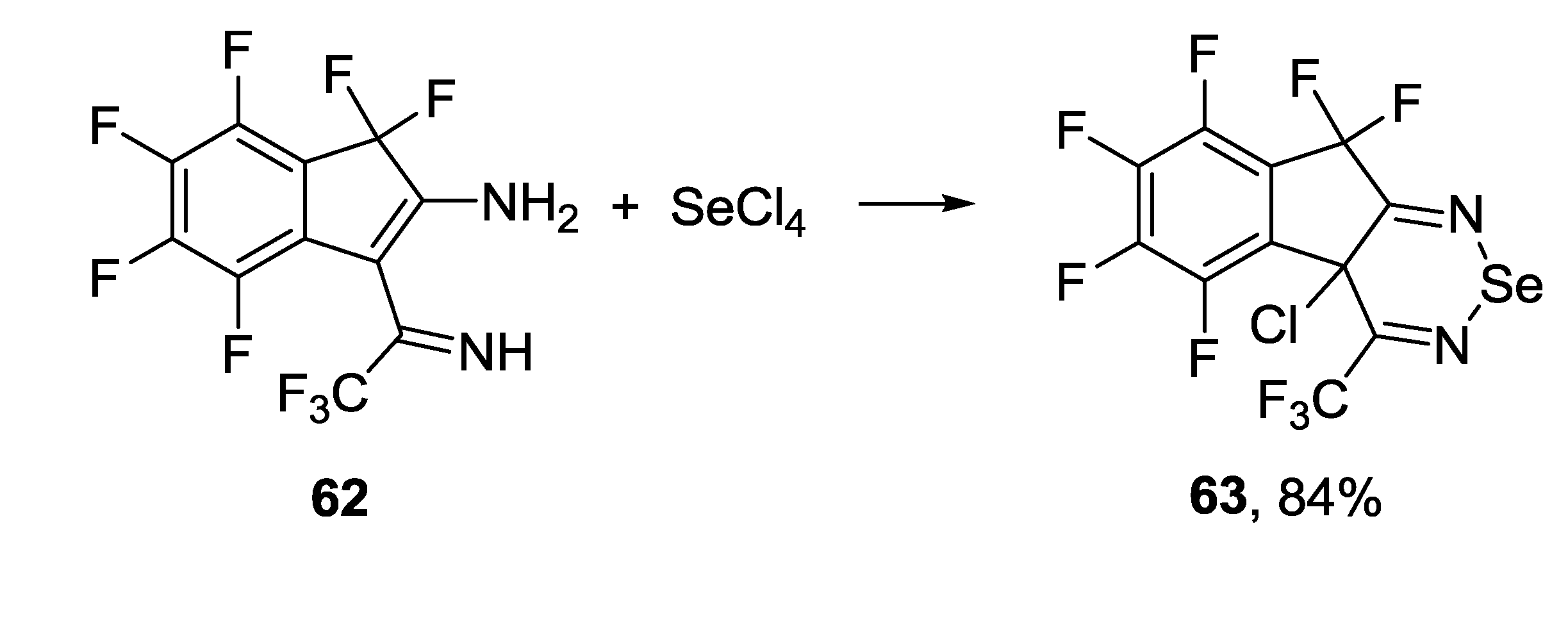
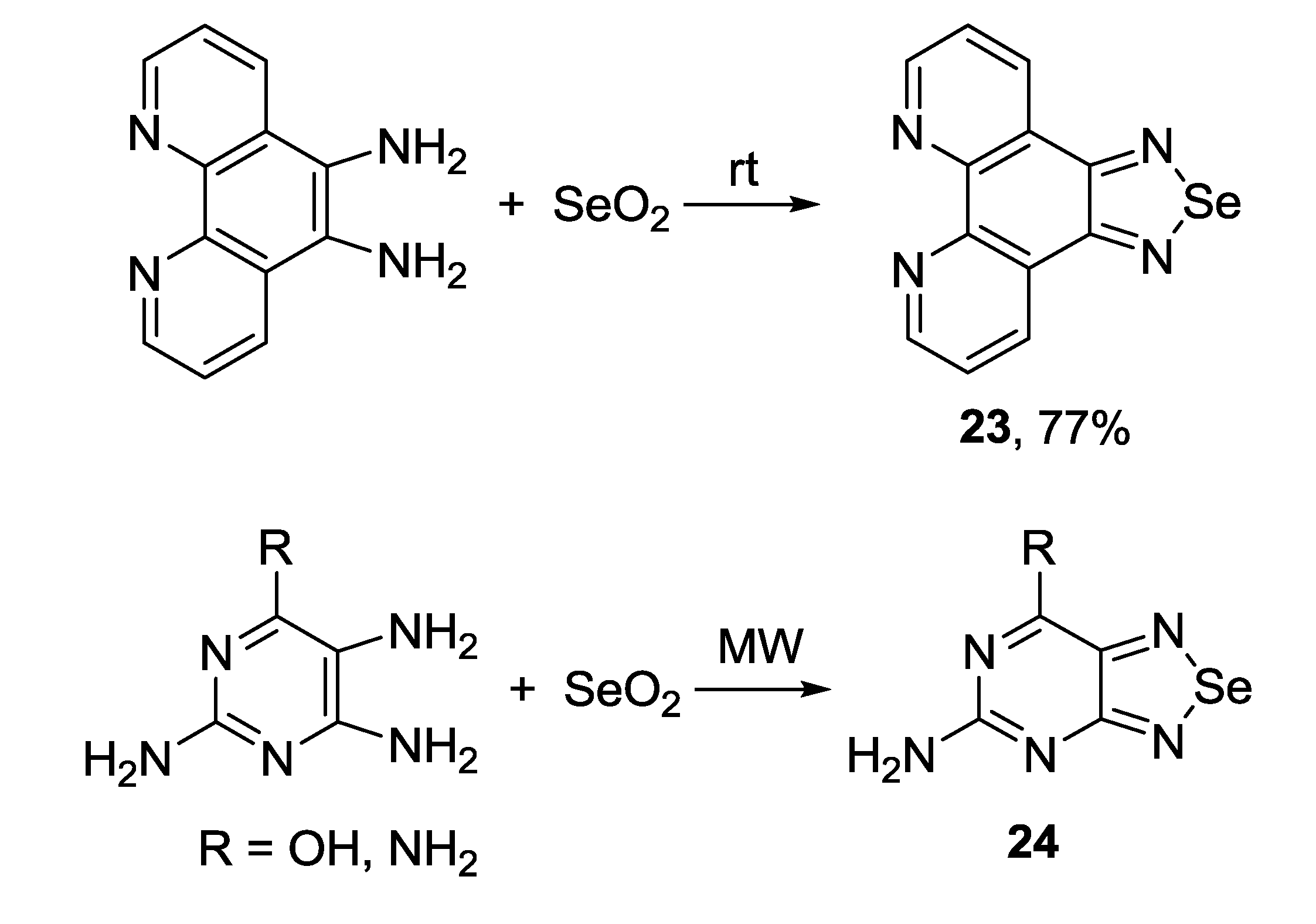
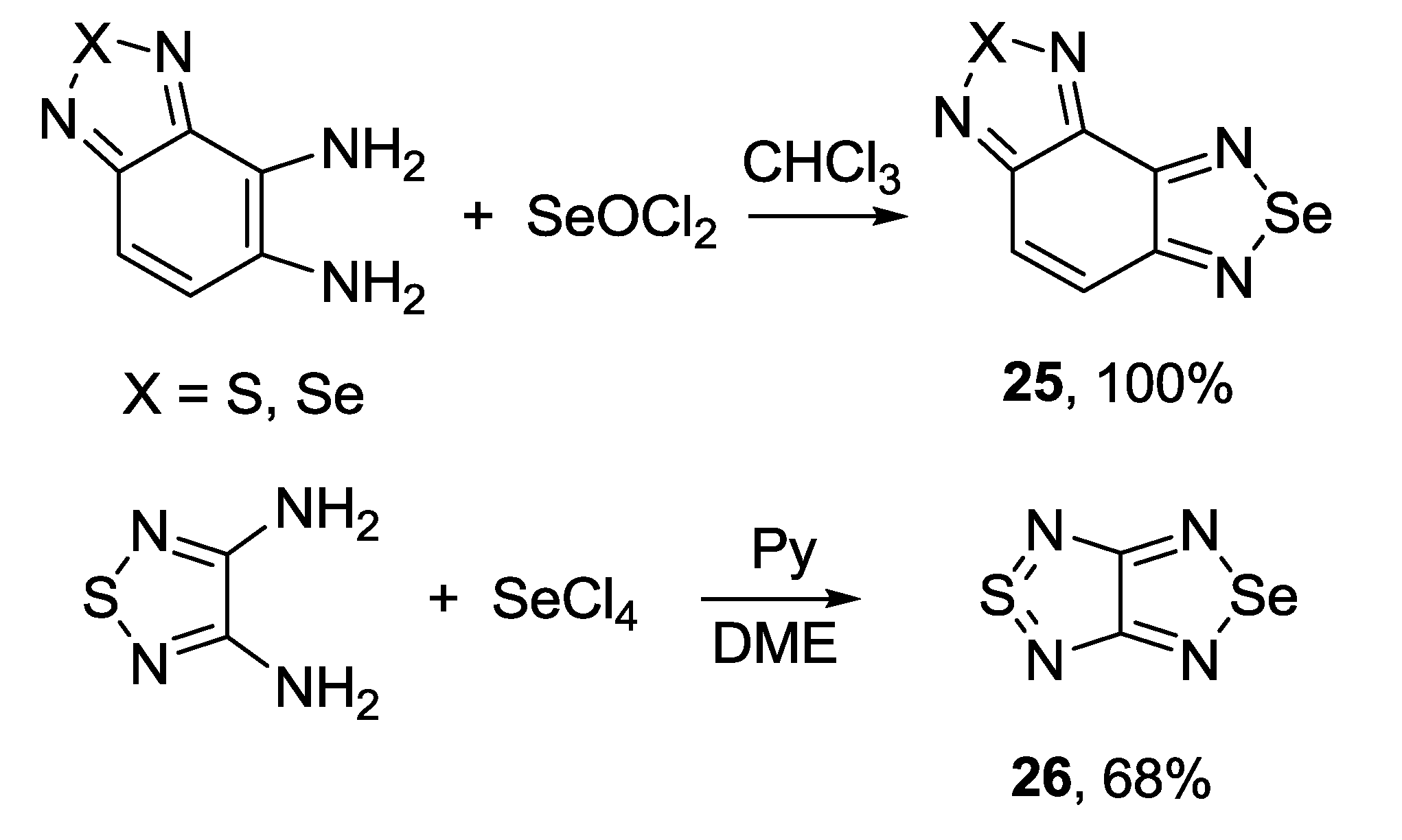
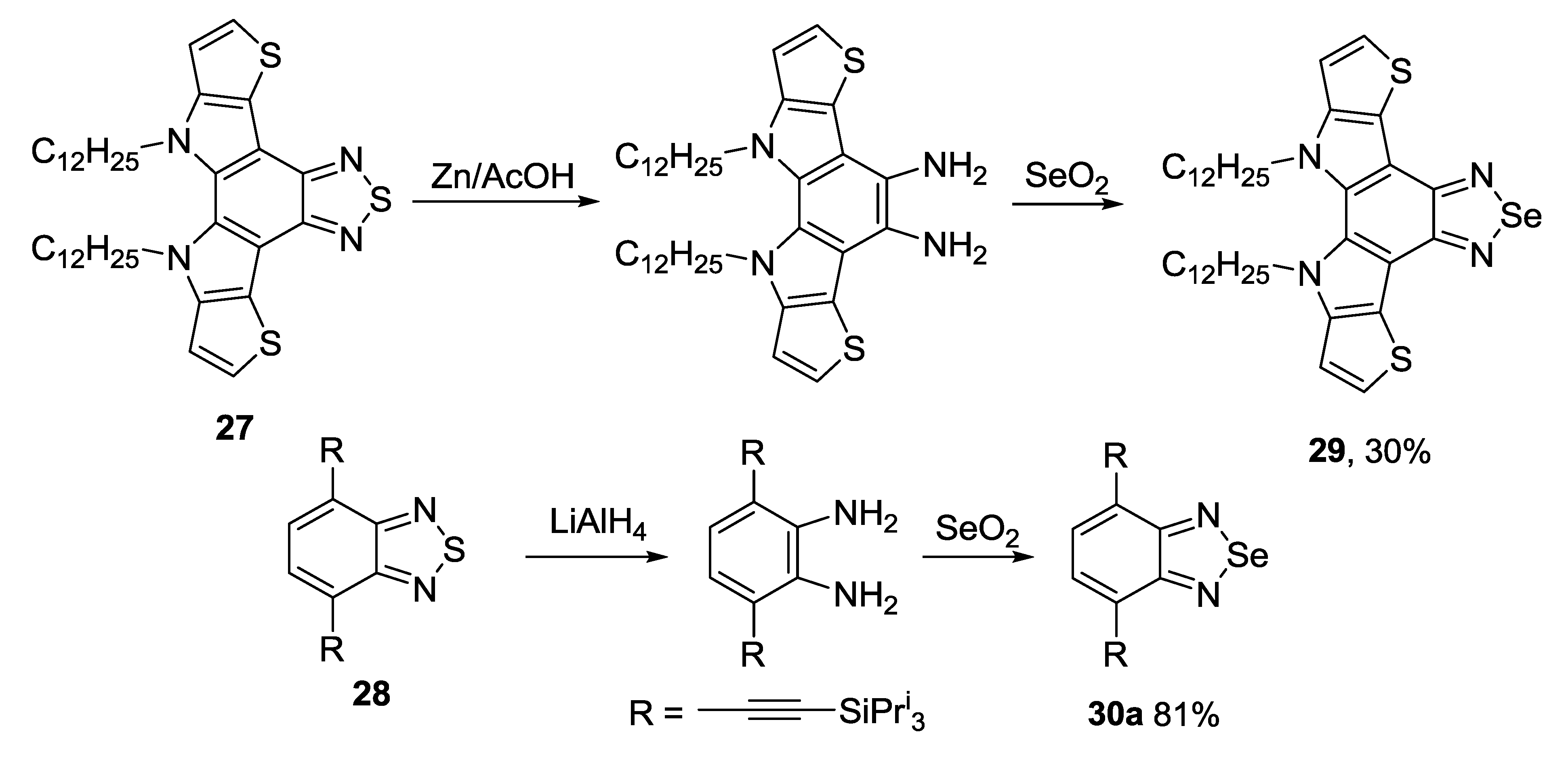
3.2. 1,2,5-Selenadiazoles


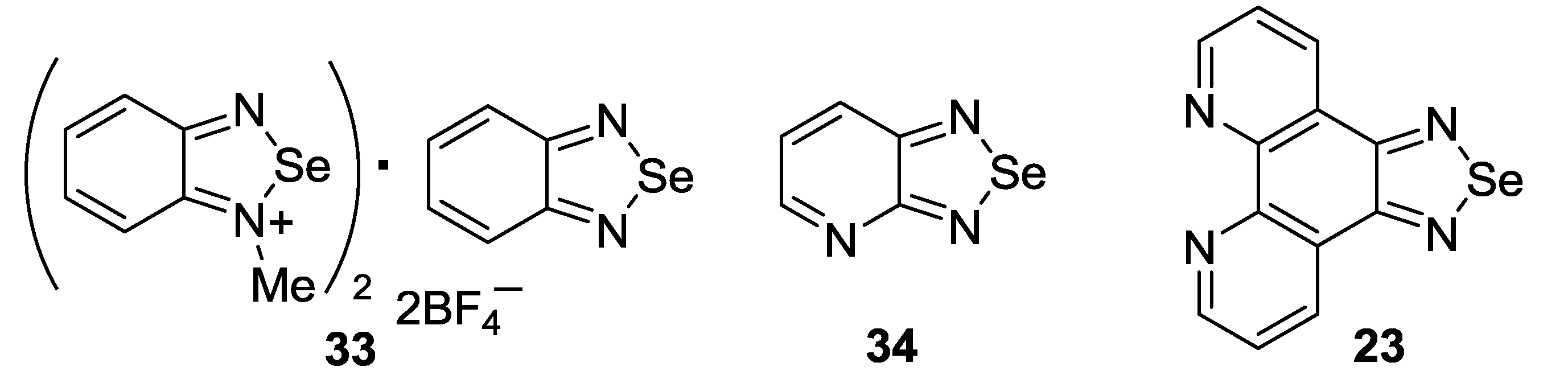
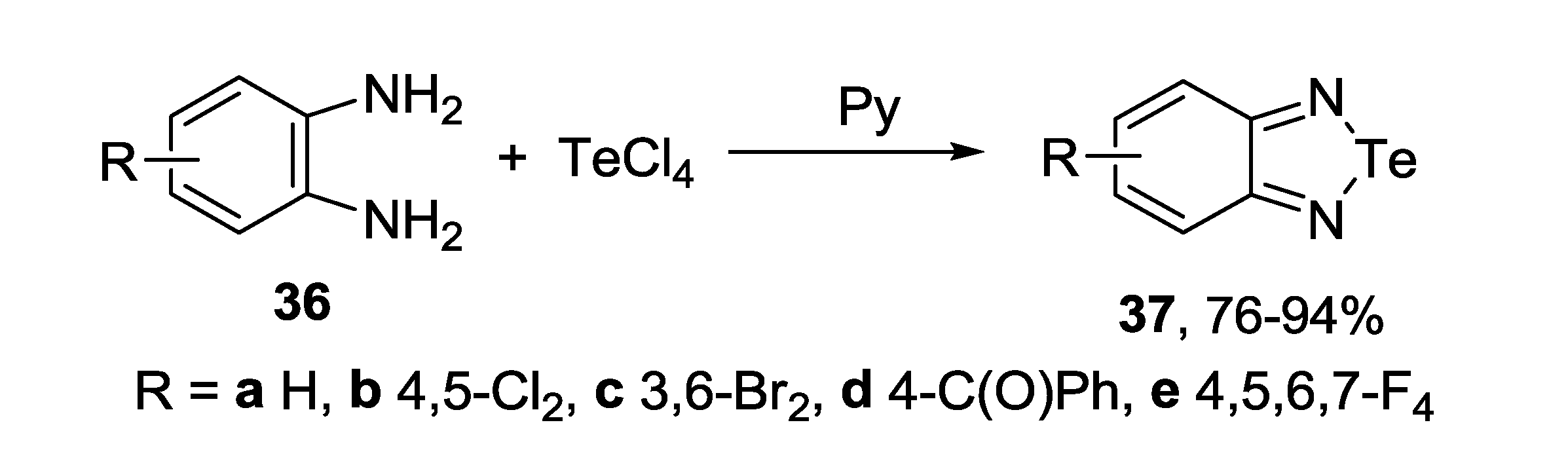
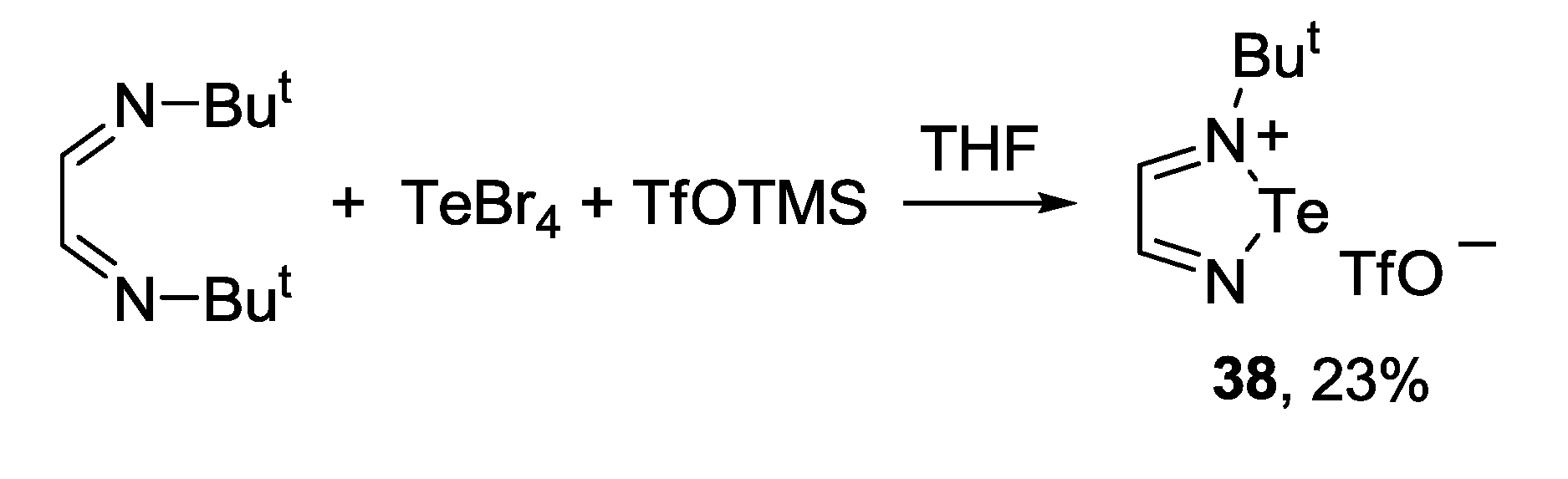
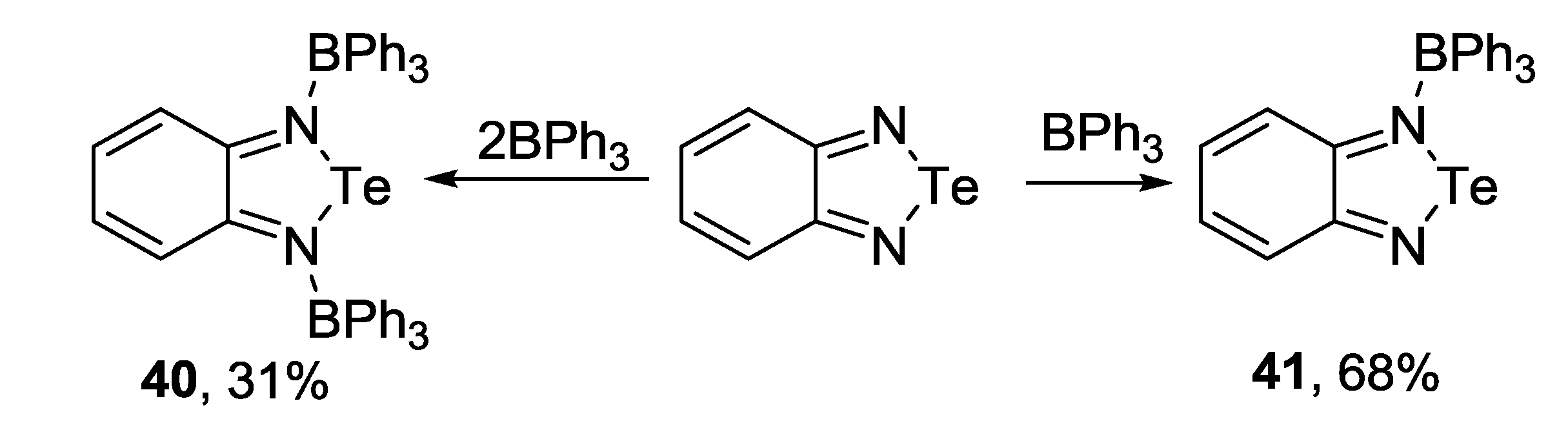
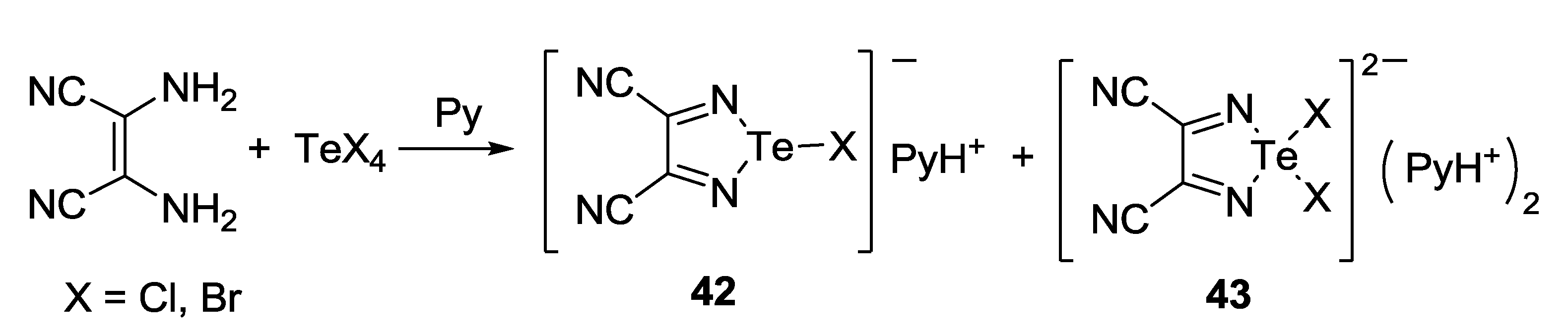
3.3. 1,2,5-Telluradiazoles
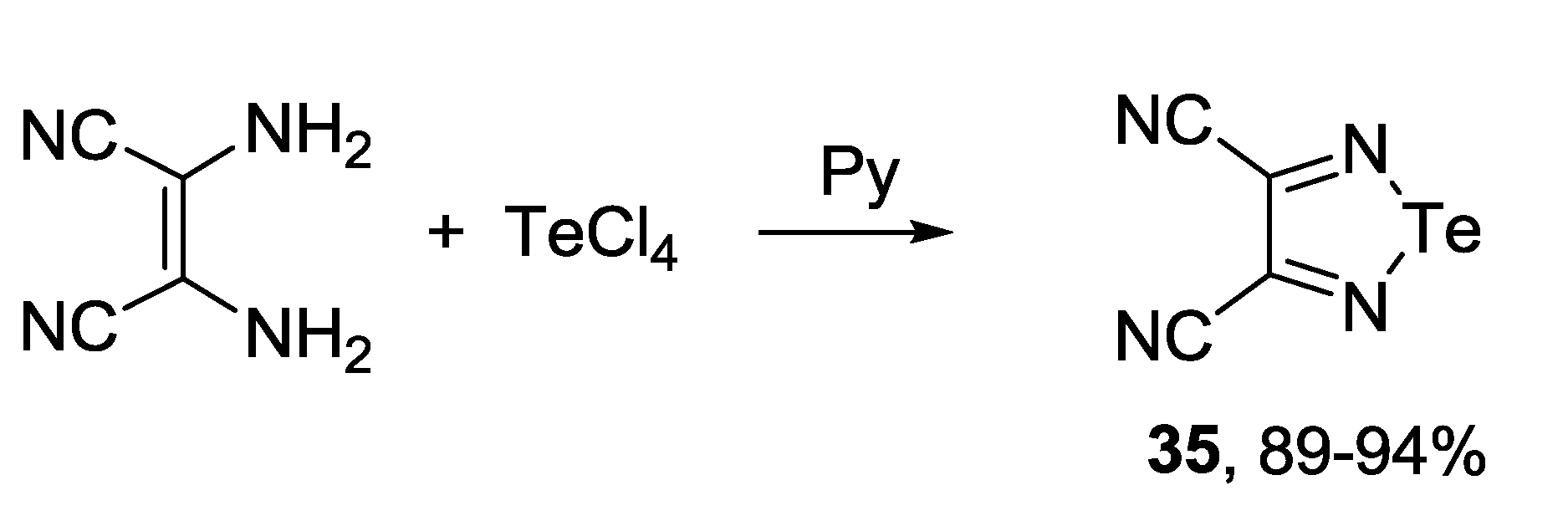

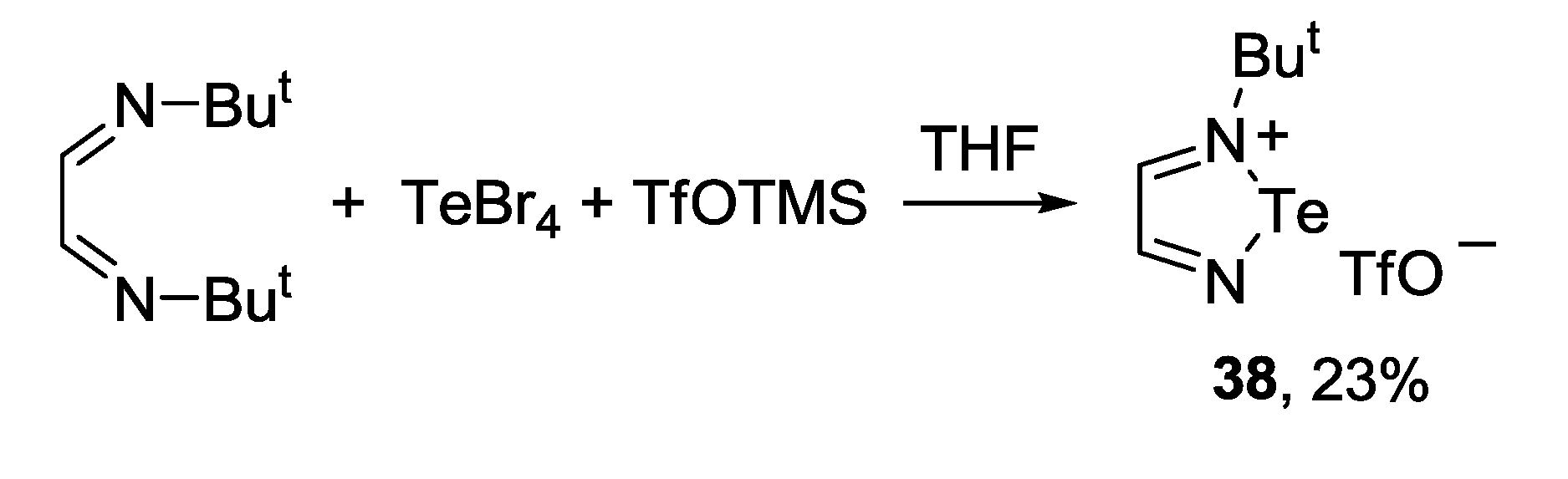
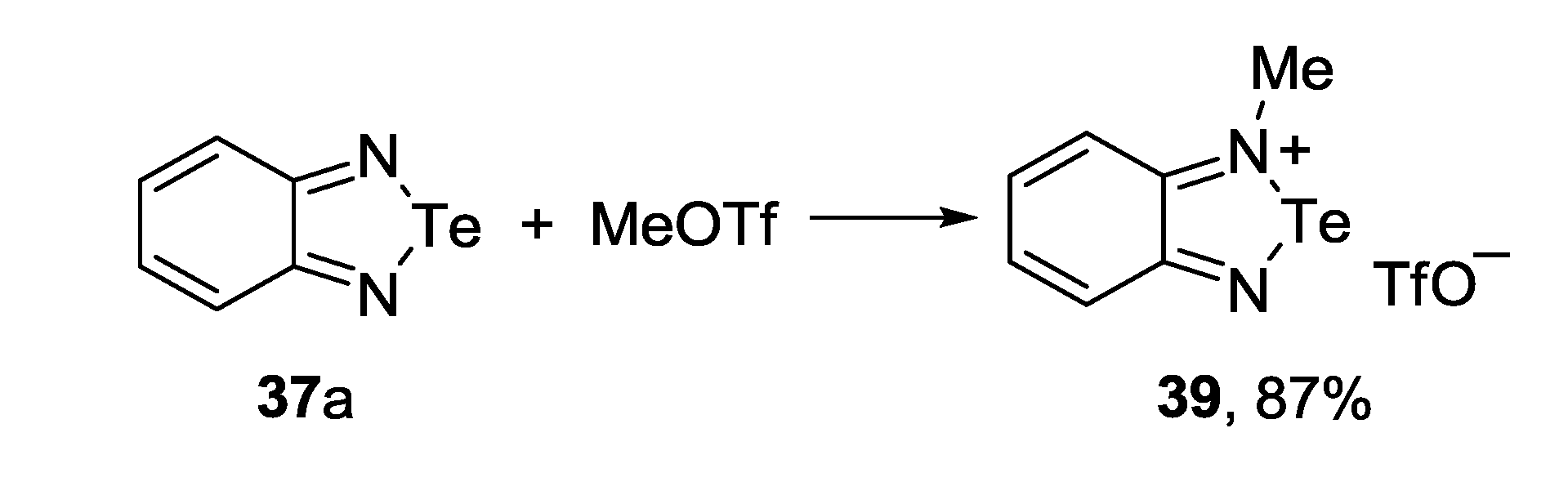
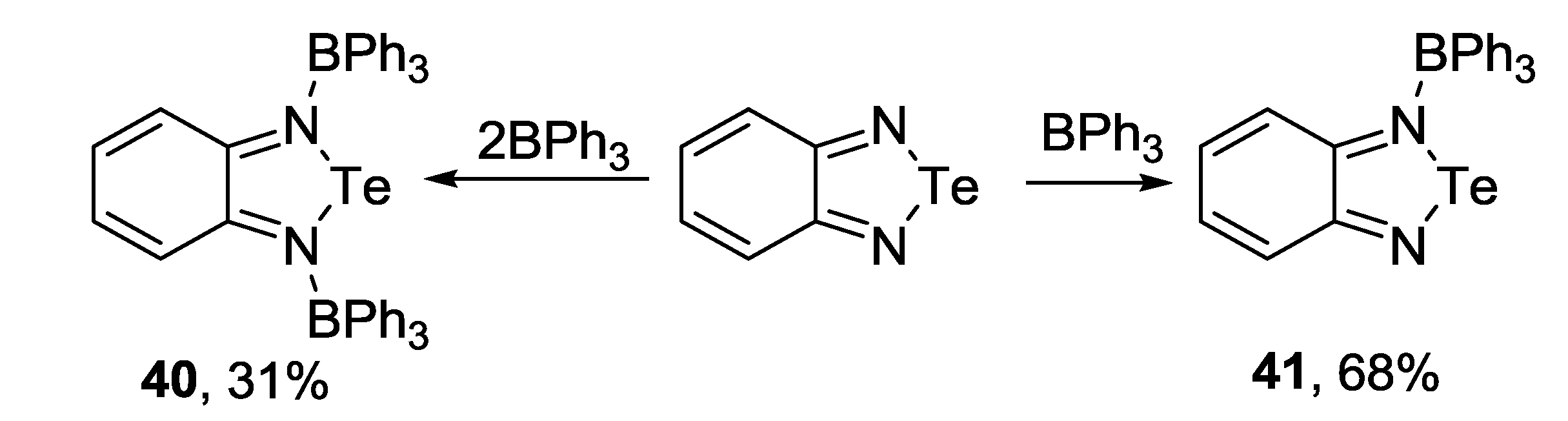
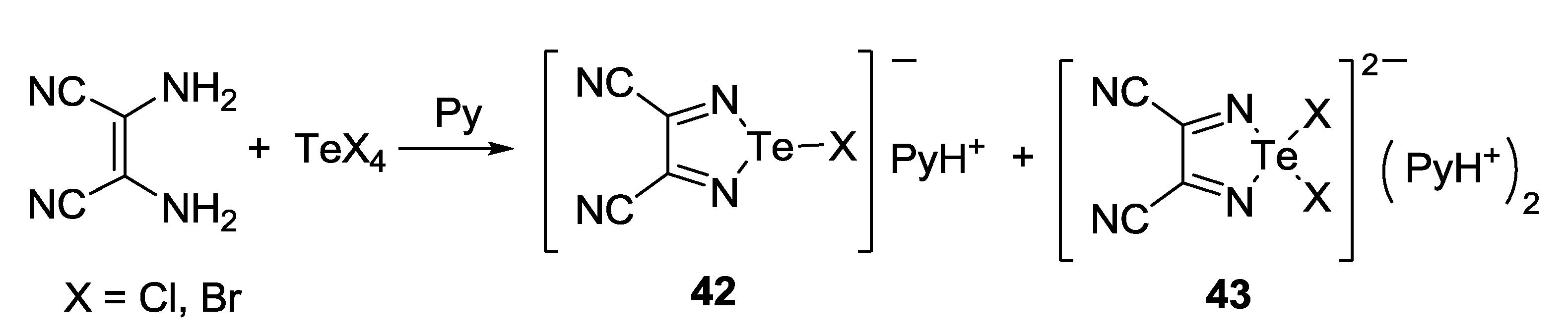

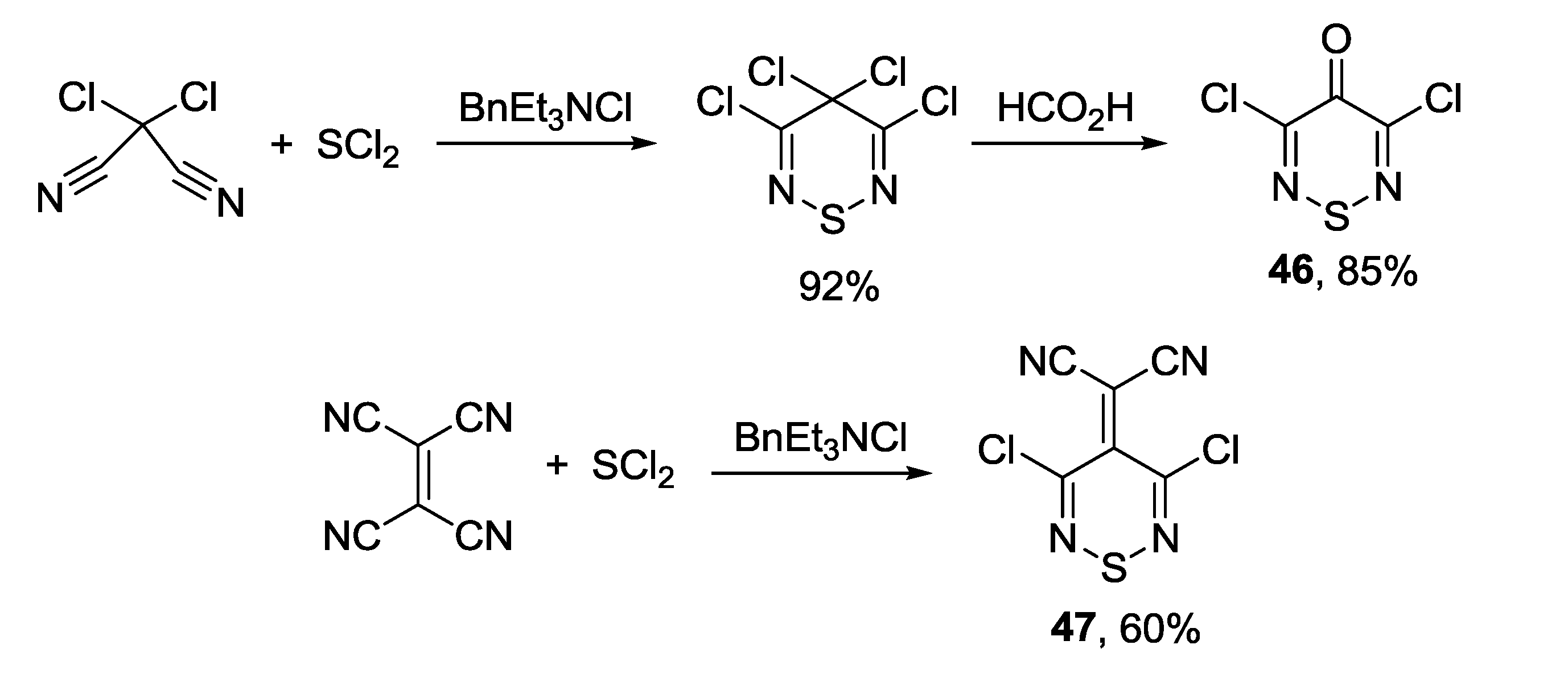
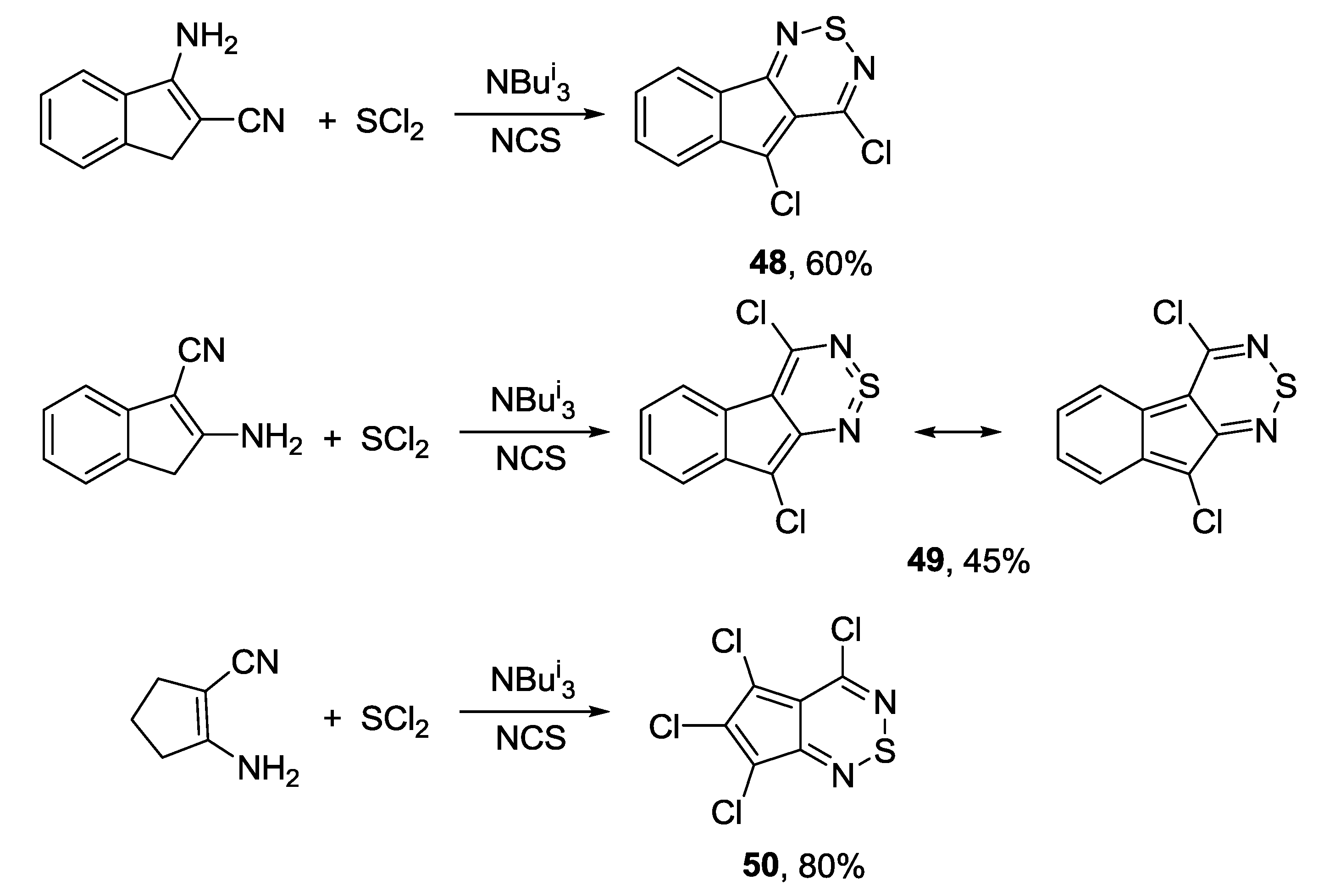
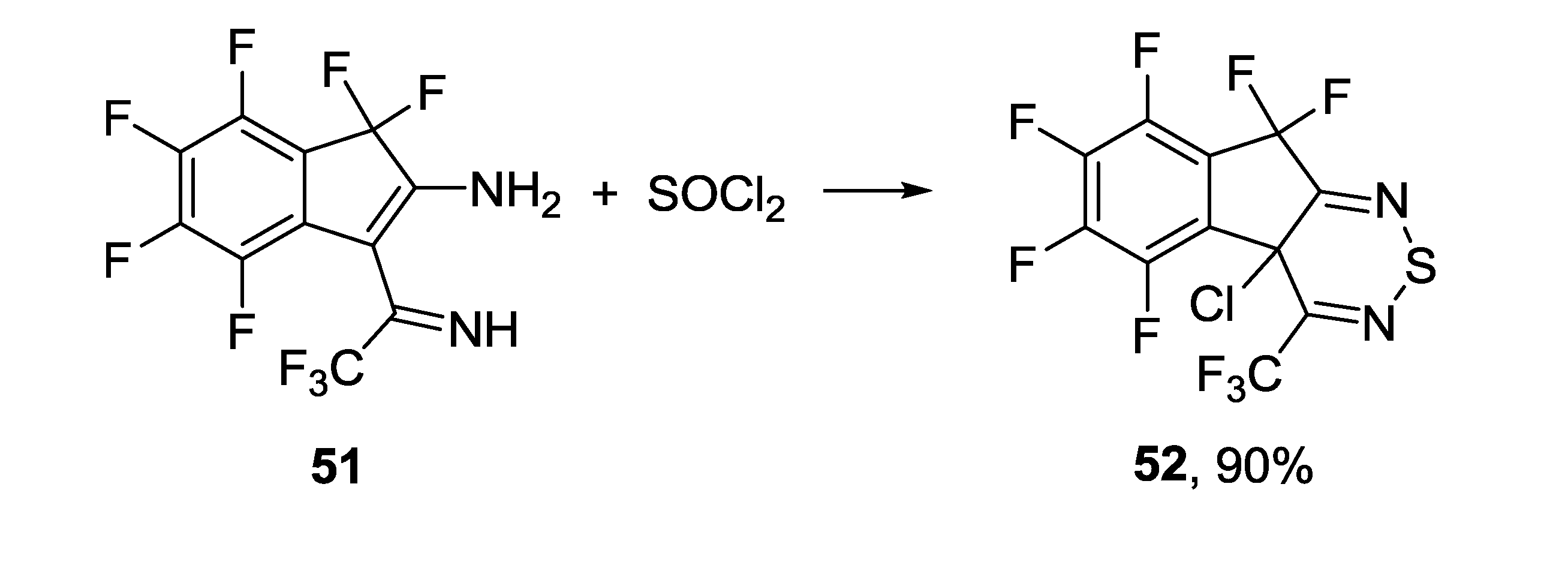
3.4. 1,2,6-Chalcogenadiazines
3.4.1. 1,2,6-Thiadiazines
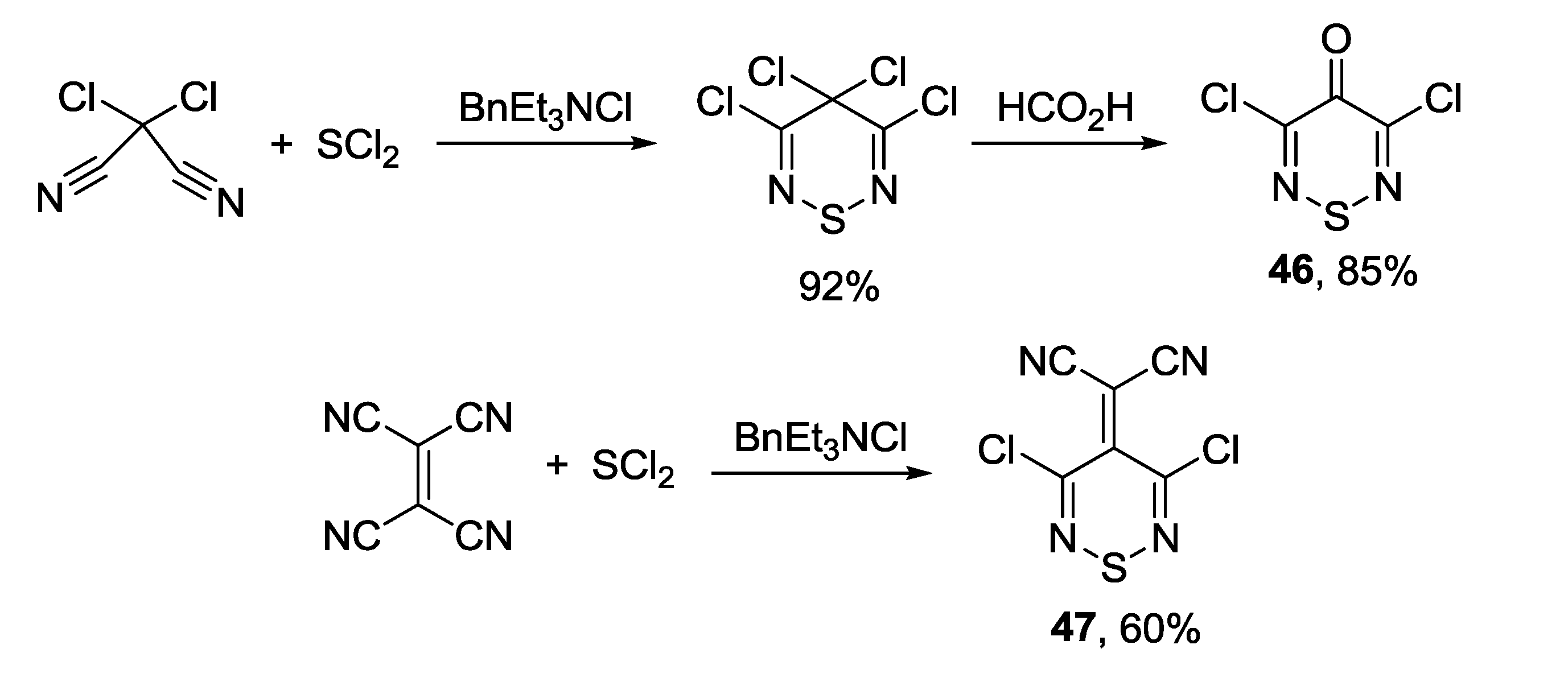
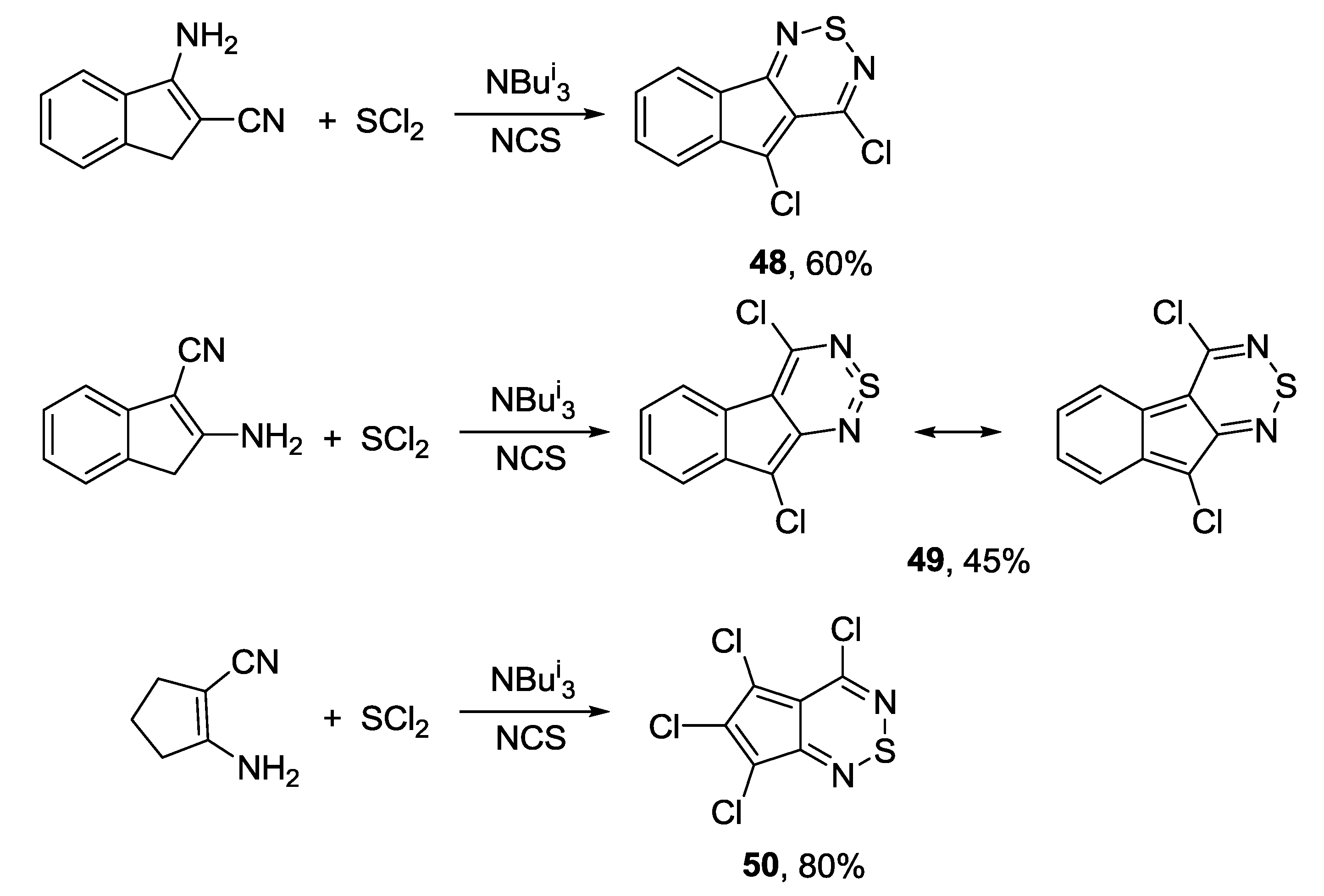
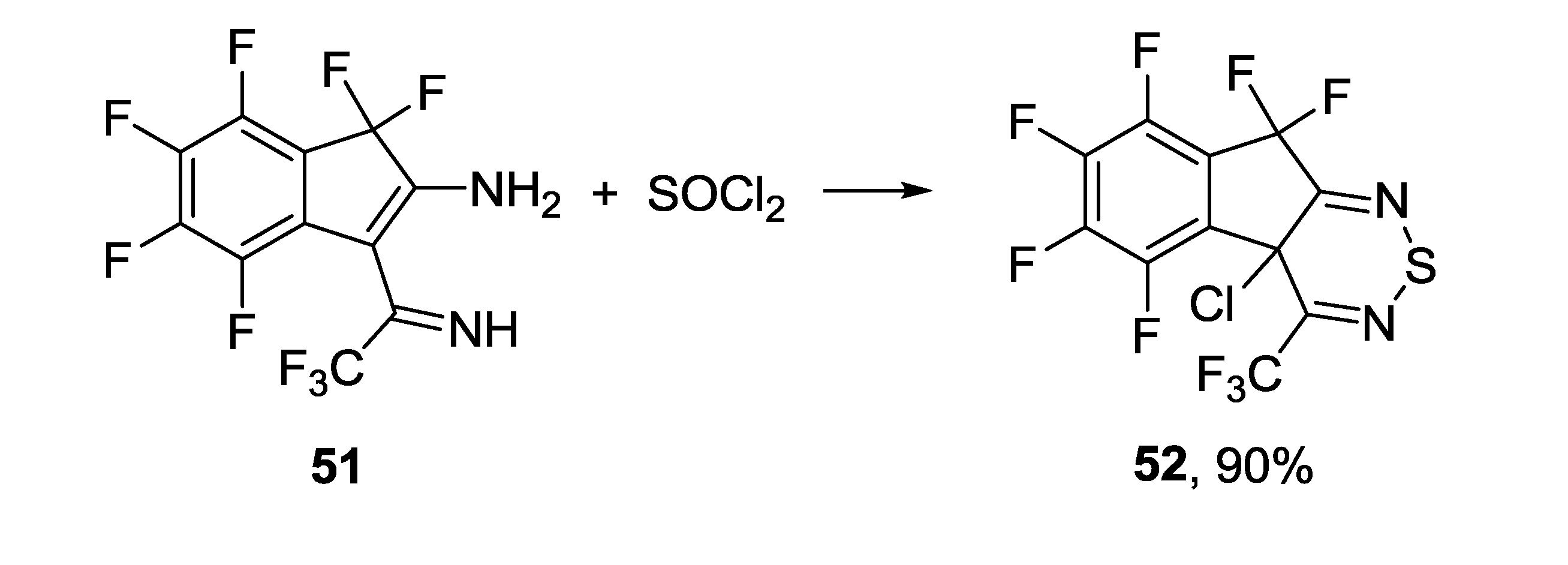
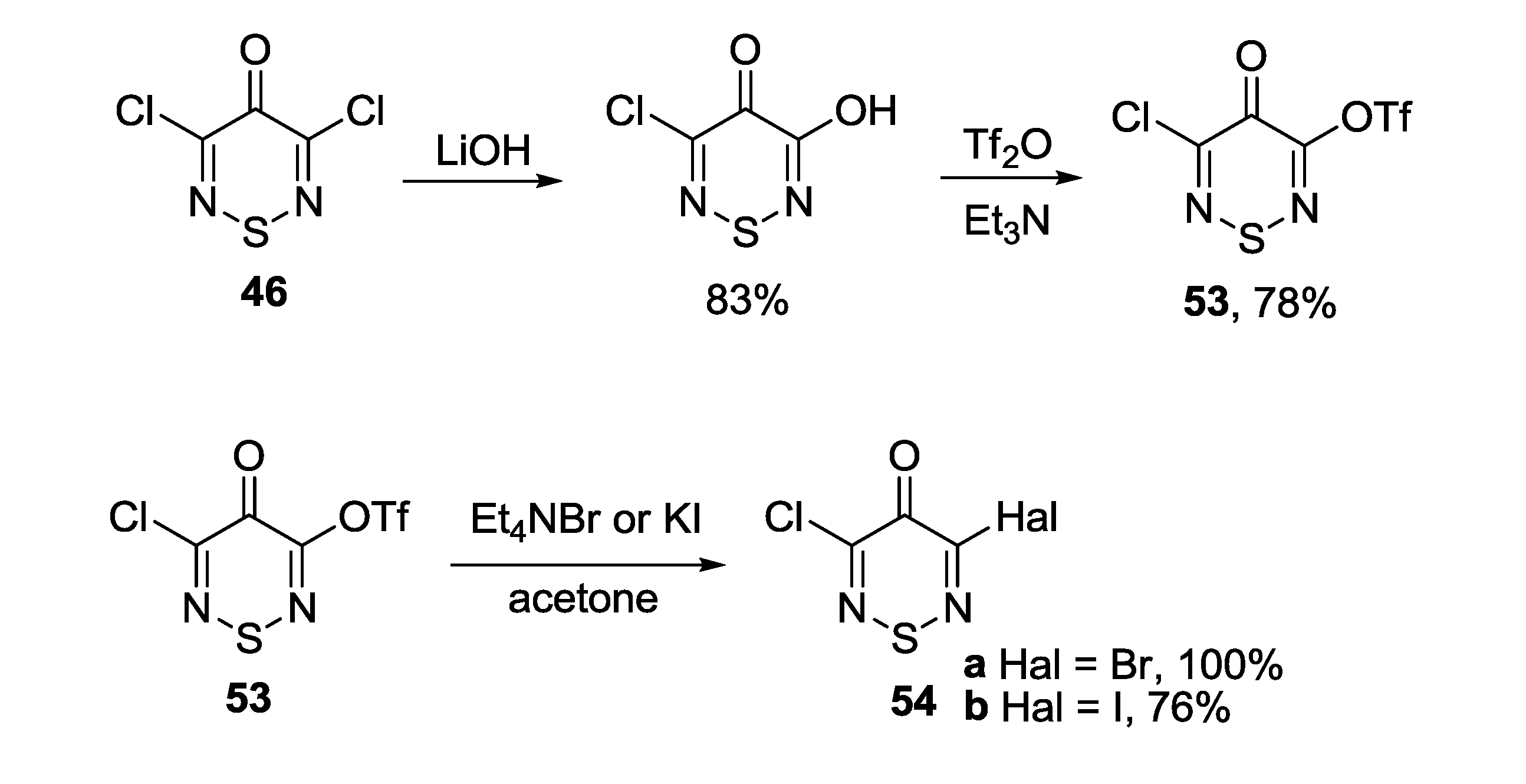
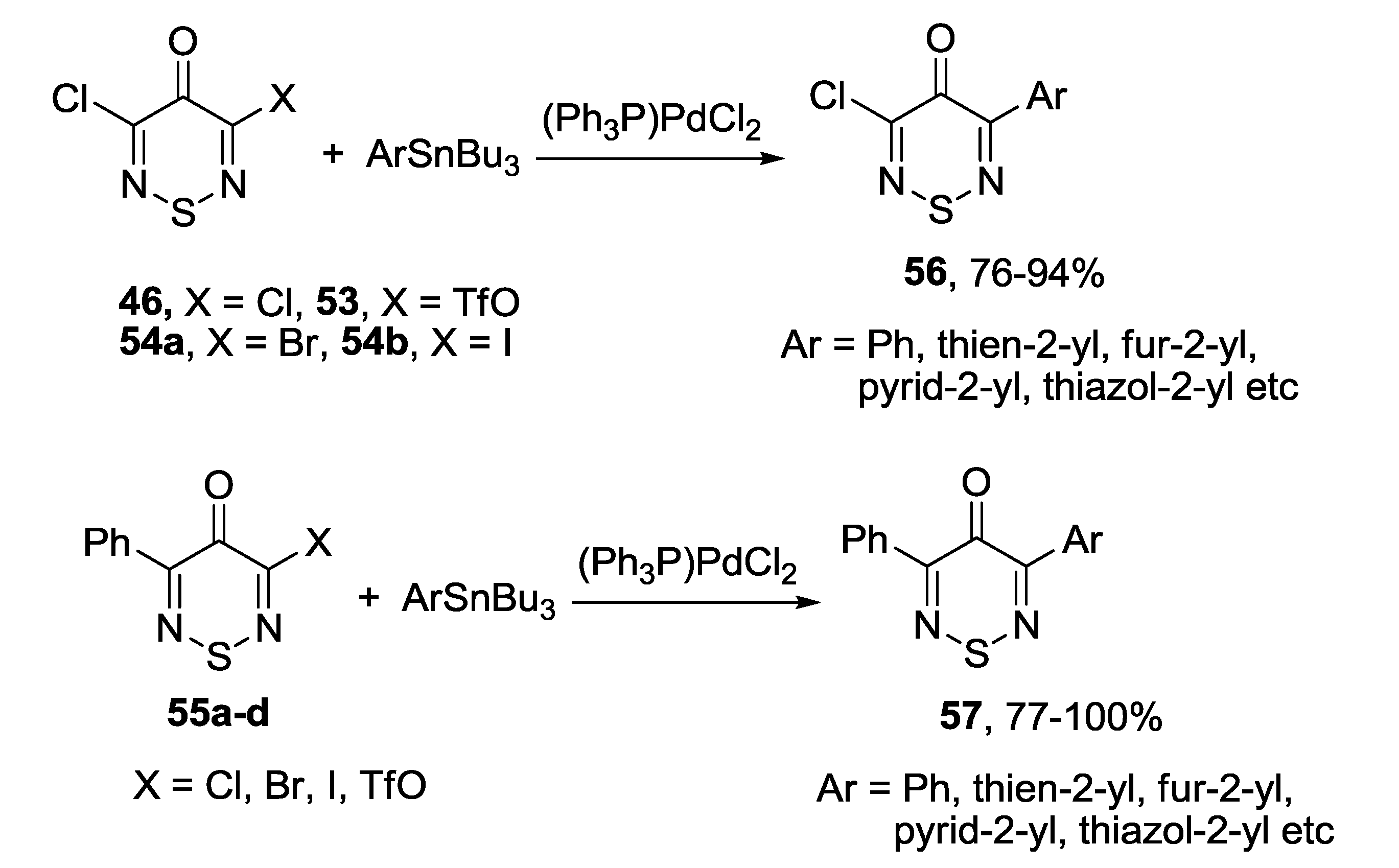
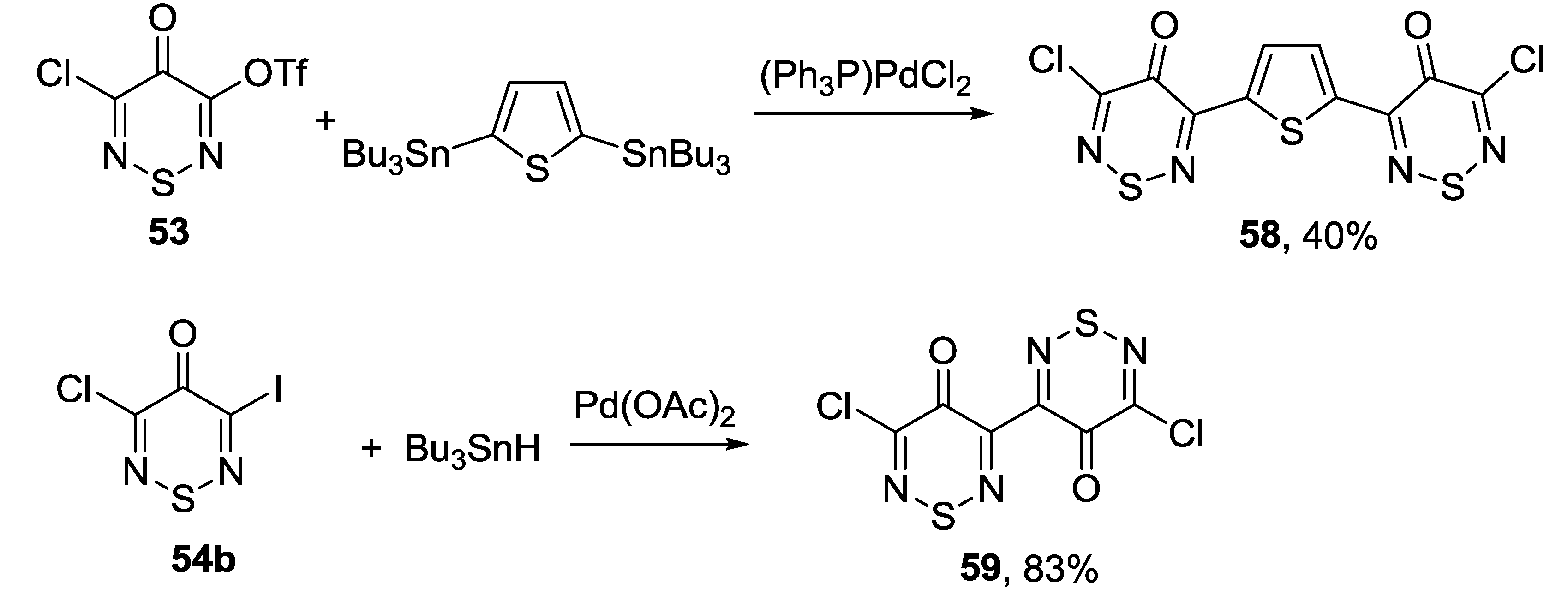
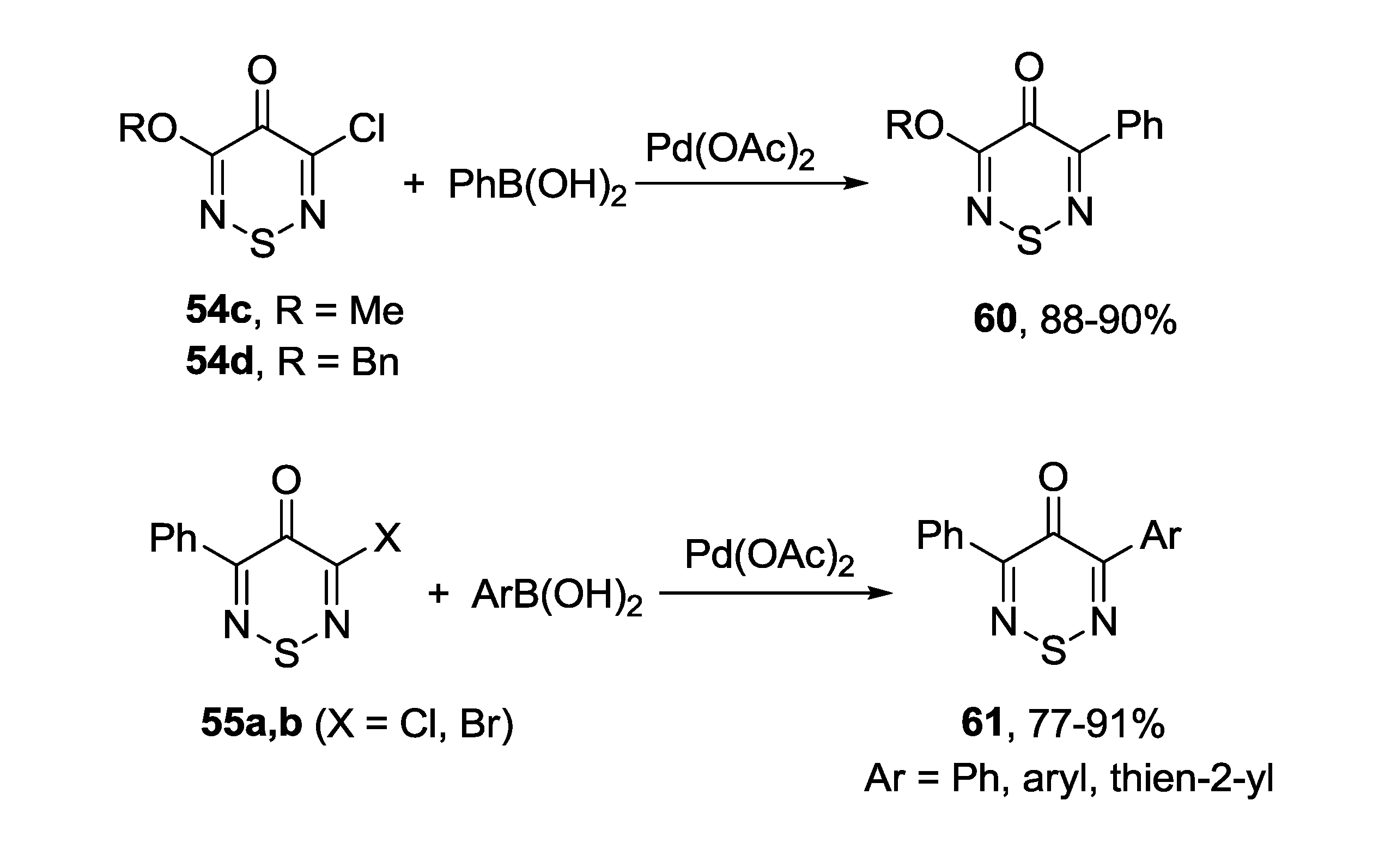
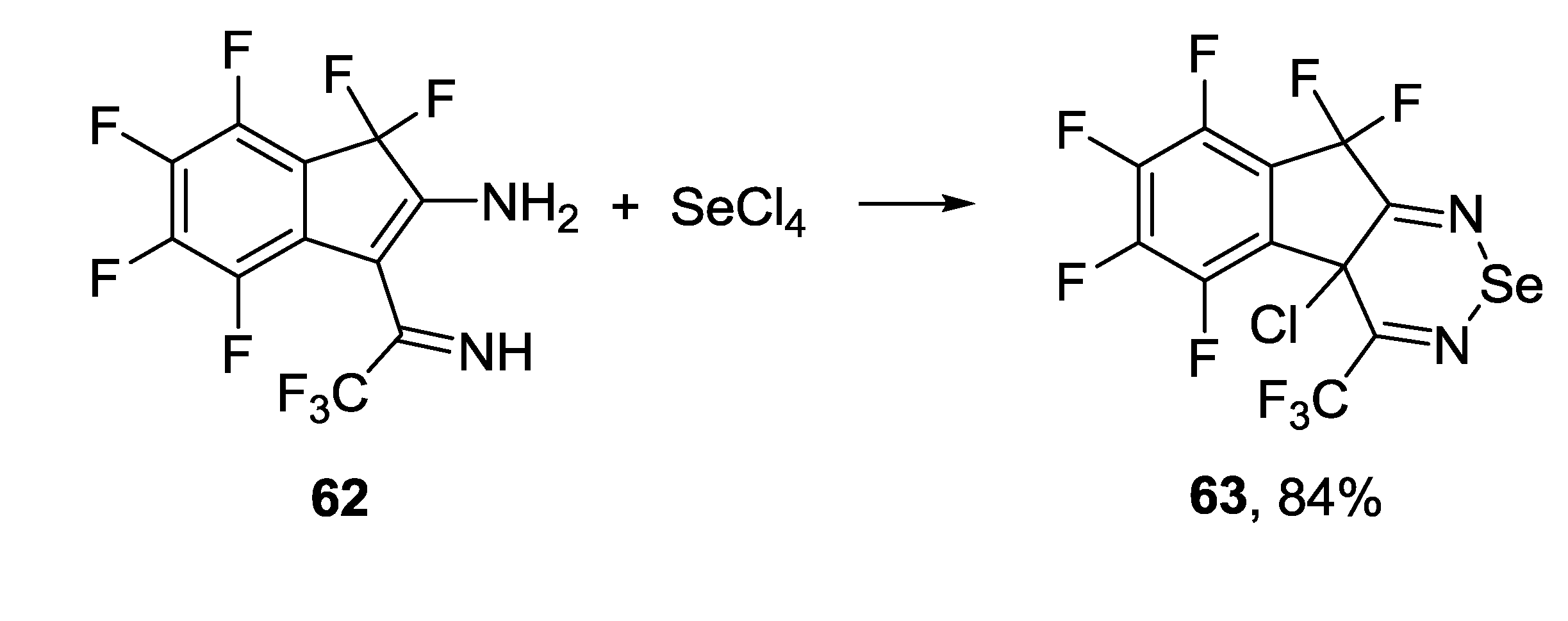
3.4.2. 1,2,6-Selenadiazines
4. Charge-Transfer Complexes and Radical-Ion Salts
4.1. Synthesis and Characterization
4.1.1. Charge-Transfer Complexes
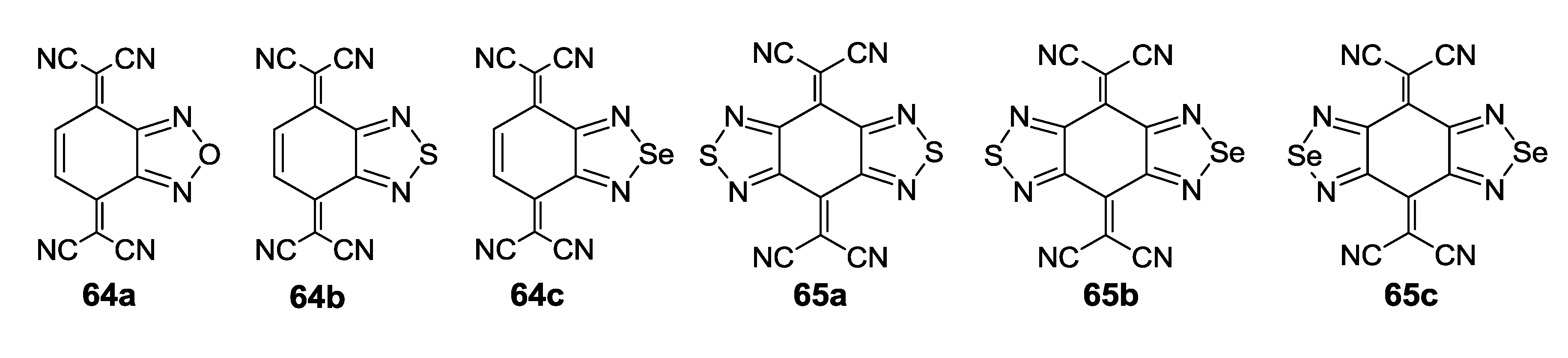
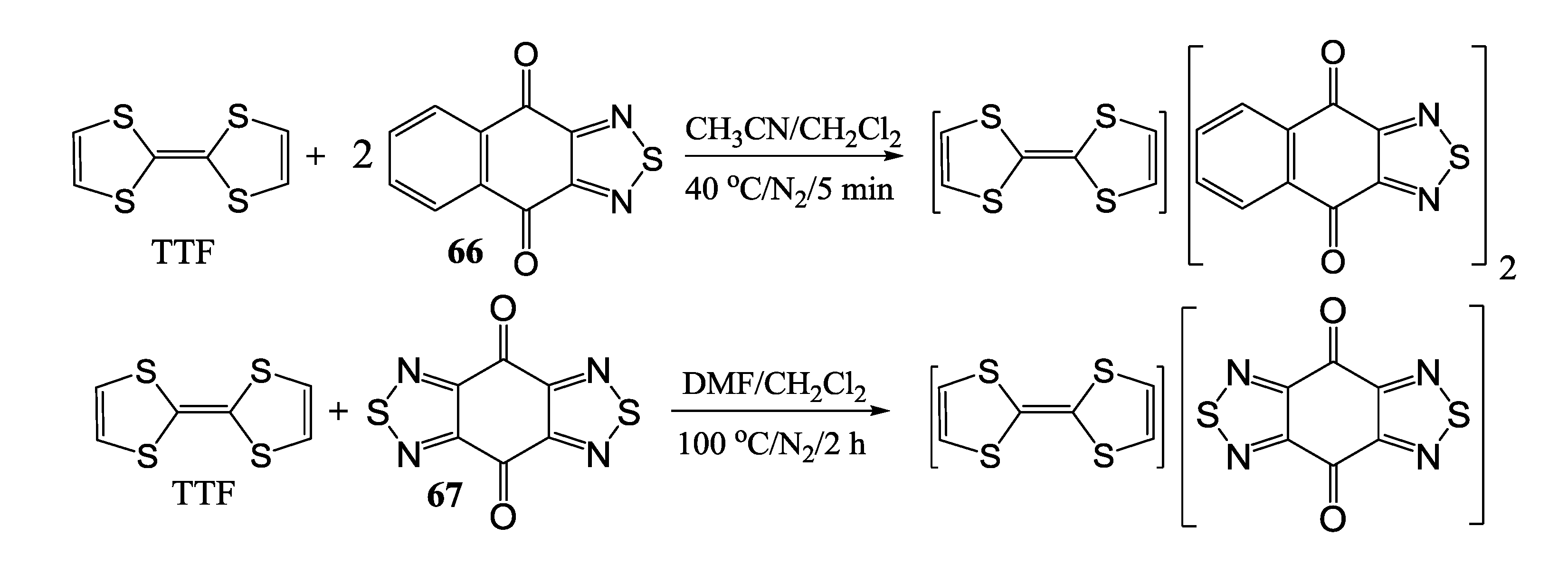
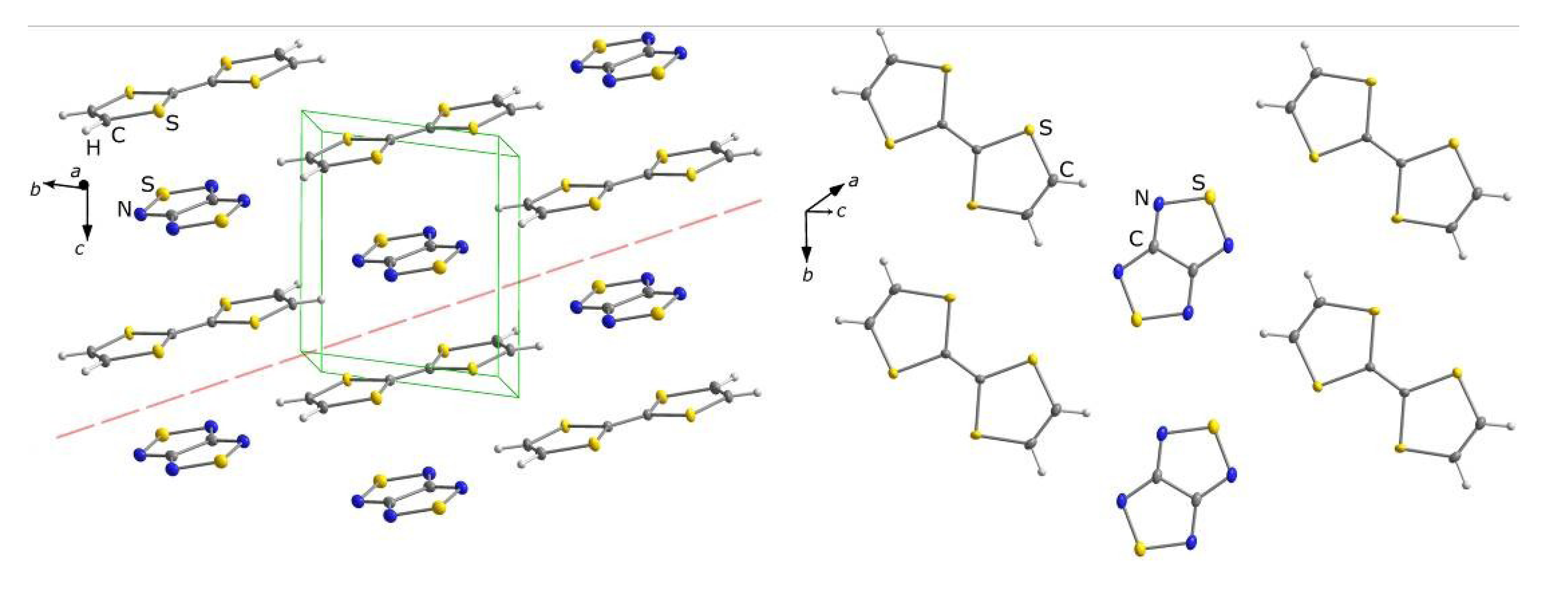
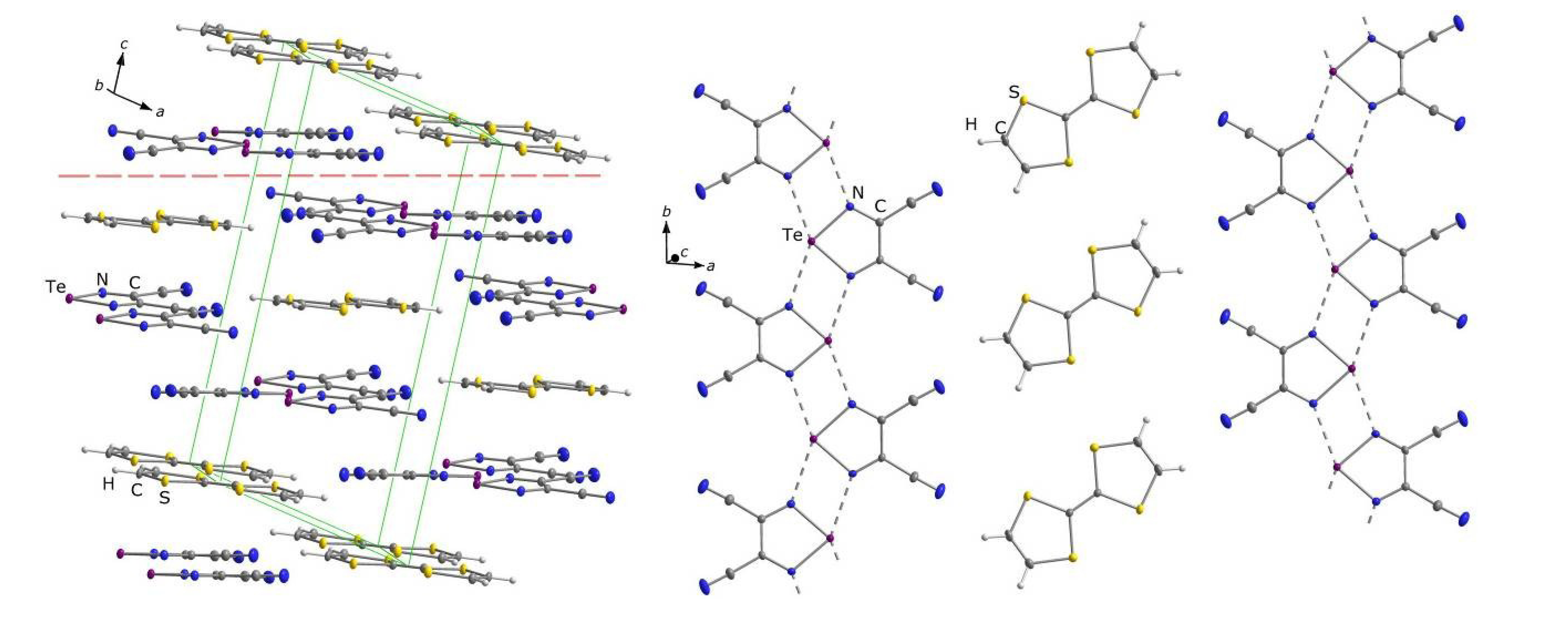
4.1.2. Radical-Ion Salts

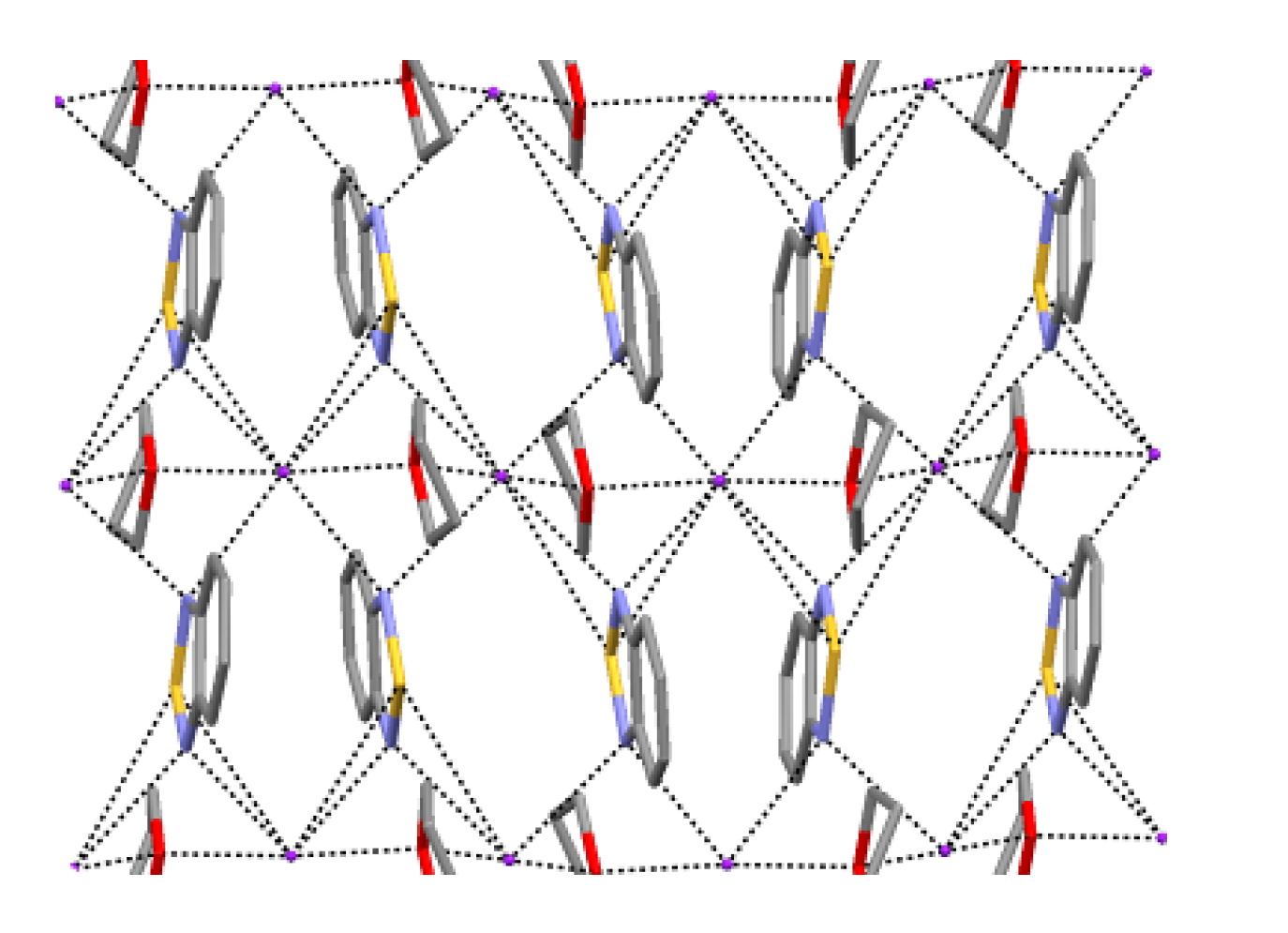


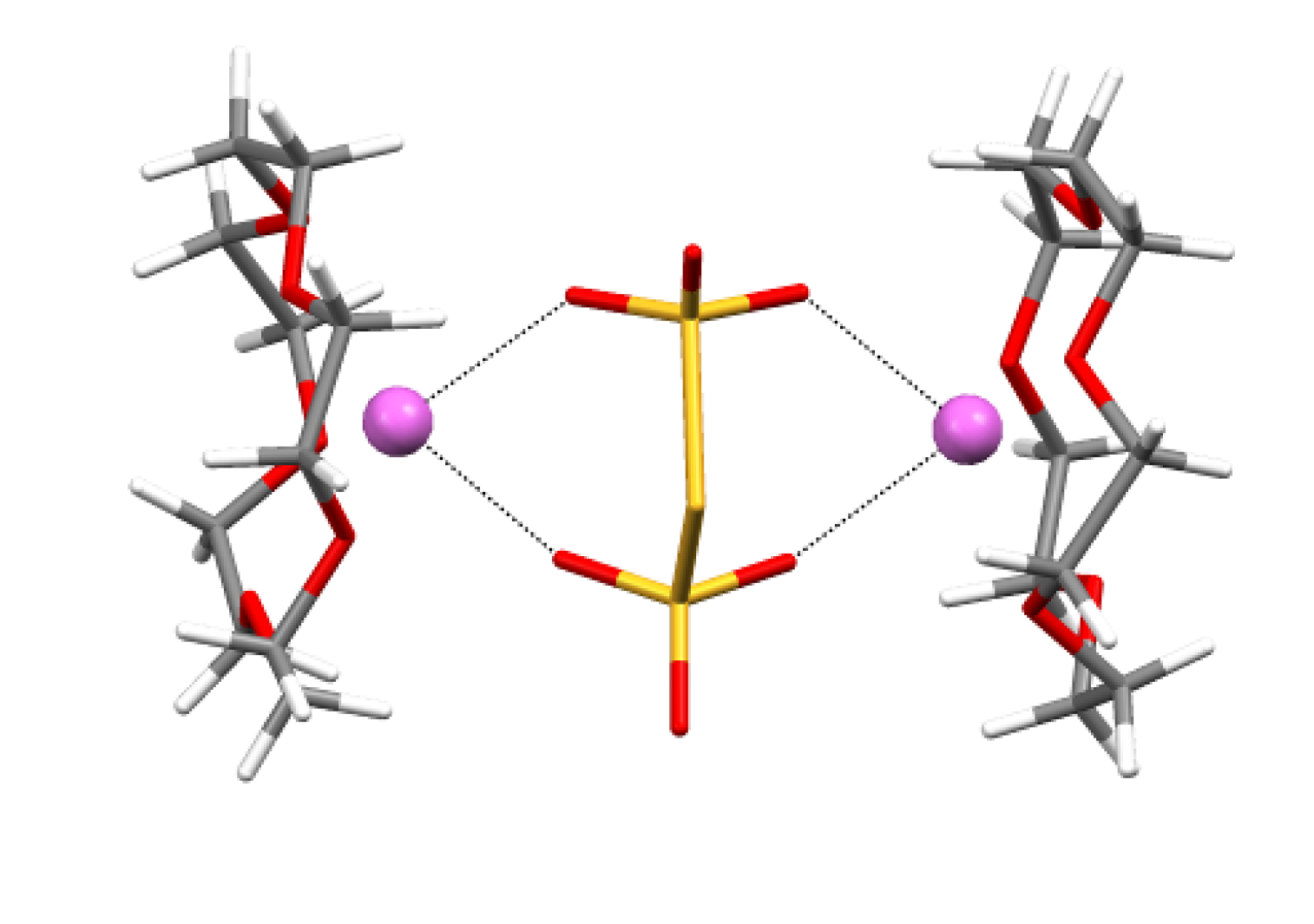
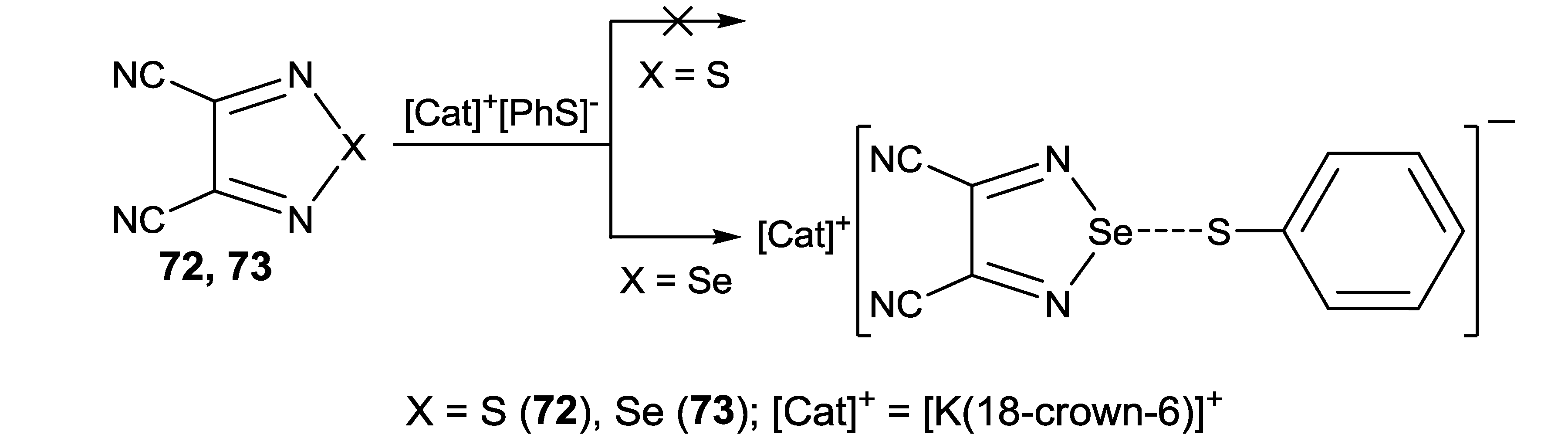
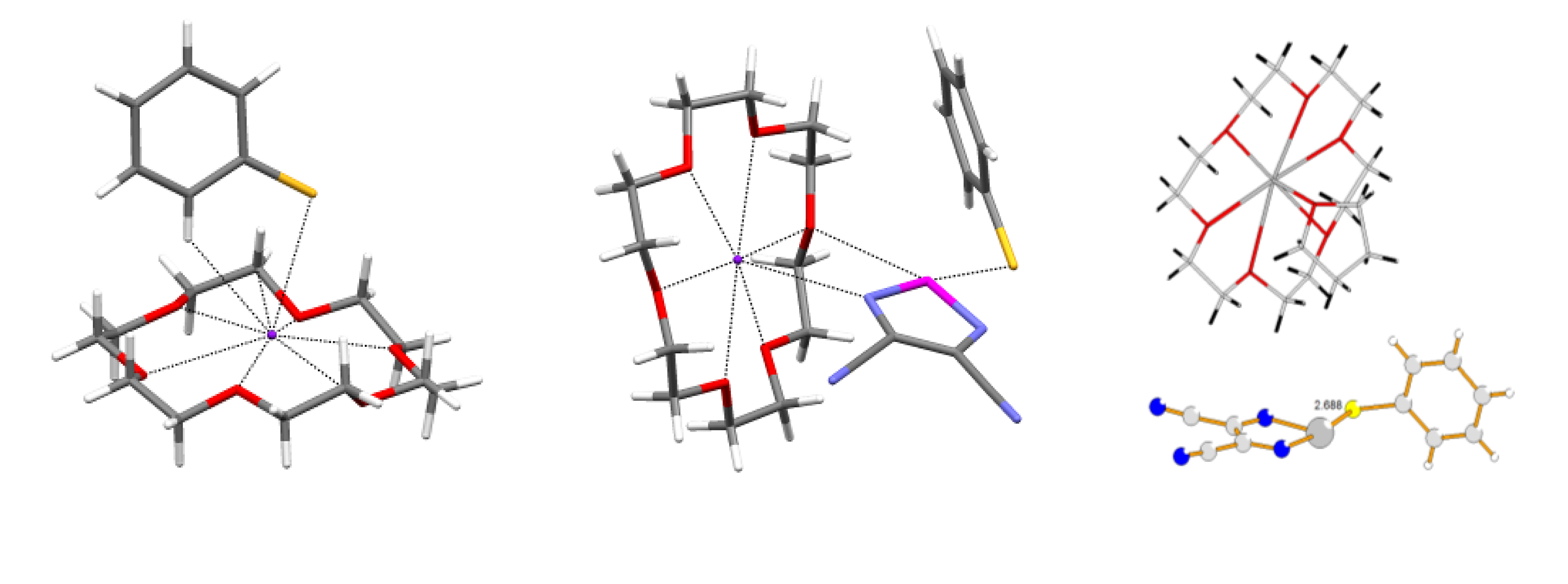

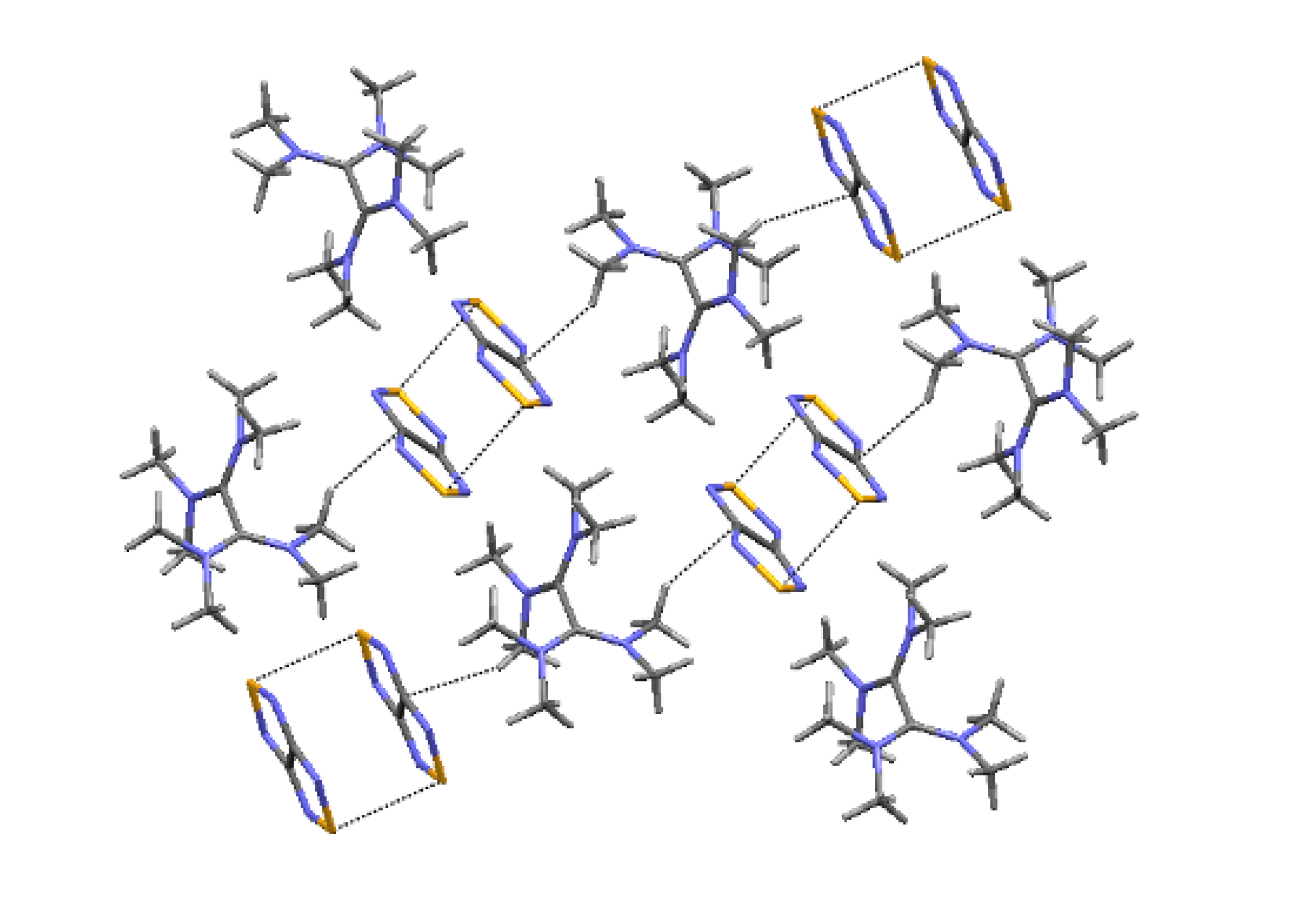
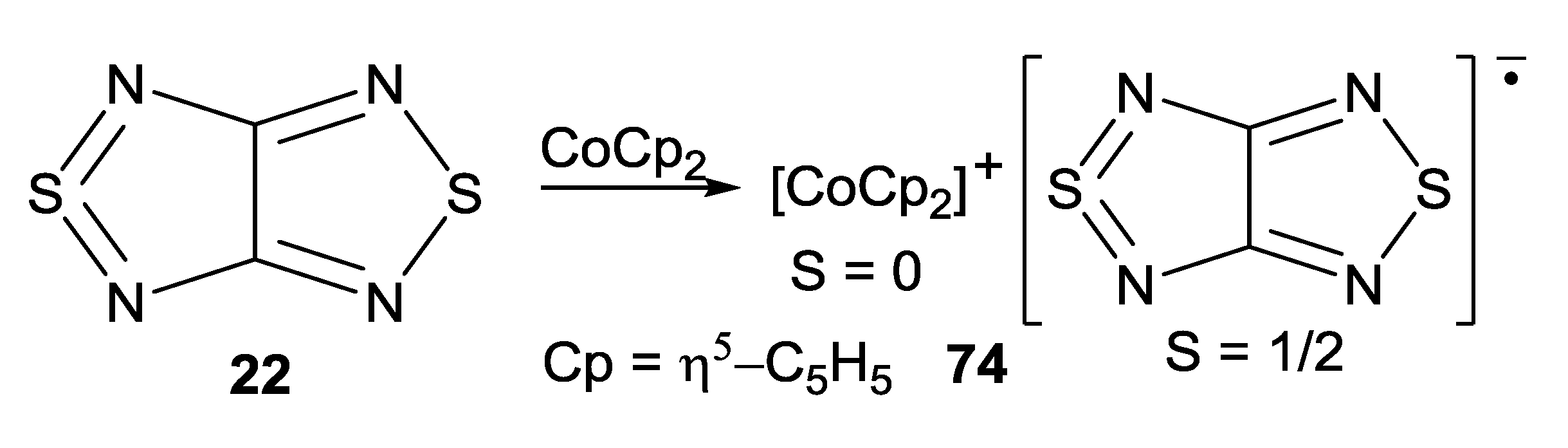
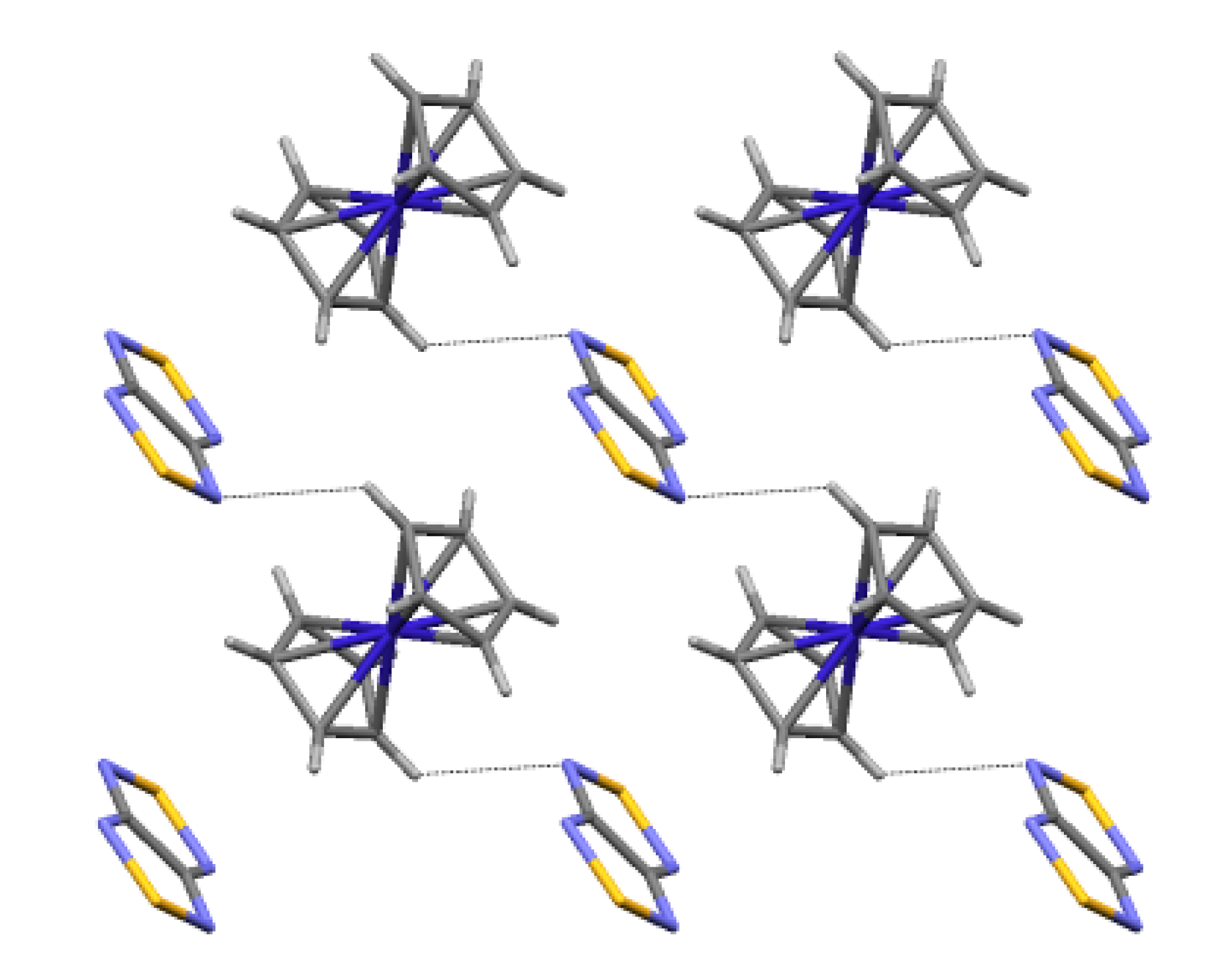
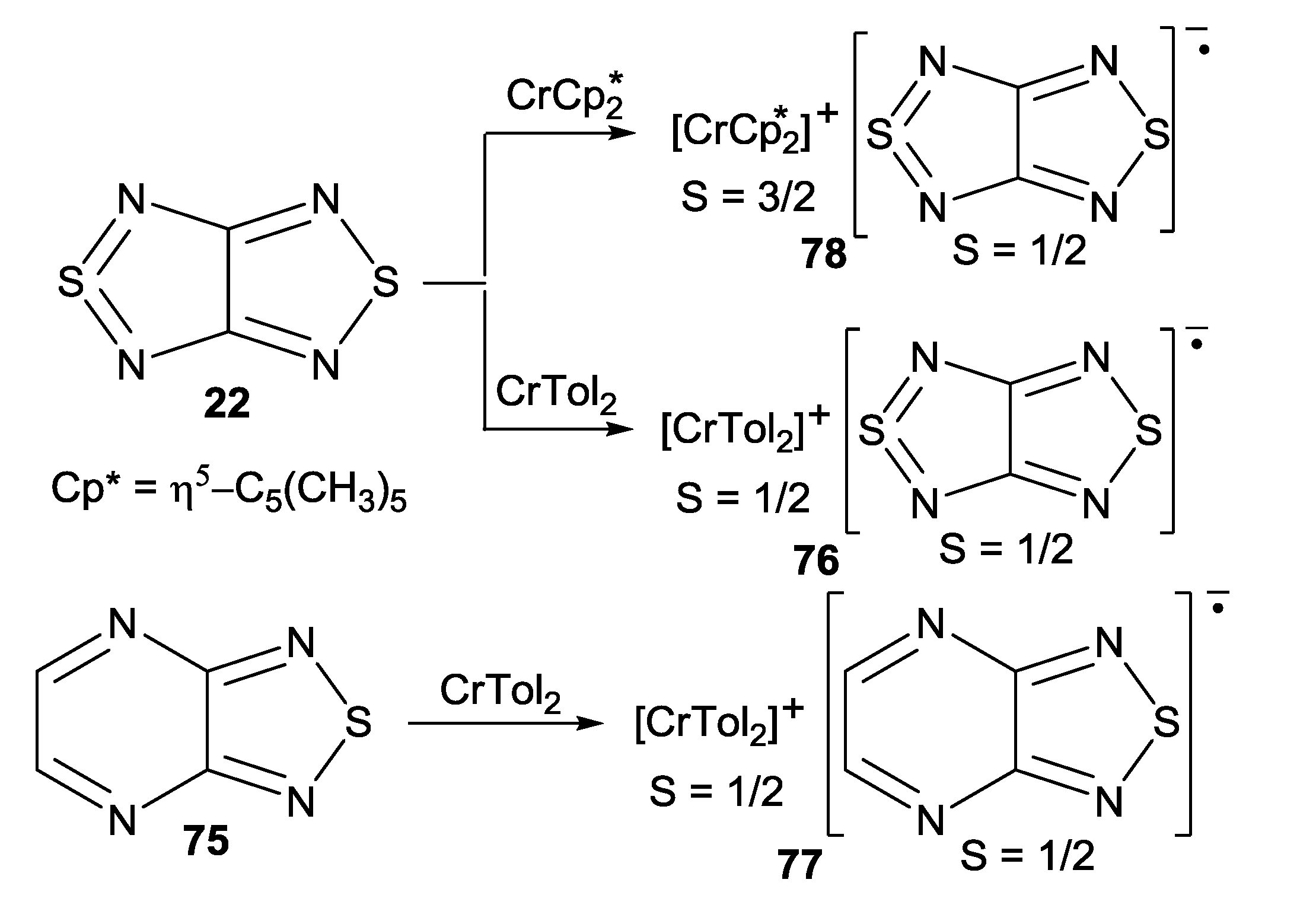
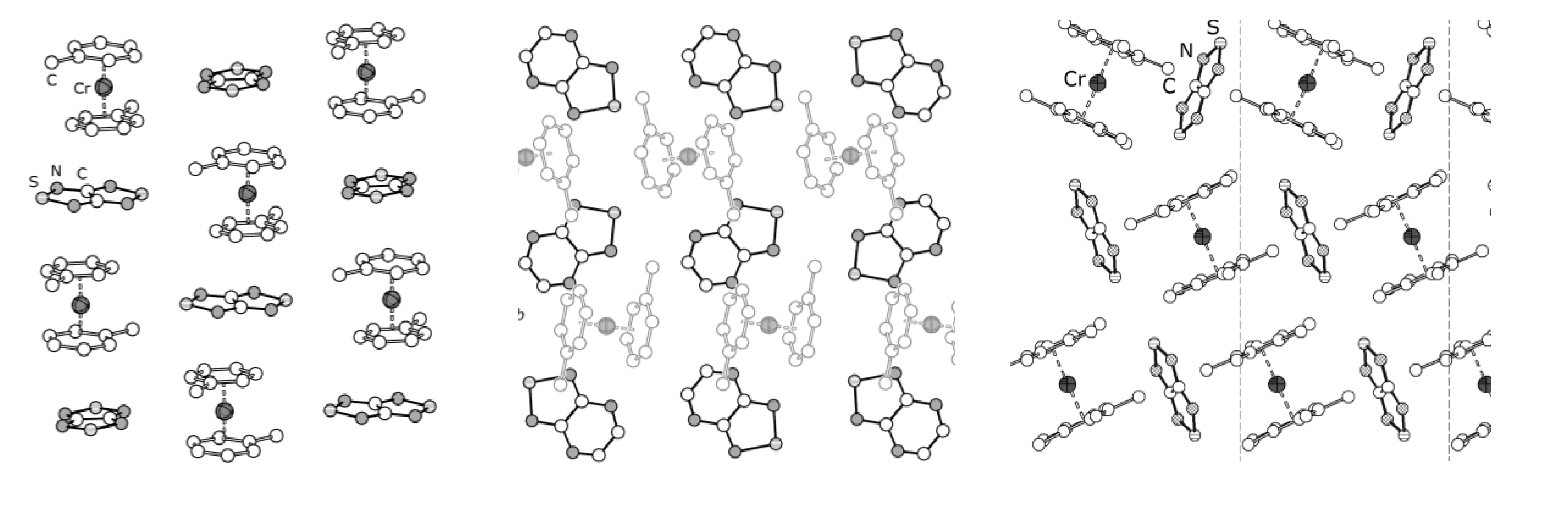
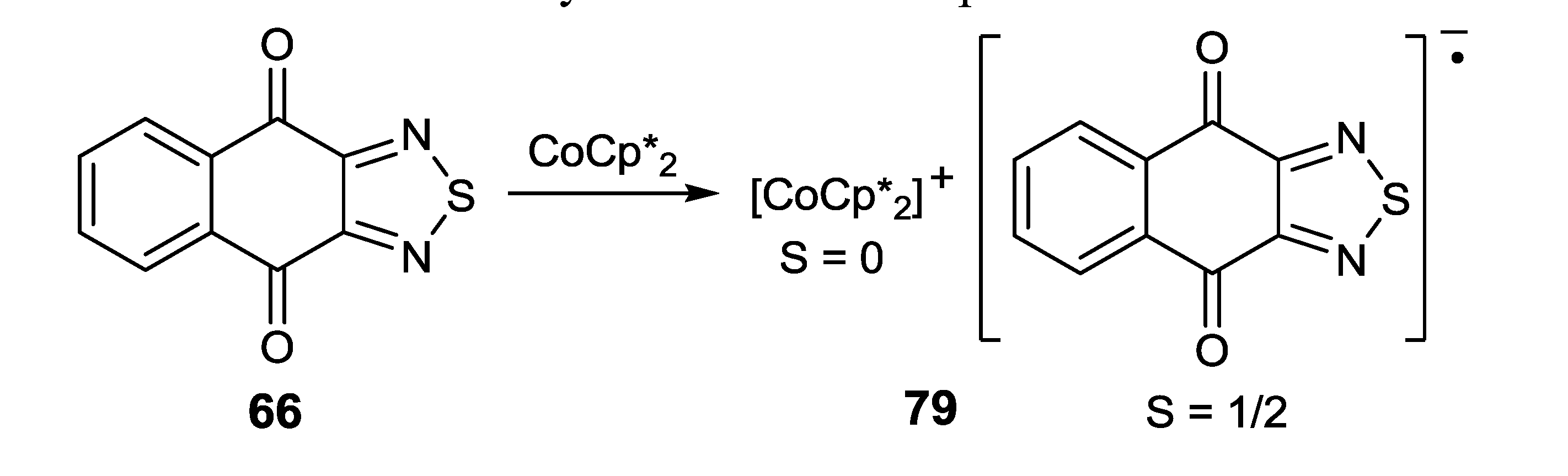

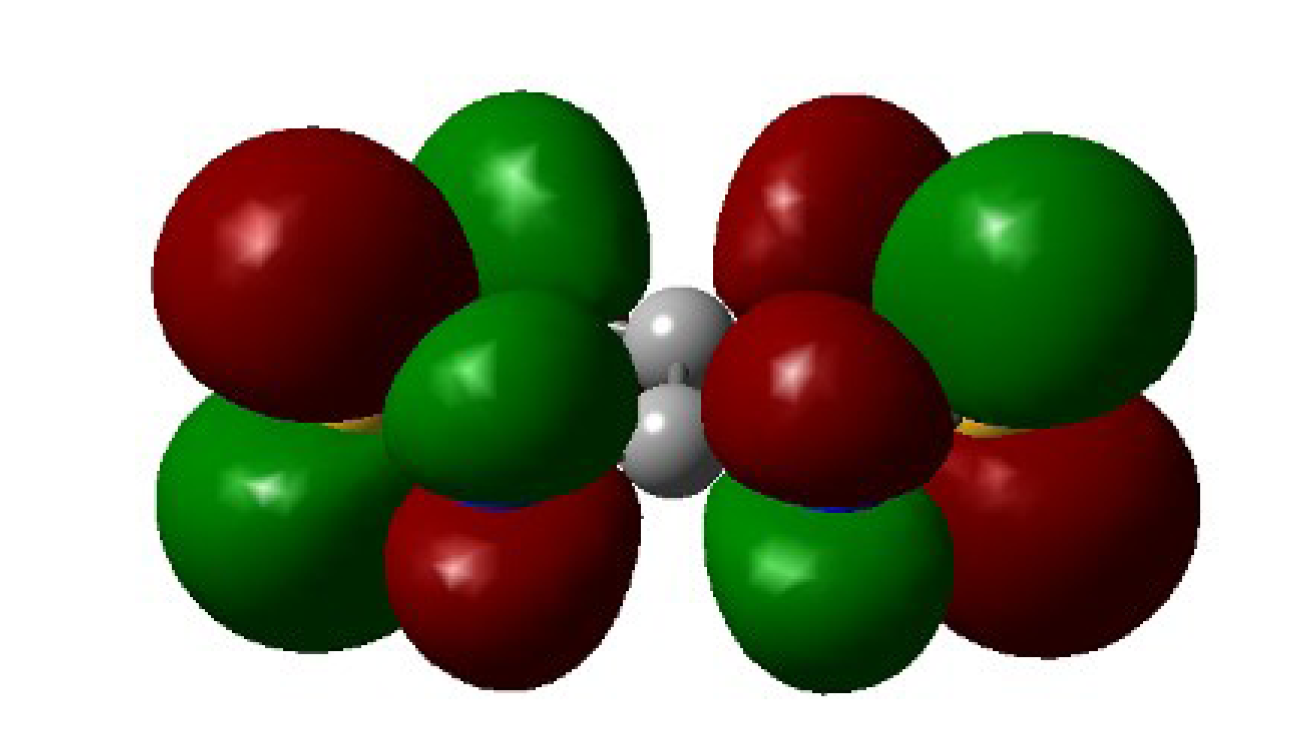
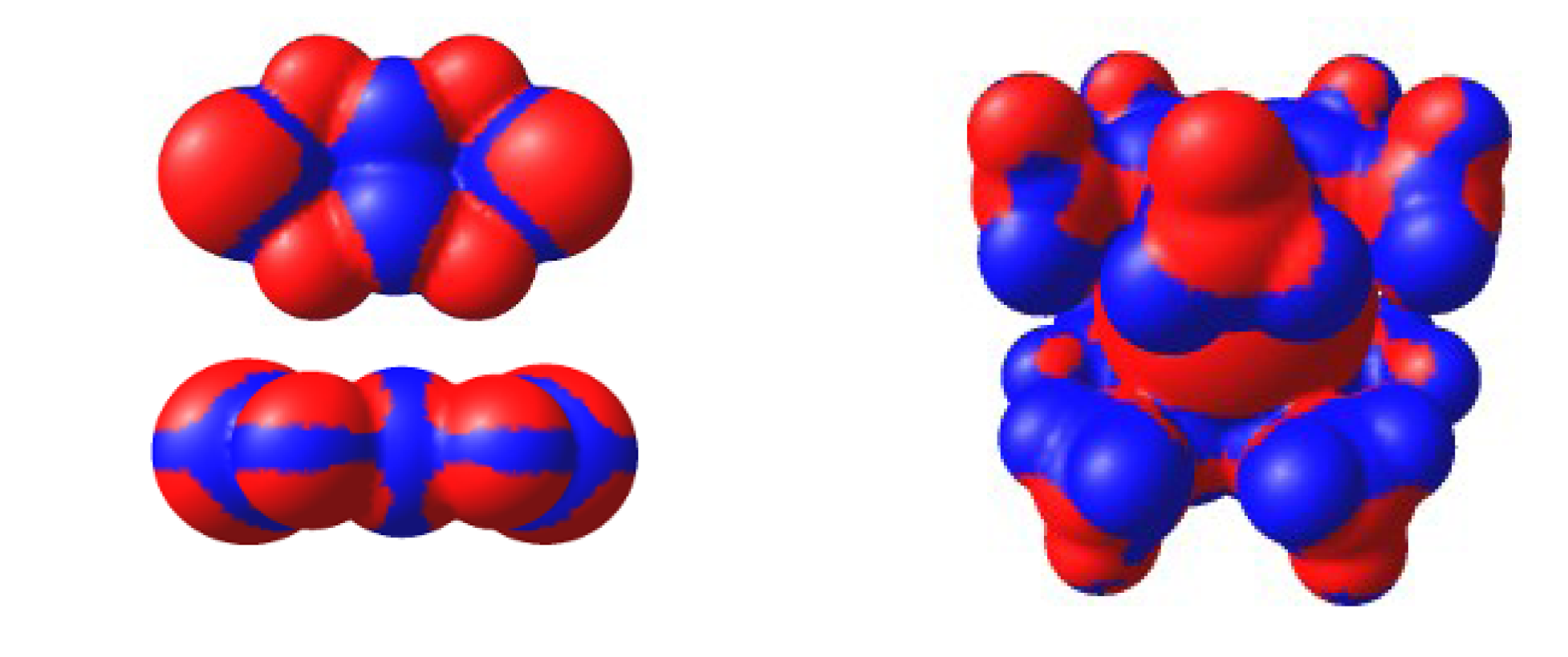
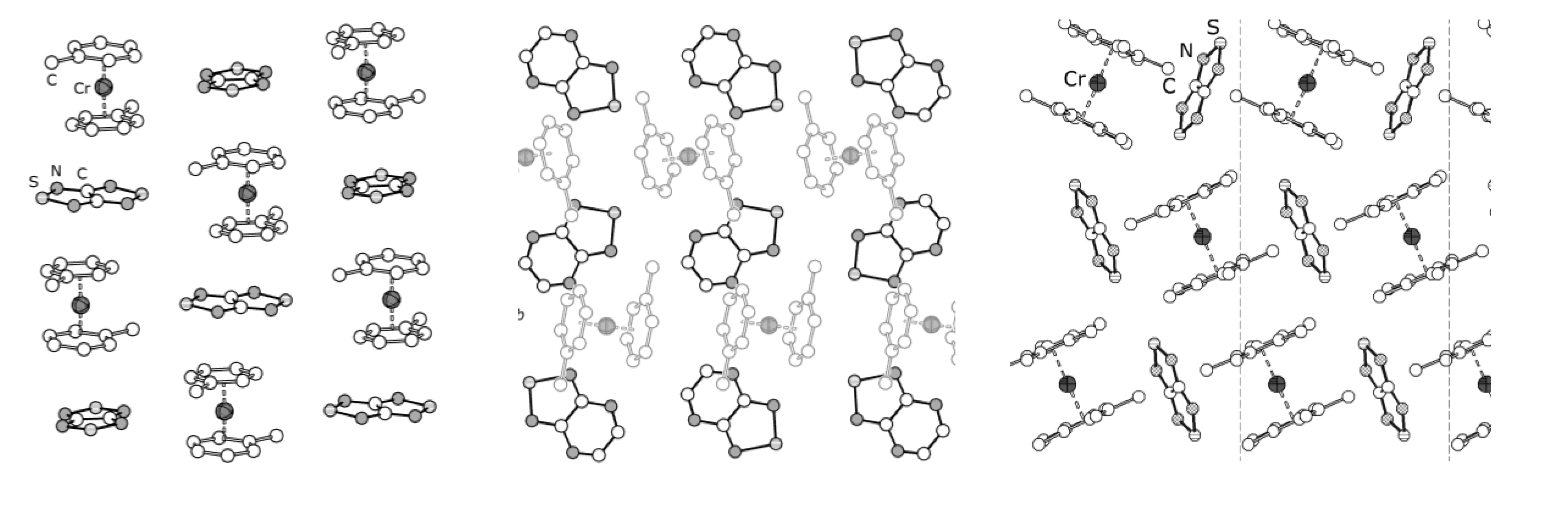
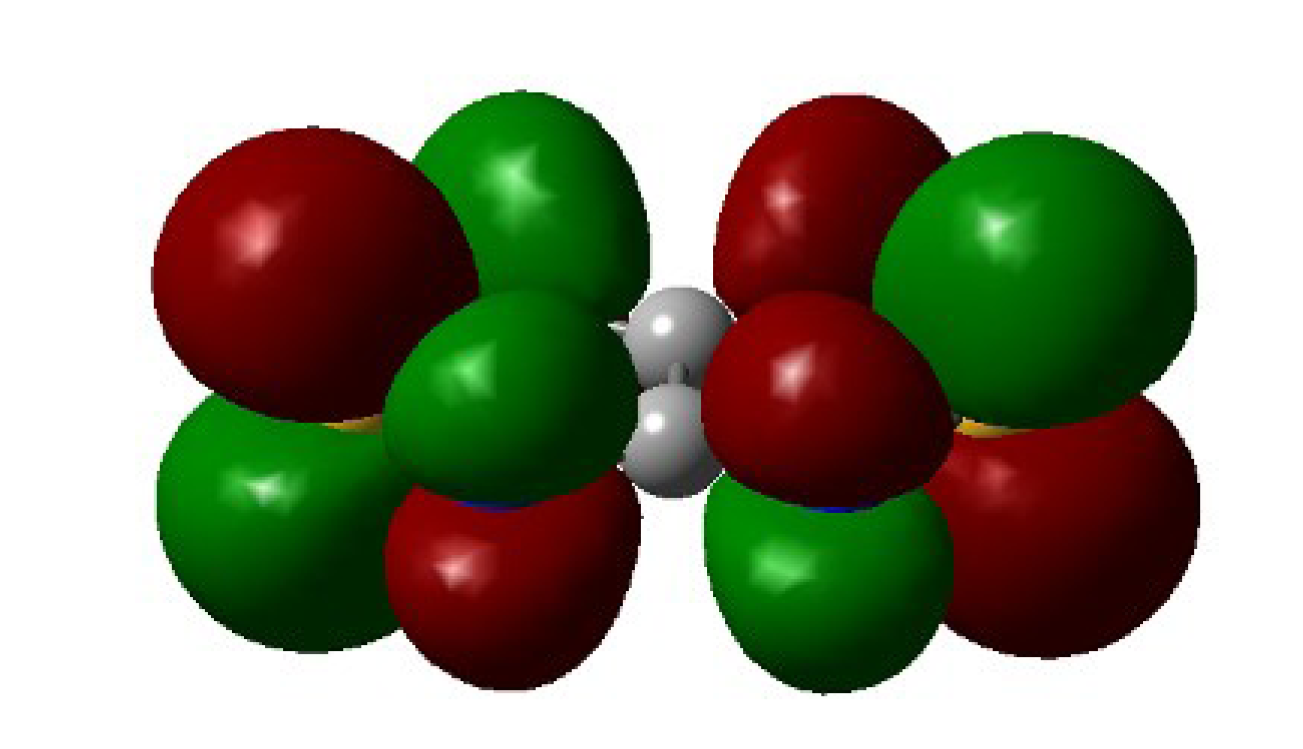
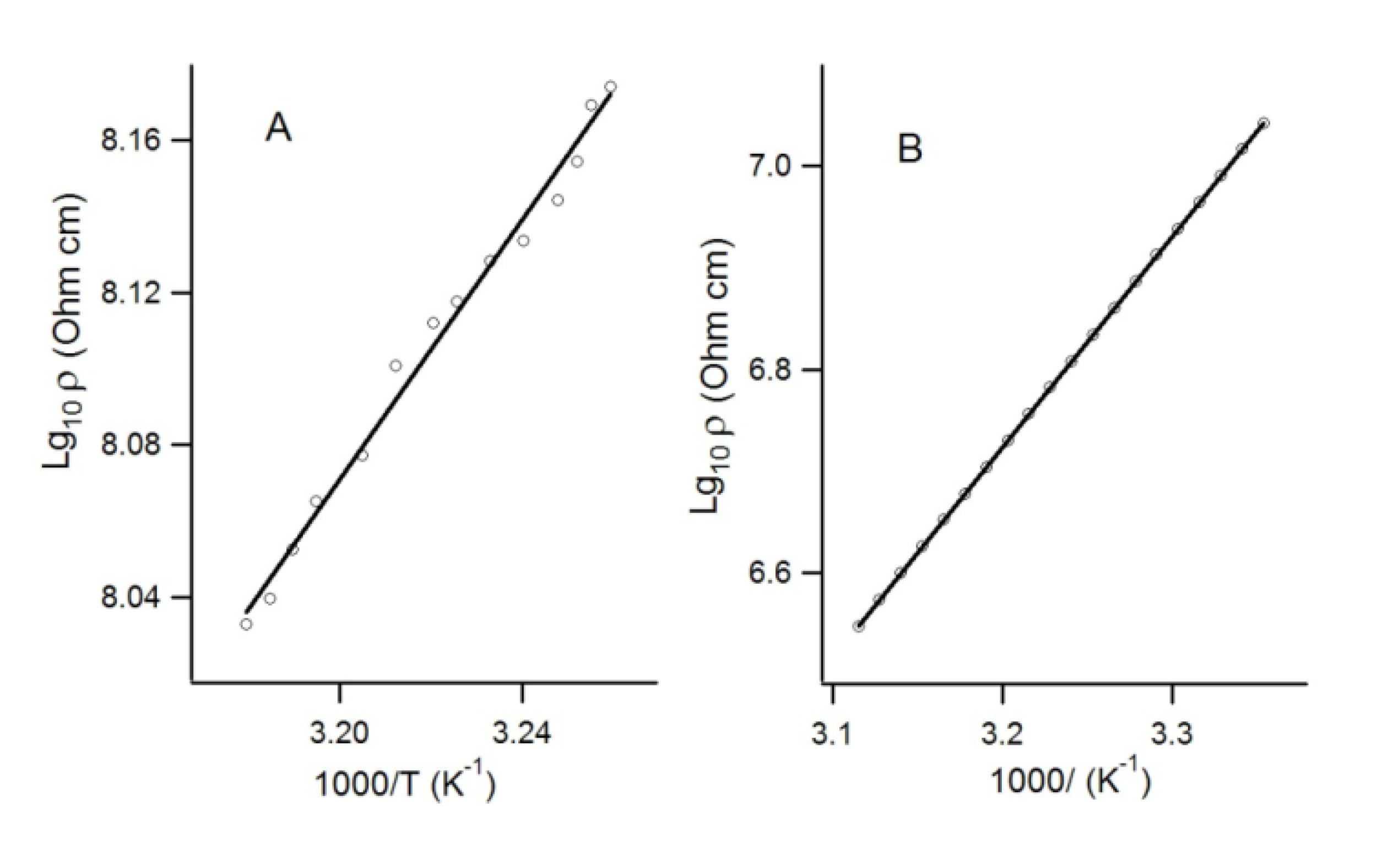
4.2. Electrical and Magnetic Properties
4.2.1. Charge-Transfer Complexes
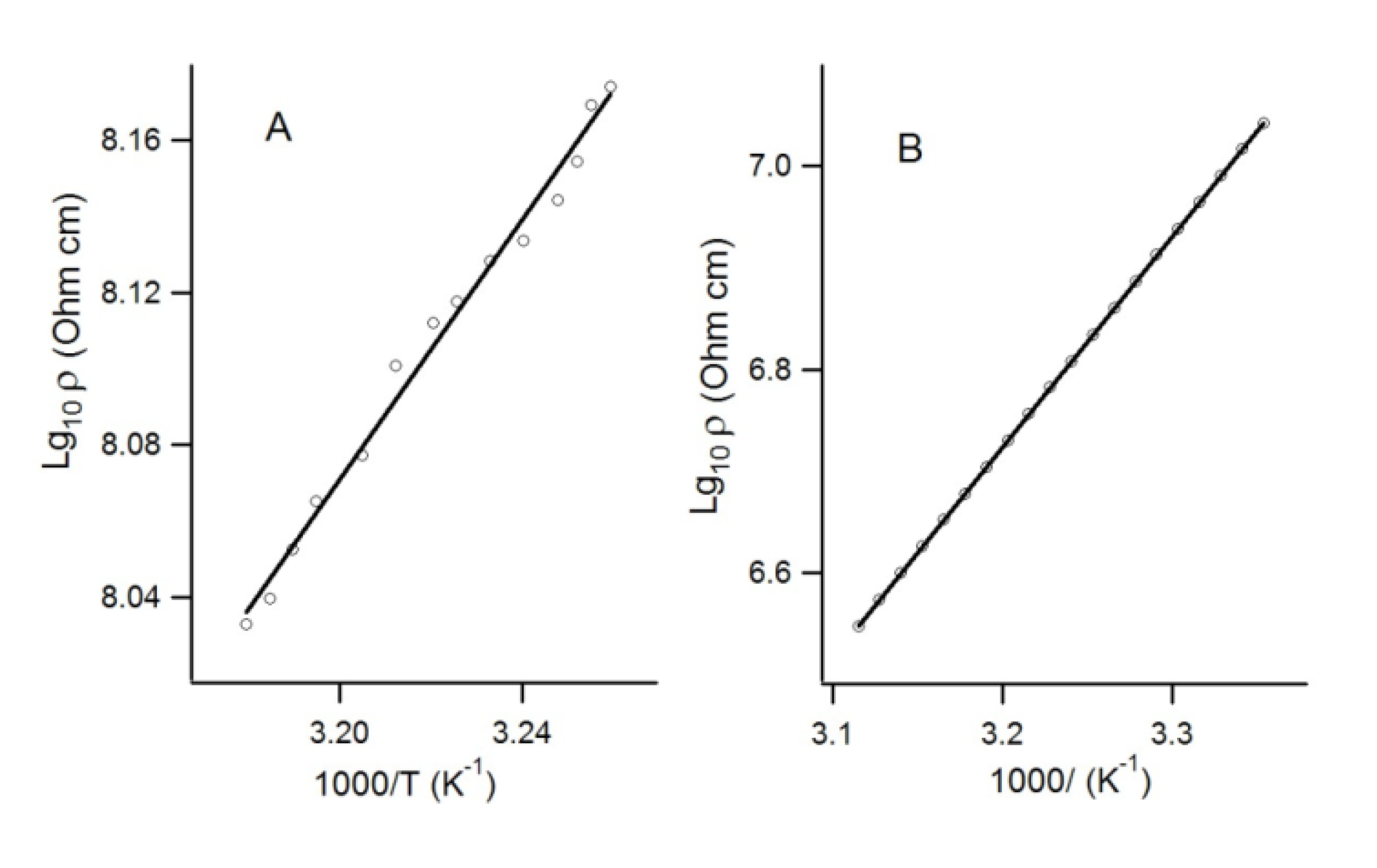
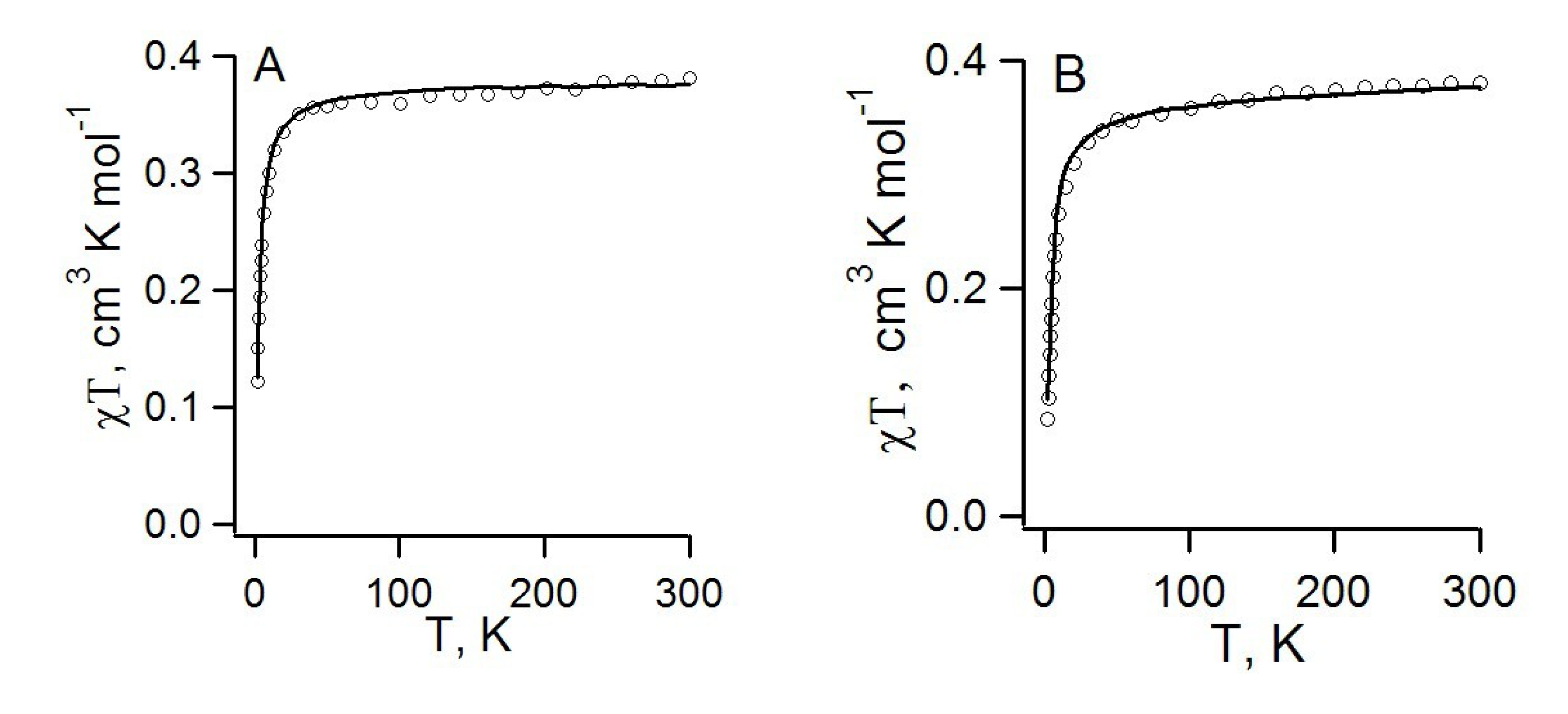
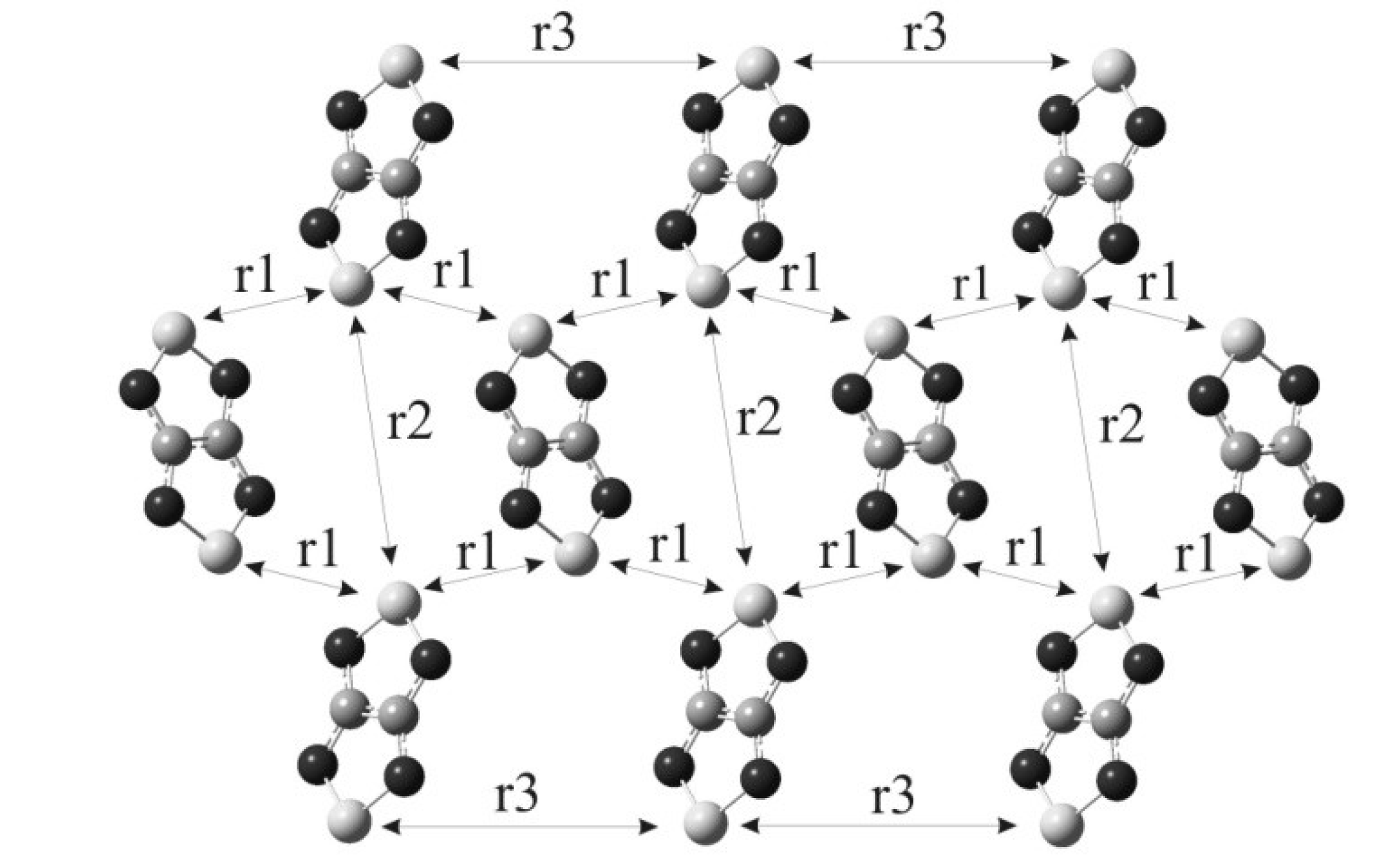
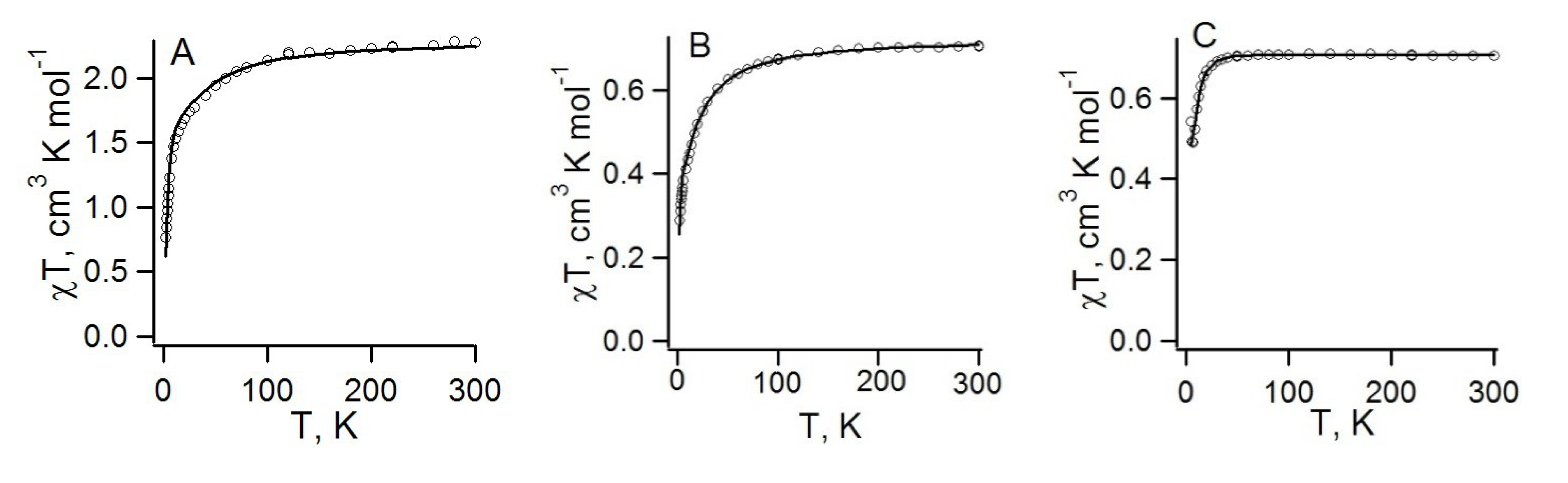
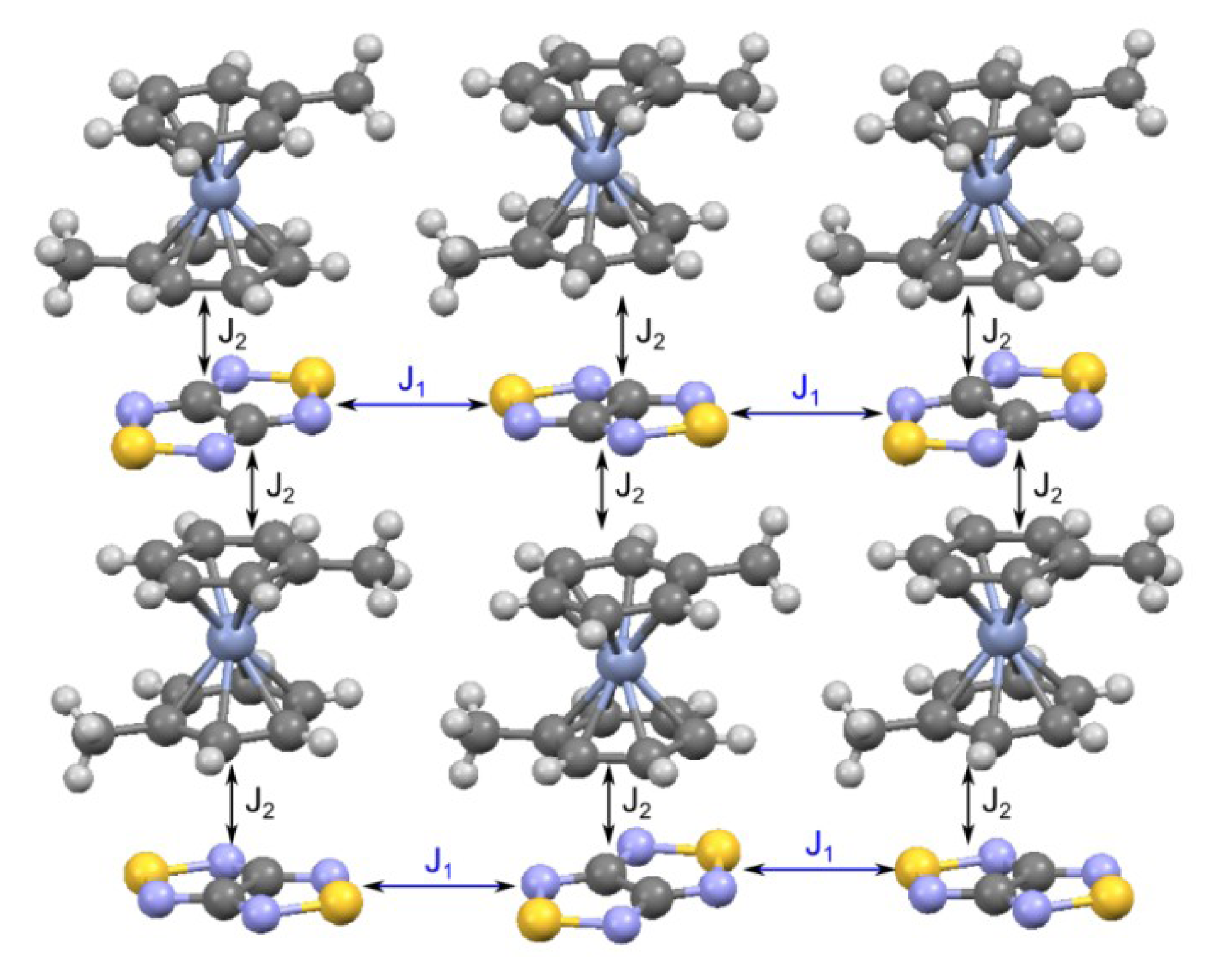
4.2.2. Radical-Anion Salts
| RA salt | TN, K | J, cm−1 | Refs. | |
|---|---|---|---|---|
| Experiment | Calculation | |||
| 69, [K(THF)][68] | 17 | −13.5, 7.2, −2.9 | −8.1, 7.2, 3.2, −1.0; a −9.0, 7.3, 1.9, −0.05; b −9.0, 4.8, −1.9, ~0; c | [74,88] |
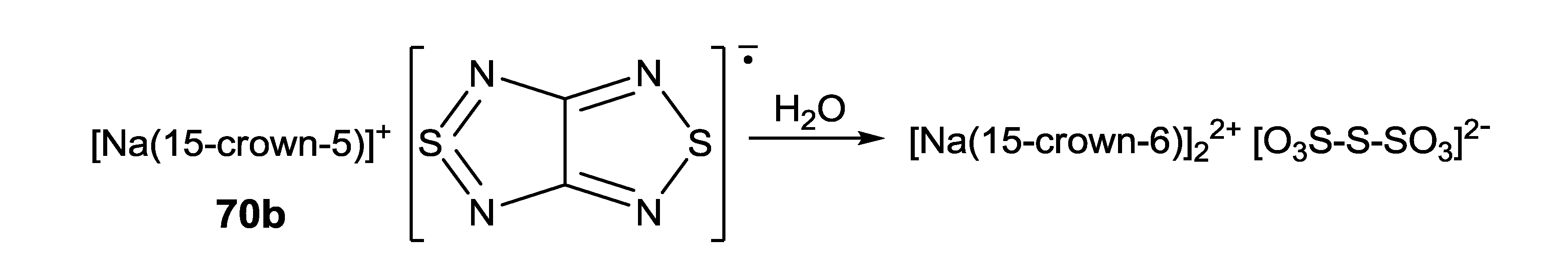
| 70b, [Na(15-crown-5)][22] | 8 | −3.42, −1.12 | - | [78] |
| 70c, [K(18-crown-6)][22] | < 2 | −1.22 | −2.13, a −1.62 b | [77,78,88] |
| 71, [K(18-crown-6)][26] | < 2 | −1.65 | −8.08, −4.02, −1.69 a <J> = 3.87 | [77] |
| 74, [CoCp2][22/] | 10 | 4.4 ± 0.6 | −2.8 a, −4.4 b | [75,88] |
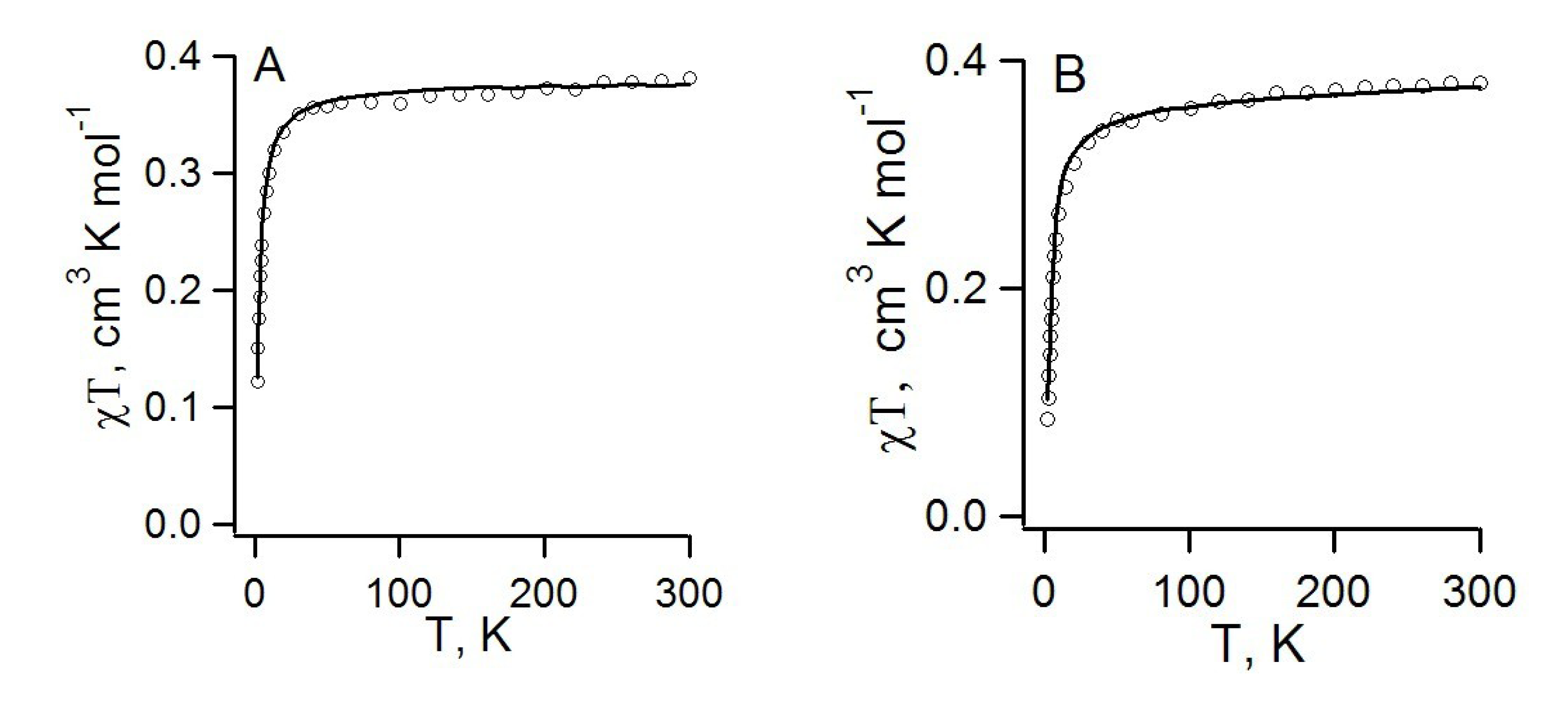
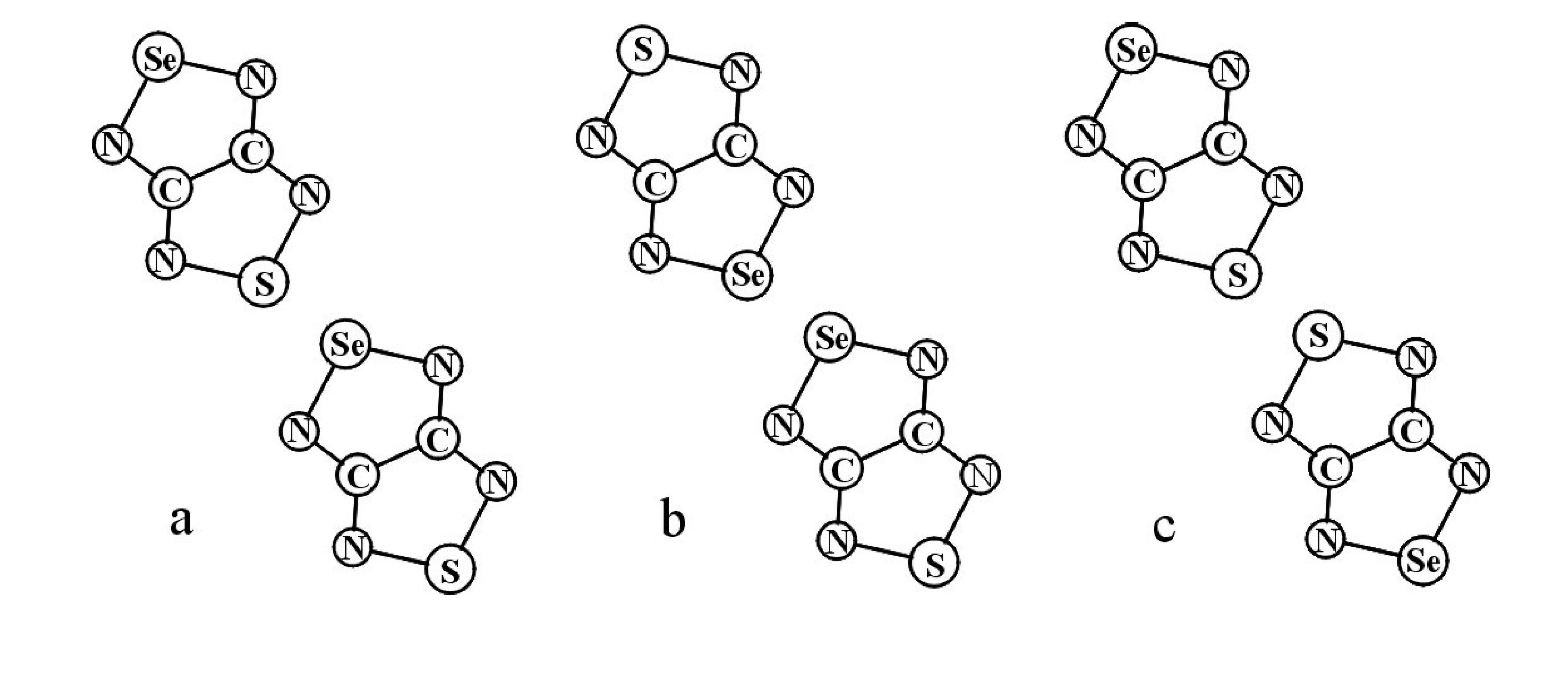


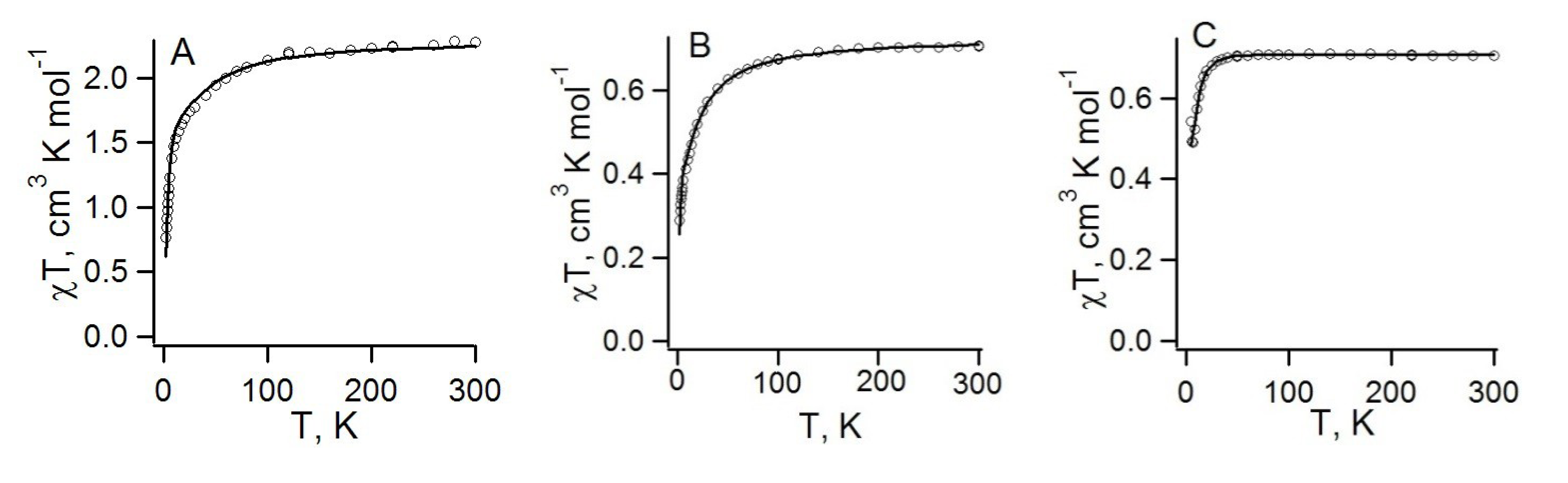
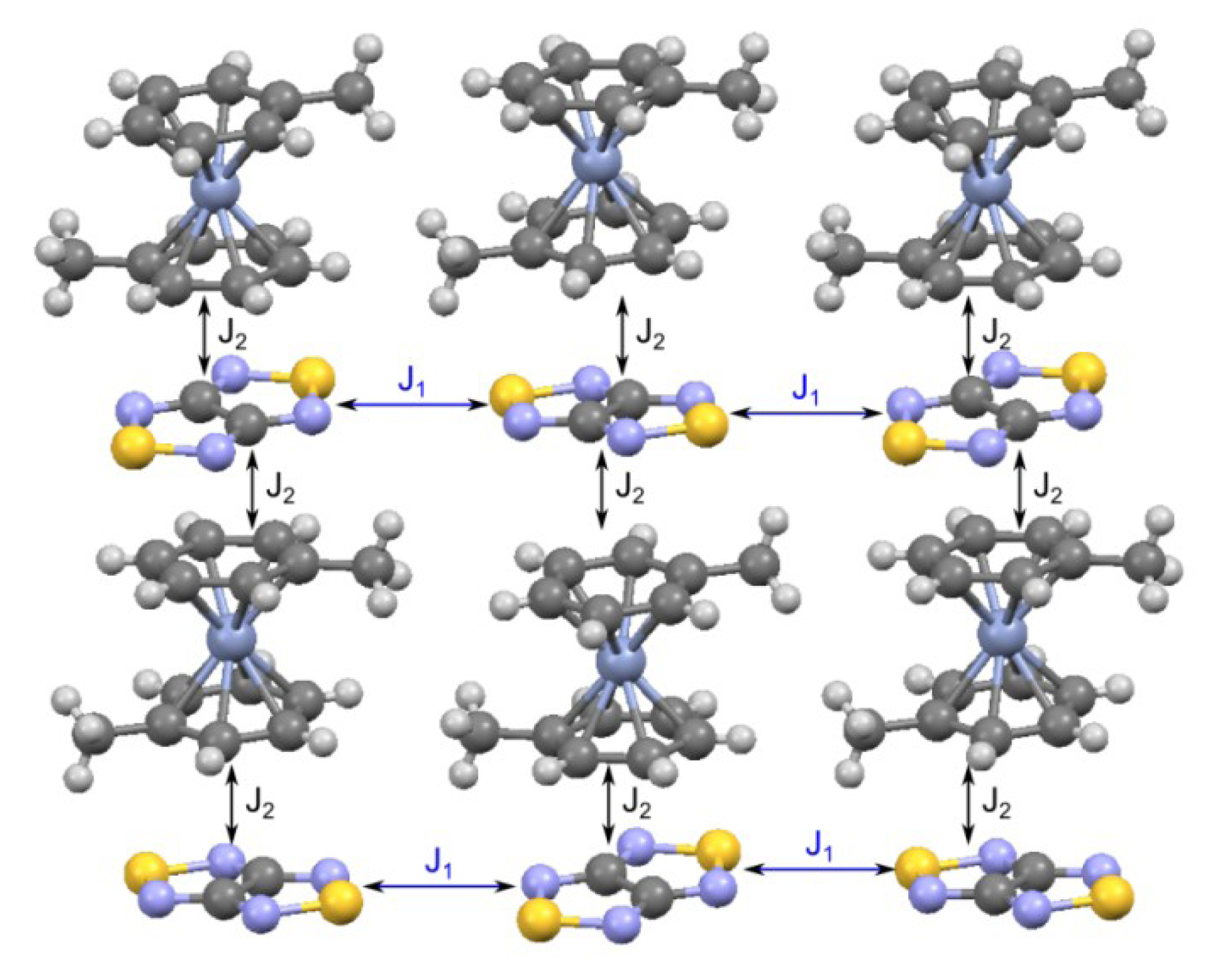
| RA salt | θW, K | J, см−1 | Refs. | |
|---|---|---|---|---|
| Experiment | Calculations | |||
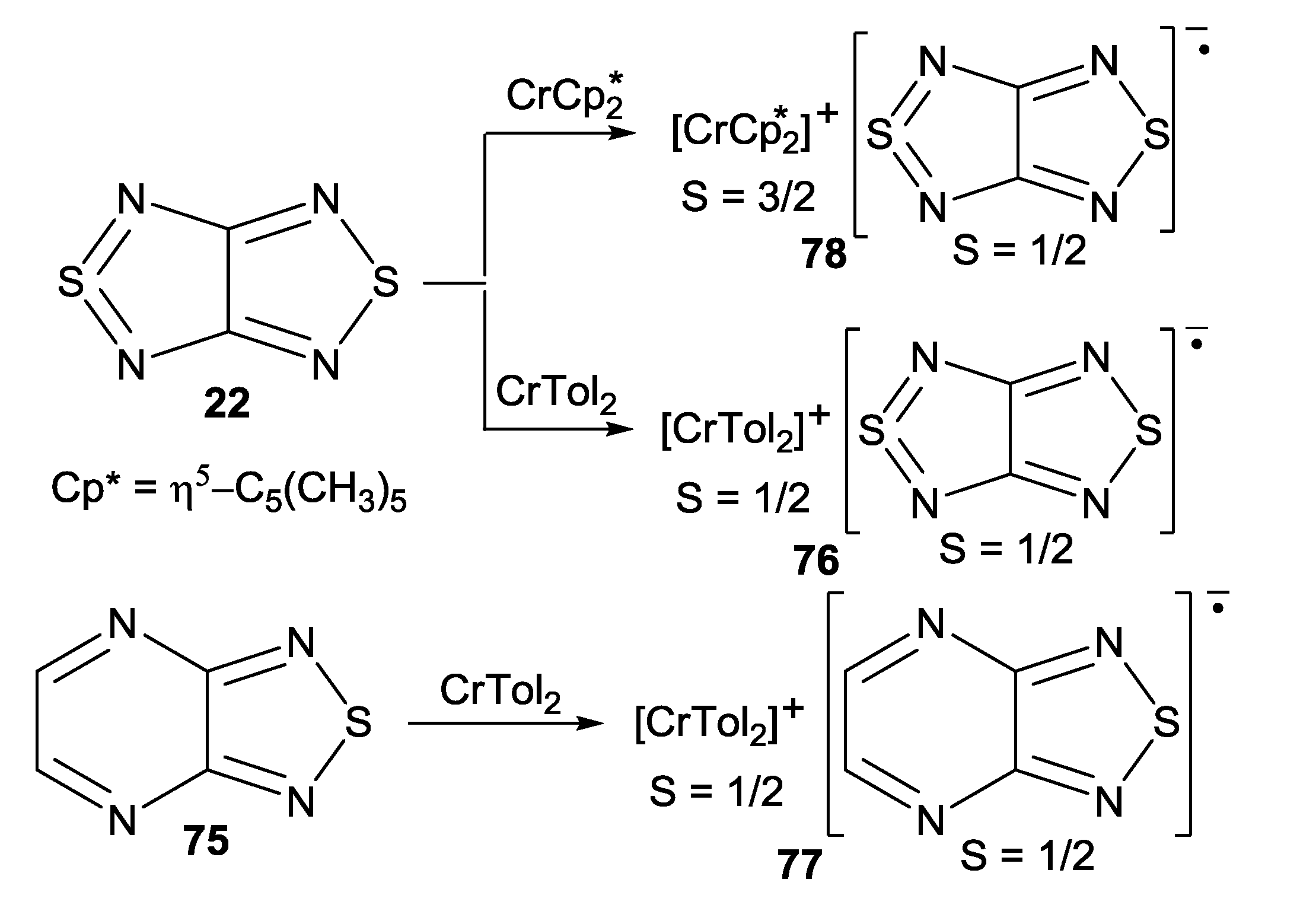
| 78, [CrCp*2][22] | −9.8 | −40 ± 9, −0.58 ± 0.03 | −61, −0.28; a −15.3, −0.08 b | [73] |
| 76, [CrTol2][22] | −7.1 K | −5.77, −0.84 | −8.25; a −7.45, −1.55 c | [71] |
| 77, [CrTol2][75] | 0.4 K | 9.72, d−7.96 d | 2.5, 0.7, −2.4, −8.3; c,e −0.60, −1.70, −2.20 c,f | [71] |
5. Summary and Future Perspectives
Acknowledgments
Conflicts of Interest
References and Notes
- Halcrow, M.A. (Ed.) Spin-Crossover Materials: Properties and Applications; Wiley: New York, NY, USA, 2013.
- Organic Spintronics; Vardeny, Z.V. (Ed.) CCR Press/Taylor & Francis: London, UK, 2010.
- Stable Radicals: Fundamentals and Applied Aspects of Odd Electron Compounds; Hicks, R.G. (Ed.) Wiley: New York, NY, USA, 2010.
- Organic Conductors, Superconductor, and Magnets: From Synthesis to Molecular Electronics; Ouahab, L.; Yagubskii, E. (Eds.) Kluwer: Dordrecht, The Netherlands, 2004.
- Ratera, I.; Veciana, J. Playing with organic radicals as building blocks for functional molecular materials. Chem. Soc. Rev. 2012, 41, 303–349. [Google Scholar] [CrossRef]
- Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Semiconducting π-conjugated systems in field-effect transistors: A molecular Odyssey of organic electronics. Chem. Rev. 2012, 112, 2208–2267. [Google Scholar] [CrossRef]
- Zhao, Y.; Ling, W.Z. Charge transfer in organic molecules for solar cells: Theoretical perspective. Chem. Soc. Rev. 2012, 41, 1075–1087. [Google Scholar] [CrossRef]
- Figueira-Duarte, T.M.; Muellen, K. Pyrene-based materials for organic electronics. Chem. Rev. 2011, 111, 7260–7314. [Google Scholar] [CrossRef]
- Anthony, J.E.; Facchetti, A.; Heeney, M.; Marder, S.T. n-Type organic semiconductors in organic electronics. Adv. Mater. 2010, 22, 3876–3892, (and other articles of this themed issue). [Google Scholar] [CrossRef]
- Miller, J.S. Zero-dimensional organic-based magnets possessing decamethylmetallocenes. J. Mater. Chem. 2010, 20, 1846–1857. [Google Scholar] [CrossRef]
- Miller, J.S. Oliver Kahn lecture: Composition and structure of the V[TCNE]x (TCNE—tetracyanoethylene) room-temperature organic-based magnet—a personal perspective. Polyhedron 2009, 28, 1596–1605. [Google Scholar] [CrossRef]
- Coropceanu, V.; Cornil, J.; Da Silva Filho, D.A.; Olivier, Y.; Silbey, R.; Bredas, J.-L. Charge transfer in organic semiconductors. Chem. Rev. 2007, 107, 926–952. [Google Scholar] [CrossRef]
- Bogani, L.; Wernsdorfer, W. Molecular spintronics using single-molecule magnets. Nature Mater. 2008, 7, 179–186. [Google Scholar] [CrossRef]
- Saito, G.; Yoshida, Y. Development of conductive organic molecular assemblies: Organic metals, superconductors, and exotic functional materials. B. Chem. Soc. Jpn 2007, 80, 1–137. [Google Scholar] [CrossRef]
- Todress, Z.V. Chalcogenadiazoles: Chemistry and Applications; CCR Press/Taylor & Francis: London, UK, 2012. [Google Scholar]
- Chivers, T.; Laitinen, R.S. Chalcogen-nitrogen chemistry. In Handbook of Chalcogen Chemistry: New Perspectives in Sulfur, Selenium and Tellurium; Devillanova, F., Ed.; RSC Press: Cambridge, UK, 2007; pp. 223–285. [Google Scholar]
- Chivers, T. A Guide to Chalcogen-Nitrogen Chemistry; World Scientific: Singapore, Singapore, 2005. [Google Scholar]
- Boere, R.T.; Roemmele, T.L. Electrochemistry of redox-active Group 15/16 heterocycles. Coordin. Chem. Rev. 2000, 210, 369–445. [Google Scholar] [CrossRef]
- Takaluoma, T.T.; Laasonen, K.; Laitinen, R.S. Molecular dynamics simulation of the solid-state topochemical polymerization of S2N2. Inorg. Chem. 2013, 52, 4648–4657. [Google Scholar] [CrossRef]
- Banister, A.J.; Gorrell, I.B. Poly(sulfur nitride): The first polymeric metal. Adv. Mater. 1998, 10, 1415–1429. [Google Scholar] [CrossRef]
- Labes, M.M.; Love, P.; Nichols, L.F. Polysulfur nitride—a metallic, superconducting polymer. Chem. Rev. 1979, 79, 1–15. [Google Scholar] [CrossRef]
- Makarov, A.Yu.; Blockhuys, F.; Bagryanskaya, I.Yu.; Gatilov, Yu.V.; Shakirov, M.M.; Zibarev, A.V. Experimental and computational study on the structure and properties of Herz cations and radicals: 1,2,3-Benzodithiazolium, 1,2,3-benzodithiazolyl and their Se congeners. Inorg. Chem. 2013, 52, 3699–3710. [Google Scholar] [CrossRef]
- Gritsan, N.P.; Makarov, A.Yu.; Zibarev, A.V. A new approach to chalcogen-nitrogen π-heterocyclic radicals. Appl. Magn. Reson. 2011, 41, 449–466. [Google Scholar] [CrossRef]
- Pivtsov, A.V.; Kulik, L.V.; Makarov, A.Yu.; Blockhuys, F. Pulse EPR and ENDOR study of 1,2,3-benzodithiazolyl, 2,1,3-benzothiaselenazolyl and 1,2,3-benzodiselenazolyl radicals. Phys. Chem. Chem. Phys. 2011, 13, 3873–3880. [Google Scholar] [CrossRef]
- Winter, S.M.; Balo, A.R.; Roberts, R.J.; Lekin, K.; Assoud, A.; Dube, P.A.; Oakley, R.T. Hybrid dithiazolothiadiazinyl radicals: Versatile building blocks for magnetic and conductive materials. Chem. Commun. 2013, 49, 1603–1605. [Google Scholar] [CrossRef]
- Lekin, K.; Leitch, A.A.; Tse, J.S.; Bao, X.; Secco, R.A.; Desgreniers, S.; Ohishi, Y.; Oakley, R.T. A pressure induced structural dichotomy in isostructural bis-1,2,3-thiaselenazolyl radical dimers. Cryst. Growth Des. 2012, 12, 4676–4684. [Google Scholar] [CrossRef]
- Mailman, A.; Winter, S.M.; Yu, X.; Robertson, C.M.; Yong, W.; Tse, J.S.; Secco, R.A.; Liu, Z.; Dube, P.A.; Howard, J.A.K.; Oakley, R.T. Crossing the insulator-to-metal barrier with a thiazyl radical conductor. J. Am. Chem. Soc. 2012, 134, 9886–9889, (and previous publications of this group). [Google Scholar] [CrossRef]
- Haynes, D.A. Crystal engineering with dithiadiazolyl radicals. CrystEngComm 2011, 13, 4793–4805. [Google Scholar] [CrossRef]
- Rawson, J.M.; Alberola, A. Stable chalcogen radicals. In Handbook of Chalcogen Chemistry: New Perspectives in Sulfur, Selenium and Tellurium; Devillanova, F., Ed.; RSC Press: Cambridge, UK, 2007; pp. 733–762. [Google Scholar]
- Awaga, K.; Tanaka, T.; Shirai, T.; Umezono, Y.; Fujita, W. Spin, charge and lattice correlation in thiazyl radicals and their molecular compounds. C. R. Chimie 2007, 10, 52–59. [Google Scholar] [CrossRef]
- Preuss, K.E. Metal complexes of thiazyl radicals. Dalton Trans. 2007, 2357–2369. [Google Scholar] [CrossRef]
- Awaga, K.; Tanaka, T.; Shirai, T.; Fujimori, M.; Suzuki, Y.; Yoshikava, H.; Fujita, W. Multi-dimensional crystal structures and unique solid-state properties of heterocyclic thiazyl radicals and related materials. B. Chem. Soc. Jpn 2006, 79, 25–34. [Google Scholar] [CrossRef]
- Okamoto, K.; Tanaka, T.; Fujita, W.; Awaga, K.; Inabe, T. Low-field negative-resistance effect in a charge-ordered state of thiazyl-radical crystals. Angew. Chem. Int. Edit. 2006, 45, 4516–4518. [Google Scholar] [CrossRef]
- Fujita, W.; Awaga, K. Room-temperature magnetic bistability in organic radical crystals. Science 1999, 286, 261–262. [Google Scholar] [CrossRef]
- Clarke, C.S.; Jornet-Somoza, J.; Mota, F.; Novoa, J.J.; Deumal, M. Origin of magnetic bistability in molecule-based magnets: A first-principles bottom-up study of the TTTA crystal. J. Am. Chem. Soc. 2010, 132, 17817–17830. [Google Scholar]
- Deumal, M.; Rawson, J.M.; Goetha, A.E.; Howard, J.A.K.; Copley, R.C.B.; Robb, M.A.; Novoa, J.J. Studying the origin of the antiferromagnetic to spin-canting transition in the β-NCC6F4CNSSN molecular magnet. Chem. Eur. J. 2010, 16, 2741–2750. [Google Scholar] [CrossRef]
- Deumal, M.; LeRoux, S.; Rawson, J.M.; Robb, M.A.; Novoa, J.J. A theoretical study of the magnetism of the α-p-cyano-tetrafluorophenyl-dithiadiazolyl radical using a first principles bottom-up procedure. Polyhedron 2007, 26, 1949–1958. [Google Scholar] [CrossRef]
- Rawson, J.M.; Alberola, A.; Whalley, A. Thiazyl radicals: Old materials for new molecular devices. J. Mater. Chem. 2006, 16, 2560–2575. [Google Scholar] [CrossRef]
- Rawson, J.M.; Luzon, J.; Palacio, F. Magnetic exchange interactions in perfluorophenyl dithiadiazolyl radicals. Coordin. Chem. Rev. 2005, 249, 2631–2641. [Google Scholar] [CrossRef]
- Shuvaev, K.V.; Passmore, J. RCNSSS+·: A novel class of stable sulfur rich radical cations. Coordin. Chem. Rev. 2013, 257, 1067–1091. [Google Scholar] [CrossRef]
- Cameron, T.S.; Decken, A.; Grein, F.; Knapp, C.; Passmore, J.; Rautiainen, J.M.; Shuvaev, K.V.; Thompson, R.C.; Wood, D.J. Preparation and characterization of (CNSSS)2(A)2 (A = AsF6−, SbF6−, Sb2F11−) containing the O2-like 5,5'-bis(1,2,3,4-trithiazolium) dication: The second example of a simple nonsterically hindered main-group diradical that retains its paramagnetism in the solid state. Inorg. Chem. 2010, 49, 7861–7879, (and previous publications of this group). [Google Scholar] [CrossRef]
- Rakitin, O.A. Stable heterocyclic radicals. Russ. Chem. Rev. 2011, 80, 647–659, (Usp. Khim. 2011, 80, 679–692). [Google Scholar] [CrossRef]
- Hicks, R.G. The synthesis and characterization of stable radicals containing the thiazyl (SN) fragment and their use as building blocks for advanced functional materials. In Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds; Hicks, R.G., Ed.; Wiley: New York, NY, USA, 2010; pp. 317–380. [Google Scholar]
- Constantinides, C.P.; Ioannou, T.A.; Koutentis, P.A. Manipulating the singlet-triplet energy gaps of arene-fused bis(1,2,3-dithiazoles): A computational study. Polyhedron. in press. [CrossRef]
- Boere, R.T.; Tuononen, H.; Chivers, T.; Roemmele, T.L. Structures and EPR spectra of binary sulfur-nitrogen radicals from DFT calculations. J. Organomet. Chem. 2007, 692, 2683–2696. [Google Scholar] [CrossRef]
- Carrington, A.; Howard, B.J.; Levy, D.H.; Robertson, J.C. Gas phase electron resonance spectrum of NS. Mol. Phys. 1968, 15, 187–192. [Google Scholar] [CrossRef]
- Uehara, H.; Morino, Y. Molecular structure of the NS free radical by gas-phase electron paramagnetic resonance. Mol. Phys. 1969, 17, 239–244. [Google Scholar] [CrossRef]
- Robertson, C.M.; Myles, D.J.T.; Leitch, A.A.; Reed, R.W.; Dooley, B.M.; Frank, N.L.; Dube, P.A.; Thompson, L.K.; Oakley, R.T. Ferromagnetism in a heavy atom heterocyclic radical conductor. J. Am. Chem. Soc. 2007, 129, 12688–12689. [Google Scholar] [CrossRef]
- Leitch, A.A.; Brusso, J.L.; Cvrkalj, K.; Reed, R.W.; Robertson, C.M.; Dube, P.A.; Oakley, R.T. Spin-canting in heavy atom heterocyclic radicals. Chem. Commun. 2007, 3368–3370. [Google Scholar]
- Mito, M.; Kawae, T.; Takeda, K.; Takagi, S.; Matsushida, Y.; Daguchi, H.; Rawson, J.M.; Palacio, F. Pressure-induced enhancement of the transition temperature of a genuine organic weak-ferromagnet up to 65 K. Polyhedron 2001, 20, 1509–1512. [Google Scholar] [CrossRef]
- Rawson, J.M.; Hayward, J.L. Reversible spin pairing in crystalline organic radicals. In Spin-Crossover Materials: Properties and Applications; Halcrow, M.A., Ed.; Wiley: New York, NY, USA, 2013; pp. 225–237. [Google Scholar]
- Shuku, Y.; Suizu, R.; Awaga, K.; Sato, U. Fe(II) spin crossover complex of [1,2,5]thiadiazolo[3,4-f][1,10]phenanthroline. CrystEngComm 2009, 11, 2065–2068. [Google Scholar] [CrossRef]
- Fujita, W.; Awaga, K. Paramagnetic-diamagnetic phase transition and magnetic ordering in thiazyl radicals. Synthetic Met. 2003, 137, 1263–1265. [Google Scholar] [CrossRef]
- Risto, M.; Assoud, A.; Winter, S.M.; Oilunkaniemi, R.; Laitinen, R.S.; Oakley, R.T. Heavy atom analogues of 1,2,3-dithiazolylium salts: Preparation, structures and redox chemistry. Inorg. Chem. 2008, 47, 10100–10109. [Google Scholar] [CrossRef]
- Cozzolino, A.F.; Gruhn, N.E.; Lichtenberer, D.L.; Vargas-Baca, I. Valence electronic structure of benzo-2,1,3-chalcogenadiazoles studied by photoelectron spectroscopy and density functional theory. Inorg. Chem. 2008, 47, 6220–6226. [Google Scholar] [CrossRef]
- Vasilieva, N.V.; Irtegova, I.G., Gritsan; Lonchakov, A.V.; Makarov, A.Yu.; Shundrin, L.A.; Zibarev, A.V. Redox properties and radical anions of fluorinated 2,1,3-benzothia(selena)diazoles and related compounds. J. Phys. Org. Chem. 2010, 23, 536–543. [Google Scholar] [CrossRef]
- Bock, H.; Haenel, P.; Neidlein, R. Radikalionen 80. Mehrfach thiadiazol-ueberbruekte benzol- und p-benzochinon-radikal anionen und ihre Cr-, Mo- und W-pentacarbonyl-komplexe. Phosphorus Sulfur 1988, 39, 235–252. [Google Scholar]
- Kaim, W. Mono- and binuclear pentacarbonyl complexes of chromium, molybdenum and tungsten with the anion radicals of 2,1,3-benzoxazole, 2,1,3-benzothiadiazole and 2,1,3-benzoselenadiazole. J. Organomet. Chem. 1984, 264, 317–326. [Google Scholar] [CrossRef]
- Hanson, P. Heteroaromatic radicals, part II: Radicals with Group VI and Groups V and VI ring heteroatoms. Adv. Heterocycl. Chem. 1980, 27, 31–149. [Google Scholar] [CrossRef]
- Kwan, C.L.; Carmack, M.; Kochi, J.K. Electron spin resonance spectra of the thiadiazolothiadiazole radical anion and related sulfur-nitrogen heterocycles. J. Phys. Chem. 1976, 80, 1786–1792. [Google Scholar] [CrossRef]
- Kamiya, M.; Akahori, Y. An electron spin resonance study of the anion radicals of 2,1,3-benzoselenadiazole and related compounds. B. Chem. Soc. Jpn. 1970, 43, 268–271. [Google Scholar] [CrossRef]
- Fajer, J.; Bielski, B.H.J.; Felton, R.H. Electronic and electron spin resonance spectra of the perfluoro-2,1,3-benzoselenadiazole anion radical. J. Phys. Chem. 1968, 72, 1281–1288. [Google Scholar] [CrossRef]
- Atherton, N.M.; Ockwell, J.N.; Dietz, R. Electron spin resonance and polarographic studies of the radical anions of some nitrogen- and sulphur-containing heterocyclic molecules. J. Chem. Soc. A 1967, 771–777. [Google Scholar] [CrossRef]
- Strom, E.T.; Russell, G.A. The electron spin resonance spectra of 2,1,3-benzoxadiazole, -benzothiadiazole, and -benzoselenadiazole radical anions. Electron withdrawal by Group VI elements. J. Am. Chem. Soc. 1965, 87, 3326–3329. [Google Scholar]
- Gerson, F.; Plattner, G.; Bartetzko, R.; Gleiter, R. The radical ions of naphtho[1,8-c,d][1,2,6]thiadiazine and some of its derivatives. Helv. Chim. Acta 1980, 63, 2144–2151. [Google Scholar] [CrossRef]
- Motokawa, N.; Miyasaka, H.; Yamashita, M.; Dunbar, K.R. An electron-transfer ferromagnet with Tc = 107 K based on a three-dimensional [Ru2]2/TCNQ system. Angew. Chem. Int. Ed. 2008, 47, 7760–7763. [Google Scholar] [CrossRef]
- Yamashida, Y.; Tomura, T. Highly polarized electron donors, acceptors and donor-acceptor compounds for organic conductors. J. Mater. Chem. 1998, 8, 1933–1944. [Google Scholar] [CrossRef]
- Tsubata, Y.; Suzuki, T.; Yamashita, Y.; Mikai, T.; Miyashi, T. Tetracyanoquinodimethanes fused with 1,2,5-thiadiazole and pyrazine units. Heterocycles 1992, 33, 337–348. [Google Scholar] [CrossRef]
- Yamashita, Y.; Y. Mikai, Y.; Miyashi, T.; Saito, G. A new type of two-dimensional organic conductors: Alkylammonium bis[1,2,5]thiadiazolotetracyanoquinonodimethanides. B. Chem. Soc. Jpn. 1988, 61, 483–493. [Google Scholar] [CrossRef]
- Zibarev, A.V.; Mews, R. A new class of paramagnetics: 1,2,5-Chalcogenadiazolydil salts as potential building blocks for molecular magnets and conductors. In Selenium and Tellurium Chemistry: From Small Molecules to Biomolecules and Materials; Woollins, J.D., Laitinen, R.S., Eds.; Springer: Berlin, Germany, 2011; pp. 123–149. [Google Scholar]
- Semenov, N.A.; Pushkarevsky, N.A.; Suturina, E.A.; Chulanova, E.A.; Kuratieva, N.V.; Bogomyakov, A.S.; Irtegova, I.G.; Vasilieva, N.V.; Konstantinova, L.S.; Gritsan, N.P.; Rakitin, O.A.; Ovcharenko, V.I.; Konchenko, S.N.; Zibarev, A.V. Bis(toluene)chromium(I) [1,2,5]thiadiazolo[3,4-c][1,2,5]thiadiazolidyl and [1,2,5]thiadiazolo[3,4-b]pyrazinidyl: New heterospin (S1 = S2 = 1/2) radical-ion salts. Inorg. Chem. 2013, 52, 6654–6663. [Google Scholar] [CrossRef]
- Gritsan, N.P.; Zibarev, A.V. Synthesis and properties of chalcogen-nitrogen π-heterocyclic radical-anion salts. Russ. Chem. B. 2011, 60, 2131–2140, (Izv. Akad. Nauk. Ser. Khim. 2011, 2091–2100). [Google Scholar] [CrossRef]
- Semenov, N.A.; Pushkarevsky, N.A.; Lonchakov, A.V.; Bogomyakov, A.S.; Pritchina, E.A.; Suturina, E.A.; Gritsan, N.P.; Konchenko, S.N.; Mews, R.; Ovcharenko, V.I.; Zibarev, A.V. Heterospin π-heterocyclic radical-anion salt: Synthesis, structure and magnetic properties of decamethylchromocenium [1,2,5]thiadiazolo[3,4-c][1,2,5]thiadiazolidyl. Inorg. Chem. 2010, 49, 7558–7564. [Google Scholar] [CrossRef]
- Konchenko, S.N.; Gritsan, N.P.; Lonchakov, A.V.; Radius, U.; Zibarev, A.V. Isolation of 2,1,3-benzothiadiazolidyl radical anion: X-ray structure and properties of a [K(THF)][C6H4N2S] salt. Mendeleev Commun. 2009, 19, 7–9. [Google Scholar] [CrossRef]
- Konchenko, S.N.; Gritsan, N.P.; Lonchakov, A.V.; Irtegova, I.G.; Mews, R.; Ovcharenko, V.I.; Radius, U.; Zibarev, A.V. Cobaltocenium [1,2,5]thiadiazolo[3,4-c][1,2,5]thiadiazolidyl: Synthesis, structure and magnetic properties. Eur. J. Inorg. Chem. 2008, 3833–3838. [Google Scholar]
- Gritsan, N.P.; Lonchakov, A.V.; Lork, E.; Mews, R.; Pritchina, E.A.; Zibarev, A.V. Diamagnetic π-dimers of [1,2,5]thiadiazolo[3,4-c][1,2,5]thiadiazolidyl radical anion in the crystalline state: Preparation and X-ray crystal structure of a [(Me2N)2CC(NMe2)2]2+[(C2N4S2)2]2− salt. Eur. J. Inorg. Chem. 2008, 1994–1998. [Google Scholar]
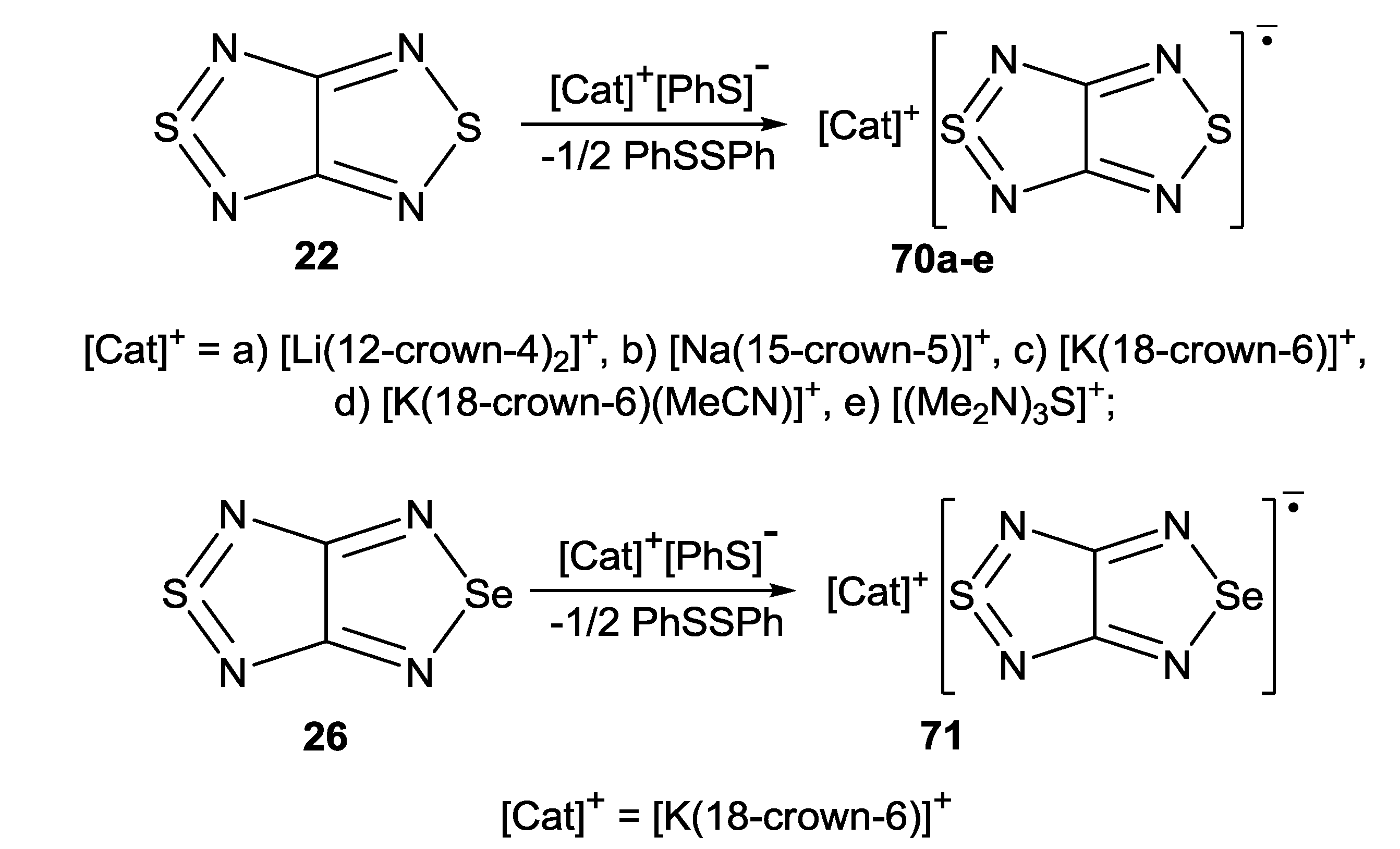
- Bagryanskaya, I.Yu.; Gatilov, Yu.V.; Gritsan, N.P.; Ikorskii, V.N.; Irtegova, I.G.; Lonchakov, A.V.; Lork, E.; Mews, R.; Ovcharenko, V.I.; Semenov, N.A.; Vasilieva, N.V.; Zibarev, A.V. [1,2,5]Selenadiazolo[3,4-c][1,2,5]thiadiazole and [1,2,5]selenadiazolo[3,4-c][1,2,5]thiadiazolidyl — a synthetic, structural and theoretical study. Eur. J. Inorg. Chem. 2007, 4751–4761. [Google Scholar]
- Ikorskii, V.N.; Irtegova, I.G.; Lork, E.; Makarov, A.Yu.; Mews, R.; Ovcharenko, V.I.; Zibarev, A.V. Early alkali metal (Li, Na, K) and tris(dimethylamino)sulfonium (TAS) salts of [1,2,5]thiadiazolo[3,4-c][1,2,5]thiadiazolidyl radical anion: Rational syntheses, structures and magnetic propertie. Eur. J. Inorg. Chem. 2006, 3061–3067. [Google Scholar]
- Makarov, A.Yu.; Irtegova, I.G.; Vasilieva, N.V.; Bagryanskaya, I.Yu.; Borrmann, T.; Gatilov, Yu.V.; Lork, E.; Mews, R.; Stohrer, W.D.; Zibarev, A.V. [1,2,5]Thiadiazolo[3,4-c][1,2,5]thiadiazolidyl: A long-lived radical anion and its stable salts. Inorg. Chem. 2005, 44, 7194–7199. [Google Scholar] [CrossRef]
- Makarov, A.Yu.; Chulanova, E.A.; Pushkarevsky, N.A.; Semenov, N.A.; Lonchakov, A.V.; Bogomyakov, A.S.; Irtegova, I.G.; Vasilieva, N.V.; Lork, E.; Gritsan, N.P.; Konchenko, S.N.; Ovcharenko, V.I.; Zibarev, A.V. A novel sulfur-nitrogen π-heterocyclic radical anion, (6H-1,2,3-benzodithiazol-6-ylidene)malononitrilidyl, and its homo- and heterospin salts. Polyhedron. to be submitted..
- Hunter, J.A.; King, B.; Lindsell, W.E.; Neish, M.A. One-electron reduction of di-imidosulphur compounds, S(NR)2, and some complexes of group 6A metal carbonyl derivatives containing S(NR)2 ligands: Studies of the radical products by electron spin resonance spectroscopy. J. Chem. Soc., Dalton 1980, 880–887. [Google Scholar]
- Rees, C.W. Polysulfur-nitrogen heterocyclic chemistry. J. Heterocycl. Chem. 1992, 29, 639–651. [Google Scholar] [CrossRef]
- Jones, R.; Morris, J.L.; Rees, C.W.; Williams, D.J. Tetrathiatetraza-azulene: Synthesis and X-ray crystal structure. Chem. Commun. 1985, 1654–1656. [Google Scholar]
- Koutentis, P.A.; Rees, C.W. Reaction of Herz salts with malononitrile: A general route to (6H-1,2,3-benzodithiazol-6-ylidene)malononitriles. J. Chem. Soc., Perkin Trans. 1 2002, 315–319. [Google Scholar] [CrossRef]
- English, R.F.; Morris, J.L.; Rees, C.W. Tetrathiatetraza-azulene. ARKIVOC 2000, 3, 228–239. [Google Scholar] [CrossRef]
- Morris, J.L.; Rees, C.W. Electron reach 10π and 14π heteroaromatic systems. Pure Appl. Chem. 1986, 58, 197–210. [Google Scholar] [CrossRef]
- Rienstra-Kiracofe, J.C.; Tschumper, G.S.; Schaefer, H.F.; Nandi, S.; Ellison, G.B. Atomic and molecular electron affinities: Photoelectron experiments and theoretical computations. Chem. Rev. 2002, 102, 231–282. [Google Scholar] [CrossRef]
- Lonchakov, A.V. Molecular design of precursors of the chalcogen-nitrogen heterocyclic radical anions and theoretical analysis of the magnetic properties of their salts. Ph. D. Thesis, Institute of Chemical Kinetics and Combustion, Russian Academy of Sciences, Novosibirsk, Russia, 2013. [Google Scholar]
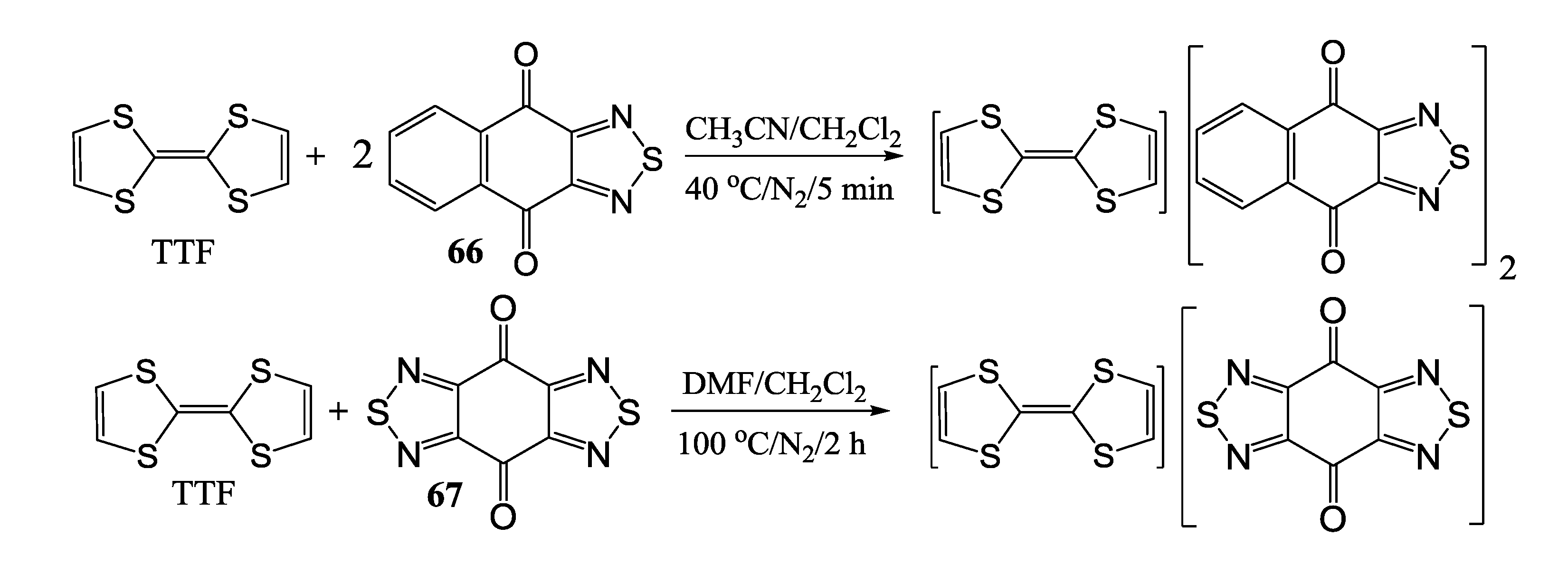
- Pushkarevsky, N.A.; Lonchakov, A.V.; Semenov, N.A.; Lork, E.; Buravov, L.I.; Konstantinova, L.S.; Silber, T.G.; Robertson, N.; Gritsan, N.P.; Rakitin, O.A.; et al. First charge-transfer complexes between tetrathiafulvalene and 1,2,5-chalcogenadiazole derivatives: Design, synthesis, crystal structures, electronic and electrical properties. Synthetic Met. 2012, 162, 2267–2276. [Google Scholar] [CrossRef]
- Kebarle, P.; Chowdhury, S. Electron affinities and electron-transfer reactions. Chem. Rev. 1987, 87, 513–534. [Google Scholar] [CrossRef]
- Yamashita, Y.; Saito, K.; Suzuki, T.; Kabuto, C.; Mikai, T.; Miyashi, T. Bis([1,2,5]thiadiazolo)[3,4-b;3’,4’-e]pyrazine: A novel 14π-electron heterocycle with high electron affinity. Angew. Chem. Int. Edit. 1988, 27, 434–435. [Google Scholar] [CrossRef]
- Strunskaya, E.I.; Yanilkin, V.V.; Bredikhina, Z.A.; Nastapova, N.V.; Morozov, V.I.; Maksimyuk, N.I.; Sharafutdinova, D.R.; Bredikhin, A.A. Electrochemical reduction and oxidation of 3,4-disubstituted 1,2,5-thiadiazoles. Rus. J. Gen. Chem. 2003, 73, 806–815. [Google Scholar] [CrossRef]
- Suzuki, T.; Tsuji, T.; Okubo, T.; Okada, A.; Obana, Y.; Fukushima, T.; Miyashi, T.; Yamashita, Y. Preparation, structure, and amphoteric redox properties of p-phenylenediamine-type dyes fused with a chalcogenadiazole unit. J. Org. Chem. 2001, 66, 8954–8960. [Google Scholar] [CrossRef]
- Sherman, E.O.; Lambert, S.M.; Pilgram, K. Polarography of some 2,1,3-benzothiadiazoles, benzofurazans, 2,1,3-benzoselenadiazoles, and 3,4-disubstituted and fused 1,2,5-thiadiazoles. J. Heterocycl. Chem. 1974, 11, 763–796. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamashita, Y.; Fukushima, T.; Miyashi, T. Tetracyanoquinodimethanes fused with a [1,2,5]chalcogenadiazole ring. Mol. Cryst. Liq. Cryst. 1997, 296, 165–180. [Google Scholar] [CrossRef]
- Suzuki, T.; Fujii, H.; Yamashita, Y.; Kabuto, C.; Tanaka, S.; Harasawa, M.; Mukai, T.; Miyashi, T. Clathrate formation and molecular recognition by novel chalcogen-cyano interactions in tetracyanoquinodimethanes fused with thiadiazole and selenadiazole rings. J. Am. Chem. Soc. 1992, 114, 3034–3043. [Google Scholar]
- Semenov, N.A.; Pushkarevsky, N.A.; Beckmann, J.; Finke, P.; Lork, E.; Mews, R.; Bagryanskaya, I.Y.; Gatilov, Yu.V.; Konchenko, S.N.; Vasiliev, V.G.; et al. Tellurium-nitrogen π-heterocyclic chemistry — synthesis, structure, and reactivity toward halides and pyridine of 3,4-dicyano-1,2,5-telluradiazole. Eur. J. Inorg. Chem. 2012, 3693–3703. [Google Scholar]
- Gieren, A.; Lamm, V.; Huebner, T.; Rabben, M.; Neidlein, R.; Droste, D. Charge-transfer-komplexe von tetrathiafulvalen (TTF) mit 1,2,5-thiadiazolchinonen. Chem. Ber. 1984, 117, 1940–1953. [Google Scholar] [CrossRef]
- Morgan, J.S.; Jennings, M.; Vindigni, A.; Clerac, R.; Preuss, K.E. TDNQ][CoCp*2] and [TDNQ]3[CoCp2]2: Radical anions of a 1,2,5-thiadiazolo-naphthoquinone. Cryst. Growth Des. 2011, 11, 2520–2527. [Google Scholar] [CrossRef]
- Weinstock, L.M.; Davis, P; Handelsmann, B.; Tull, R. General synthetic system for 1,2,5-thiadiazoles. J. Org. Chem. 1967, 32, 2823–2840. [Google Scholar] [CrossRef]
- Koutentis, P.A. 1,2,5-Thiadiazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: Oxford, 2008; Volume 5, pp. 516–564. [Google Scholar]
- Koutentis, P.A. 1,2,5-Thiadiazoles and related compounds. In Science of Synthesis; Storr, R.C., Gilchrist, T.L., Eds.; Thieme: Stuttgart, Germany, 2003; Volume 13, pp. 297–348. [Google Scholar]
- Neto, B.A.D.; Lapis, A.A.M.; da Silva Júnior, E.N.; Dupont, J. 2,1,3-Benzothiadiazole and derivatives: Synthesis, Properties, Reactions, and applications in light technology of small molecules. Eur. J. Org. Chem. 2013, volume, 228–235. [Google Scholar]
- Xiaoxia, S.; Xiaoxia, S.; Yali, R. Synthesis of a novel acceptor unit for D-A type polymers: 4,8-Dibromobenzo[1,2-c;4,5-c']bis[1,2,5]thiadiazole. Adv. Mater. Res. 2012, 482-484, 1221–1224. [Google Scholar] [CrossRef]
- Du, X.; Qi, J.; Zhang, Z.; Ma, D.; Wang, Z.Y. Efficient non-doped near infrared organic light-emitting devices based on fluorophores with aggregation-induced emission enhancement. Chem. Mater. 2012, 24, 2178–2185. [Google Scholar] [CrossRef]
- Bashirov, D.A.; Sukhikh, T.S.; Kuratieva, N.V.; Naumov, D.Yu.; Konchenko, S.N.; Semenov, N.A.; Zibarev, A.V. Iridium complexes with 2,1,3-benzothiadiazole and related ligands. Polyhedron 2012, 42, 168–174. [Google Scholar] [CrossRef]
- Ioannou, T.A.; Koutentis, P.A.; Krassos, H.; Loizou, G.; Lo Re, D. Some cyclization reactions of 1,3-diphenylbenzo[e][1,2,4]triazin-7(1H)-one: Preparation and computational analysis of non- symmetrical zwitterionic biscyanines. Org. Biomol. Chem. 2012, 10, 1339–1348. [Google Scholar] [CrossRef]
- Noujeim, N.; Zhu, K.; Vukotic, V.N.; Loeb, S.J. [2]Pseudorotaxanes from T-shaped benzimidazolium axles and [24]crown-8 wheels. Org. Lett. 2012, 14, 2484–2487. [Google Scholar] [CrossRef]
- Shin, W.; Jo, M.Y.; You, D.S.; Jeong, Y.S.; Yoon, D.Y.; Kang, J.W.; Cho, J.H.; Lee, G.D.; Hong, S.S.; Kim, J.H. Improvement of efficiency of polymer solar cell by incorporation of the planar shaped monomer in low band gap polymer. Synthetic Met. 2012, 162, 768–774. [Google Scholar] [CrossRef]
- Sonar, P.; Foong, T.R.B.; Singh, S.P.; Li, Y.; Dodabalapur, A. A furan-containing conjugated polymer for high mobility ambipolar organic thin film transistors. Chem. Commun. 2012, 48, 8383–8385. [Google Scholar]
- Friebe, C.; Wild, A.; Perelaer, J.; Schubert, U.S. Inkjet printing of zinc(II) bis-2,2':6',2''-terpyridine metallopolymers: Printability and film-forming studies by a combinatorial thin-film library approach. Macromol. Rapid Comm. 2012, 33, 503–509. [Google Scholar] [CrossRef]
- Hemgesberg, M.; Ohlmann, D.M.; Schmitt, Y.; Wolfe, M.R.; Mueller, M.K.; Erb, B.; Sun, Y.; Goossen, L.J.; Gerhards, M.; Thiel, W.R. Simple access to sol-gel precursors bearing fluorescent aromatic core units. Eur. J. Org. Chem. 2012, 2142–2151. [Google Scholar]
- Osaka, I.; Shimawaki, M.; Mori, H.; Doi, I.; Miyazaki, E.; Koganezawa, T.; Takimiya, K. Synthesis, characterization, and transistor and solar cell applications of a naphthobisthiadiazole-based semiconducting polymer. J. Am. Chem. Soc. 2012, 134, 3498–3507. [Google Scholar] [CrossRef]
- Lin, L.Y.; Tsai, C.H.; Lin, F.; Huang, T.W.; Chou, S.H.; Wu, C.C.; Wong, K.T. 2,1,3-Benzothiadiazole-containing donor-acceptor-acceptor dyes for dye-sensitized solar cells. Tetrahedron 2012, 68, 7509–7516. [Google Scholar] [CrossRef]
- Shi, L.; He, C.; Zhu, D.; He, Q.; Li, Y.; Chen, Y.; Sun, Y.; Fu, Y.; Wen, D.; Cao, H.; Cheng, J. High performance aniline vapor detection based on multi-branched fluorescent triphenylamine-benzothiadiazole derivatives: Branch effect and aggregation control of the sensing performance. J. Mater. Chem. 2012, 22, 11629–11635. [Google Scholar]
- Lee, K.; Lee, J.; Jeong, E.J.; Kronk, A.; Elenitoba-Johnson, K.S.J.; Lim, M.S.; Kim, J. Conjugated polyelectrolyte-antibody hybrid materials for highly fluorescent live cell-imaging. Adv. Mater. 2012, 24, 2479–2484. [Google Scholar]
- Garo, F.; Haener, R. 2,1,3-Benzothiadiazole-modified DNA. Eur. J. Org. Chem. 2012, 2801–2808. [Google Scholar] [CrossRef]
- Huber, J.; Jung, C.; Mecking, S. Nanoparticles of low optical band gap conjugated polymers. Macromolecules 2012, 45, 7799–7805. [Google Scholar] [CrossRef]
- Sun, Y.; Welch, G.C.; Leong, W.L.; Takacs, C.J.; Bazan, G.C.; Heeger, A.J. Solution-processed small-molecule solar cells with 6.7% efficiency. Nature Mater. 2012, 11, 44–48. [Google Scholar]
- Henson, Z.B.; Welch, G.C.; van der Poll, T.; Bazan, G.C. Pyridylthiadiazole-based narrow band gap chromophores. J. Am. Chem. Soc. 2012, 134, 3766–3779. [Google Scholar] [CrossRef]
- Fan, J.; Yuen, J.D.; Wang, M.; Seifter, J.; Seo, J.H.; Mohebbi, A. R.; Zakhidov, D.; Heeger, A.; Wudl, F. High-performance ambipolar transistors and inverters from an ultralow bandgap polymer. Adv. Mater. 2012, 24, 2186–2190. [Google Scholar] [CrossRef]
- Chang, D.W.; Ko, S.J.; Kim, G.H.; Bae, S.Y.; Kim, J.Y.; Dai, L.; Baek, J.B. Molecular engineering of conjugated polymers for solar cells and field-effect transistors: Side-chain versus main-chain electron acceptors. J. Polymer Sci. A 2012, 50, 271–279. [Google Scholar] [CrossRef]
- Cho, N.; Song, K.; Lee, J.K.; Ko, J. Facile synthesis of fluorine-substituted benzothiadiazole-based organic semiconductors and their use in solution-processed small-molecule organic solar cells. Chem. Eur. J. 2012, 18, 11433–11439. [Google Scholar] [CrossRef]
- Konstantinova, L.S.; Knyazeva, E.A.; Obruchnikova, N.V.; Gatilov, Yu.V.; Zibarev, A.V.; Rakitin, O.A. Reaction of vicinal nitroamines with sulfur monochloride — a short and convenient route to fused 1,2,5-thiadiazoles and their N-oxides. Tetrahedron Lett. 2013, 54, 3075–3078. [Google Scholar] [CrossRef]
- Semenov, N.A.; Bagryanskaya, I.Yu.; Alekseev, A.V.; Gatilov, Yu.V.; Lork, E.; Mews, R.; Roeschenthaler, G.V.; Zibarev, A.V. New molecular complexes of trimeric perfluoro-ortho-phenylenemercury with heterocyclic compounds. J. Struct. Chem. 2010, 51, 552–557, (Zh. Strukt. Khim. 2010, 51, 569–574). [Google Scholar] [CrossRef]
- Li, Q.; Sun, D.; Zhou, Y.; Liu, D.; Zhang, Q.; Liu, J. Anticancer activity of novel ruthenium complex with 1,10-phenanthrolineselenazole as potent telomeric G-quadruplex inhibitor. Inorg. Chem. Commun. 2012, 20, 142–146. [Google Scholar] [CrossRef]
- Goswami, S.; Hazra, A.; Das, M.K. Selenodiazole-fused diacetamidopyrimidine, a selective fluorescence sensor for aliphatic monocarboxylates. Tetrahedron Lett. 2010, 51, 3320–3323. [Google Scholar] [CrossRef]
- Goswami, S.; Hazra, A.; Chakrabarty, R.; Fun, H.K. Recognition of carboxylate anions and carboxylic acids by selenium-based new chromogenic fluorescent sensor: A remarkable fluorescence enhancement of hindered carboxylates. Org. Lett. 2009, 11, 4350–4353. [Google Scholar] [CrossRef]
- Cillo, C.M.; Lash, T.D. Benzo[1,2-c:3,4-c']bis[1,2,5]selenadiazole, [1,2,5]selenadiazolo[3,4-e]-2,1,3-benzothiadiazole, furazanobenzo-2,1,3-thiadiazole, furazanobenzo-2,1,3-selenadiazole and related heterocyclic systems. J. Heterocyclic Chem. 2004, 41, 955–962. [Google Scholar] [CrossRef]
- Cheng, Y.J.; Chen, C.H.; Ho, Y.J.; Chang, S.W.; Witek, H.A.; Hsu, C.S. Thieno[3,2-b]pyrrolo donor fused with benzothiadiazolo, benzoselenadiazolo and quinoxalino acceptors: Synthesis, characterization, and molecular properties. Org. Lett. 2011, 13, 5484–5487. [Google Scholar] [CrossRef]
- Coombs, B.A.; Lindner, B.D.; Edkins, R.M.; Rominger, F.; Beeby, A.; Bunz, U.H.F. Photophysical property trends for a homologous series of bis-ethynyl-substituted benzochalcogenadiazoles. New J. Chem. 2012, 36, 550–553. [Google Scholar] [CrossRef]
- Padhy, H.; Sahu, D.; Chiang, I.H.; Patra, D.; Lin, H.C.; Kekuda, D.; Chu, C.W. Synthesis and applications of main-chain Ru(II) metallo-polymers containing bis-terpyridyl ligands with various benzodiazole cores for solar cells. J. Mater. Chem. 2011, 21, 1196–1205. [Google Scholar] [CrossRef]
- Ishii, T.; Ikeda, K.; Kichise, Y.; Ogawa, M. Red-light-emitting system based on aggregation of donor-acceptor derivatives in polar aqueous media. Chem. Asian J. 2012, 7, 1553–1557. [Google Scholar] [CrossRef]
- Edwards, R.; Cummins, I.; Steel, P. Method and means relating to multiple herbicide resistance in plants. WO2009/34396. 2010. [Google Scholar]
- Reish, M.E.; Nam, S.; Lee, W.; Woo, H.Y.; Gordon, K.C. A spectroscopic and DFT study of the electronic properties of carbazole-based D-A type copolymers. J. Phys. Chem. C 2012, 116, 21255–21266. [Google Scholar] [CrossRef]
- Gibson, G.L.; McCormick, T.M.; Seferos, D.S. Atomistic band gap engineering in donor-acceptor polymers. J. Am. Chem. Soc. 2012, 134, 539–547. [Google Scholar] [CrossRef]
- Chen, L.; Wang, L.; Jing, X.; Wang, F. Color tuning of novel 2,1,3-naphthothiadiazole and 2,1,3-benzoselenadiazole based D-A-D' type dopants to realize highly efficient saturated red emission in non-polar solvents. J. Mater. Chem. 2011, 21, 10265–10267. [Google Scholar] [CrossRef]
- Baran, D.; Oktem, G.; Celebi, S.; Toppare, L. Neutral-state green conjugated polymers from pyrrole bis-substituted benzothiadiazole and benzoselenadiazole for electrochromic devices. Macromol. Chem. Phys. 2011, 212, 799–805. [Google Scholar] [CrossRef]
- Mikroyannidis, J.A.; Suresh, P.; Sharma, G.D. Synthesis of benzoselenadiazole-based small molecule and phenylenevinylene copolymer and their application for efficient bulk heterojunction solar cells. Org. Electron. 2010, 11, 311–321. [Google Scholar] [CrossRef]
- Zhao, W.; Cai, W.; Xu, R.; Yang, W.; Gong, X.; Wu, H.; Cao, Y. Novel conjugated alternating copolymer based on 2,7-carbazole and 2,1,3-benzoselenadiazole. Polymer 2010, 51, 3196–3202. [Google Scholar] [CrossRef]
- Icli, M.; Pamuk, M.; Algi, F.; Onal, A.M.; Cihaner, A. Donor-acceptor polymer electrochromes with tunable colors and performance. Chem. Mater. 2010, 22, 4034–4044. [Google Scholar] [CrossRef]
- Das, S.; Pati, P.B.; Zade, S.S. Cyclopenta[c]thiophene-based D-A conjugated copolymers: Effect of heteroatoms (S, Se, and N) of benzazole acceptors on the properties of polymers. Macromolecules 2012, 45, 5410–5417. [Google Scholar] [CrossRef]
- Jung, I.H.; Kim, H.; Park, M.J.; Kim, B.; Park, J.H.; Jeong, E.; Woo, H.Y.; Yoo, S.; Shim, H.K. Synthesis and characterization of cyclopentadithiophene-based low bandgap copolymers containing electron-deficient benzoselenadiazole derivatives for photovoltaic devices. J. Polymer Sci. A 2010, 48, 1423–1432. [Google Scholar]
- Berionni, G.; Pegot, B.; Marrot, J.; Goumont, R. Supramolecular association of 1,2,5-chalcogenadiazoles: An unexpected self-assembled dissymetric [Se···N]2 four-membered ring. CrystEngComm 2009, 11, 986–988. [Google Scholar] [CrossRef]
- Milios, C.J.; Ioannou, P.V.; Raptopoulou, C.P.; Papaefstathiou, G.S. Crystal engineering with 2,1,3-benzoselenadiazole and mercury(II) chloride. Polyhedron 2009, 28, 3199–3202. [Google Scholar] [CrossRef]
- Mukherjee, G.; Singh, P.; Ganguri, C.; Sharma, S.; Singh, H.B.; Goel, N.; Singh, U.P.; Butcher, R.J. Selenadiazolopyridine: A synthon for supramolecular assembly and complexes with metallophilic interactions. Inorg. Chem. 2012, 51, 8128–8140. [Google Scholar] [CrossRef]
- Yuan, F.; Chen, X.; Zhou, Y.; Yang, F.; Zhang, Q.; Liu, J. Synthesis of a ruthenium(II) polypyridine complex with 1,10-phenanthrolineselenazole as ligand and investigation of its G-quadruplex DNA-binding properties. J. Coord. Chem. 2012, 65, 1246–1257. [Google Scholar] [CrossRef]
- Cozzolino, A.F.; Yang, Q.; Vargas-Baca, I. Engineering second-order nonlinear optical activity by means of a noncentrosymmetric distortion of the [Te-N]2 supramolecular synthon. Cryst. Growth Des. 2010, 10, 4959–4964. [Google Scholar] [CrossRef]
- Cozzolino, A.F.; Britten, J.F.; Vargas-Baca, I. The effect of steric hindrance on the association of telluradiazoles through Te-N secondary bonding interactions. Cryst. Growth Des. 2006, 6, 181–186. [Google Scholar] [CrossRef]
- Cozzolino, A.F.; Bain, A.D.; Hanhan, S.; Vargas-Baca, I. N-Triphenylboryl- and N,N'-bis(triphenylboryl)benzo-2,1,3-telluradiazole. Chem. Commun. 2009, 4043–4045. [Google Scholar]
- Kovtonyuk, V.N.; Makarov, A.Yu.; Shakirov, M.M.; Zibarev, A.V. A polyfluoroaromatic tellurium-nitrogen compound: Synthesis and properties of 4,5,6,7-tetrafluoro-2,1,3-benzotelluradiazole. Chem. Commun. 1996, 1991–1992. [Google Scholar]
- Cozzolino, A. F.; Whitfield, P. S.; Vargas-Baca, I. Supramolecular chromotropism of the crystallinephases of 4,5,6,7-tetrafluorobenzo-2,1,3-telluradiazole. J. Am. Chem. Soc. 2010, 132, 17265–17270. [Google Scholar] [CrossRef]
- Badyal, K.; Herr, M.; McWhinnie, W.R.; Hamor, T.A.; Paxton, K. Investigation of heterocyclic systems containing nitrogen-tellurium bonds. Phosphorus Sulfur 1998, 141, 221–229. [Google Scholar] [CrossRef]
- Chivers, T.; Gao, X.; Parvez, M. Preparation, crystal structures, and isomerization of the tellurium diimide dimers RNTe(μ-NR’)2TeNR (R = R’ = tBu; R = PPh2NSiMe3, R’ = tBu, tOct): X-ray structure of the telluradiazole dimer [tBu2C6H2N2Te]2. Inorg. Chem. 1996, 35, 9–15. [Google Scholar] [CrossRef]
- Risto, M.; Reed, R.W.; Robertson, C.M.; Oilunkaniemi, R.; Laitinen, R.S.; Oakley, R.T. Self-association of the N-methyl benzotelluradiazolylium cation: Implications for the generation of super-heavy atom radicals. Chem. Commun. 2008, 3278–3280. [Google Scholar]
- Semenov, N.A.; Pushkarevsky, N.A.; Lonchakov, A.V.; Gritsan, N.P.; Lork, E.; Beckmann, J.; Zibarev, A.V. Hypercoordination of anions to heavier 1,2,5-chalcogenadiazoles. Manuscript in preparation. 2013. [Google Scholar]
- Dutton, J.L.; Ragogna, P.J. Donor-acceptor chemistry at heavy chalcogen centers. Inorg. Chem. 2009, 48, 1722–1730. [Google Scholar] [CrossRef]
- Stuzhin, P.A.; Mikhailov, M.S.; Yurina, E.S.; Bazanov, M.I.; Koifman, O.I.; Pakhomov, G.L.; Travkin, V.V.; Sinelshchikova, A.A. First tellurium-containing phthalocyanine analogues: Strong effect of tellurium on spectral, redox and conductivity properties of porphyrazines with annulated chalcogenadiazole ring(s). Chem. Commun. 2012, 48, 10135–10137. [Google Scholar] [CrossRef]
- Codding, P.W.; Koenig, H.; Oakley, R.T. The crystal and molecular structures of 1,2,7,9-tetrathia-3,6,8,10-tetraazacyclohepta[e]indene, a novel tricyclic carbon-sulfur-nitrogen ring. Can. J. Chem. 1983, 61, 1562–1566. [Google Scholar] [CrossRef]
- Haddon, R.C.; Kaplan, M.L.; Marshall, J.H. Naphtho[1,8-c,d:4,5-c’,d’]bis[1,2,6]thiadiazine. A compound of ambiguous aromatic character. J. Am. Chem. Soc. 1978, 100, 1235–1239. [Google Scholar]
- Kaplan, M.L.; Haddon, R.C.; Schilling, F.C.; Marshall, J.H.; Bramwell, F.B. Naphtho[1,8-c,d:4-c’,d’]bis[1,2,6]selenadiazine. J. Am. Chem. Soc. 1979, 101, 3306–3308. [Google Scholar] [CrossRef]
- Gieren, A.; Lamm, V.; Haddon, R.C.; Kaplan, M.L. Molecular, electronic and crystal structure of naphtho[1,8-c,d:4-c’,d’]bis[1,2,6]selenadiazine. J. Am. Chem. Soc. 1980, 102, 5070–5073. [Google Scholar] [CrossRef]
- Hua, G.; Li, Y.; Fuller, A.M.; Slawin, A.M.Z.; Woollins, J.D. Facile synthesis and structure of novel 2,5-disubstituted 1,3,4-selenadiazoles. Eur. J. Org. Chem. 2009, 1612–1618. [Google Scholar]
- Weintraub, P.M. 1,2,6-Oxadiazines and 1,2,6-thiadiazines. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Turnbull, K., Eds.; Elsevier: Oxford, UK, 2008; Volume 9, pp. 335–400. [Google Scholar]
- Geevers, J.; Trompen, W.P. Synthesis and reactions of 3,5-dichloro-4H-1,2,6-thiadiazin-4-one. Rec. Trav. Chim. 1974, 93, 270–272. [Google Scholar] [CrossRef]
- Koutentis, P.A.; Rees, C.W.; White, A.J.P.; Williams, D.J. Reaction of tetracyanoethylene with SCl2: New molecular rearrangements. Chem. Commun. 2000, 303–304. [Google Scholar]
- Koutentis, P.A.; Rees, C.W. Reaction of tetracyanoethylene with SCl2: New molecular rearrangements. J. Chem. Soc., Perkin Trans. 1 2000, 1089–1094. [Google Scholar] [CrossRef]
- Macho, S.; Miguel, D.; Neo, A.G.; Rodríguez, T.; Torroba, T. Cyclopentathiadiazines, new heterocyclic materials from cyclic enaminonitriles. Chem. Commun. 2005, 334–336. [Google Scholar]
- Gómez, T.; Macho, S.; Miguel, D.; Neo, A.G.; Rodríguez, T.; Torroba, T. Cyclopentathiadiazines, cyclohepta- and cyclopentadithiazoles: New materials and a rich heterocyclic chemistry of cyclic enaminonitriles. Eur. J. Org. Chem. 2005, 5055–5066. [Google Scholar]
- Karpov, V.M.; Platonov, V.E.; Rybalova, T.V.; Gatilov, Yu.V.; Shakirov, M.M. Fluorinated dihydroindeno[2,1-c][1,2,6]thiadiazines: The first synthesis, structural characterization and reactivity. J. Fluorine Chem. 2012, 135, 254–260. [Google Scholar] [CrossRef]
- Ioannidou, H.A.; Kizas, C.; Koutentis, P.A. Selective Stille coupling reactions of 3-chloro-5-halo(pseudohalo)-4H-1,2,6-thiadiazin-4-ones. Org. Lett. 2011, 13, 5886–5889. [Google Scholar] [CrossRef]
- Ioannidou, H.A.; Koutentis, P.A. Synthesis of asymmetric 3,5-diaryl-4H-1,2,6-thiadiazin-4-ones via Suzuki-Miyaura and Stille coupling reactions. Tetrahedron 2012, 68, 7380–7385. [Google Scholar] [CrossRef]
- Ioannidou, H.A.; Kizas, C.; Koutentis, P.A. Palladium catalyzed C-C coupling reactions of 3,5-dichloro-4H-1,2,6-thiadiazin-4-one. Org. Lett. 2011, 13, 3466–3469. [Google Scholar] [CrossRef]
- Haas, A.; Hoppmann, E.; Karpov, V.M.; Platonov, V.E.; Shakirov, M.M. The first synthesis of fluorine-containing indeno[2,1c][1,2,6]selenadiazines. J. Fluorine Chem. 2000, 102, 313–316. [Google Scholar] [CrossRef]
- Yamada, J.; Sugimoto, T. TTF Chemistry: Fundamentals and Applications of Tetrathiafulvalene; Springer: Berlin, Germany, 2004. [Google Scholar]
- Lichtenberger, D.L.; Johnston, R.L.; Hinkelmann, K.; Suzuki, T.; Wudl, F. Relative electron donor strengths of tetrathiafulvene derivatives: Effects of chemical substitutions and the molecular environment from a combined photoelectron and electrochemical study. J. Am. Chem. Soc. 1990, 112, 3302–3307. [Google Scholar] [CrossRef]
- Gleiter, R.; Schmidt, E.; Cowan, D.O.; Ferraris, J.P. The electronic structure of tetrathiafulvalene. J. Electron Spectrosc. 1973, 2, 207–210. [Google Scholar] [CrossRef]
- Enoki, T.; Miyazaki, A. Magnetic TTF-based charge-transfer complexes. Chem. Rev. 2004, 104, 5449–5477. [Google Scholar] [CrossRef]
- Yamashita, Y.; Suzuki, T.; Mukai, T.; Saito, G. Preparation and properties of a tetracyanoquinodimethane fused with 1,2,5-thiadiazole units. Chem. Commun. 1985, 1044–1045. [Google Scholar]
- Yamashita, Y.; Suzuki, T.; Saito, G.; Mukai, T. Highly conductive complexes of bis-1,2,5-thiadiazolotetracyanoquinodimethan (BDTA-TCNQ) with amines. Chem. Lett. 1985, 14, 1759–1762. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Rappoport, D.; Furche, F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 134105. [Google Scholar] [CrossRef]
- Schegolev, I.F.; Yagubskii, E.B. Cation-radical salts of tetrathiotetracene and tetraselenotetracene: Synthetic aspects and physical properties. In Extended Linear Chain Compounds; Miller, J.S., Ed.; Plenum Press: New York, NY, USA, 1982; Volume 2, pp. 385–416. [Google Scholar]
- Semenov, N.A.; Pushkarevsky, N.A.; Barabanov, I.I.; Gritsan, N.P.; Zibarev, A.V. Charge-transfer complexes between tetrathiatetracene and 1,2,5-chalcogenadiazole derivatives. Work in progress. 2013. [Google Scholar]
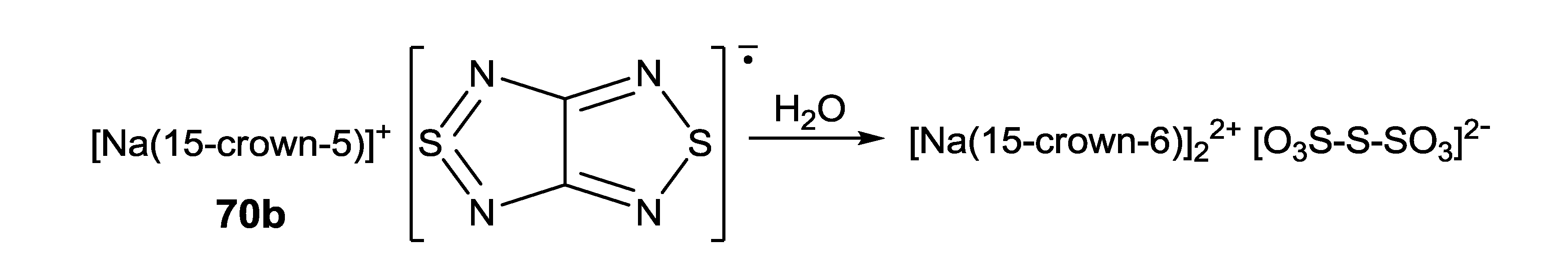
- Lork, E.; Mews, R.; Zibarev, A.V. Hydrolysis product of [Na(15-crown-5)] salt of [1,2,5]thiadiazolo[3,4-c][1,2,5]thiadiazolidyl radical anion. Mendeleev Commun. 2009, 19, 147–148. [Google Scholar] [CrossRef]
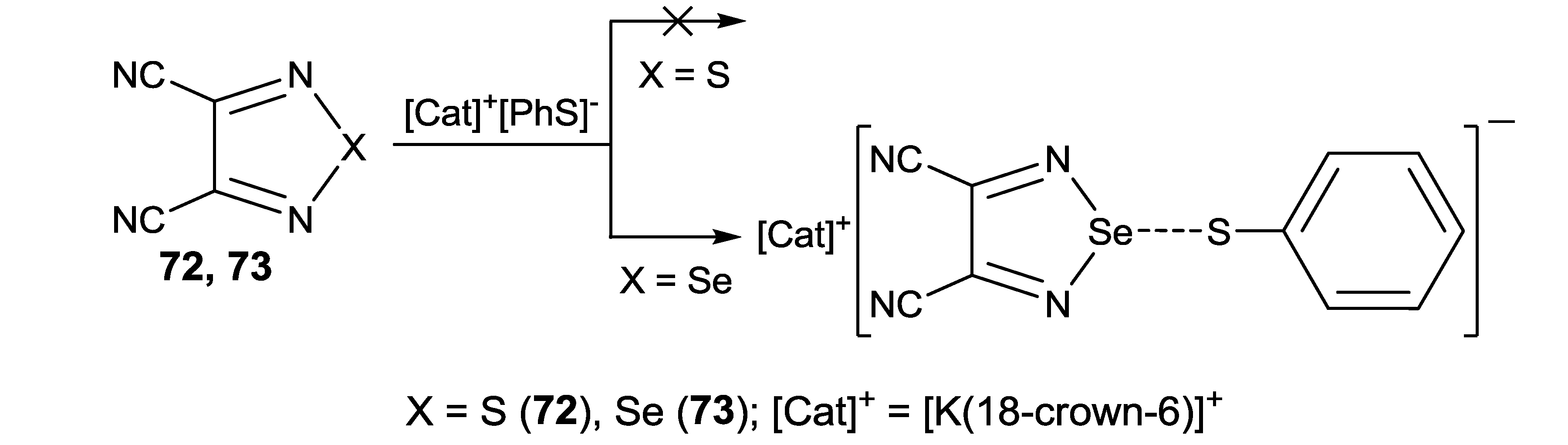
- Suturina, E.A.; Semenov, N.A.; Lonchakov, A.V.; Bagryanskaya, I.Yu.; Gatilov, Yu.V.; Irtegova, I.G.; Vasilieva, N.V.; Lork, E.; Mews, R.; Gritsan, N.P.; et al. Interaction of 1,2,5-chalcogenadiazole derivatives with thiophenolate: Hypercoordination with formation of interchalcogen bond versus reduction to radical anion. J. Phys. Chem. A 2011, 115, 4851–4860. [Google Scholar] [CrossRef]
- Kahn, O. Molecular Magnetism; VCH Publishers: New York, NY, USA, 1993. [Google Scholar]
- Novoa, J.J.; Deumal, M. The mechanism of the through-space magnetic interactions in purely organic molecular magnets. Struct. Bond. 2001, 100, 33–60. [Google Scholar] [CrossRef]
- Hirel, C.; Luneau, D.; Pecaut, J.; Öhrström, L.; Bussiere, G.; Reber, C. The cyano nitronyl nitroxide radical: Experimental and theoretical evidence for the fourth case of the McConnell-I mechanism. Chem. Eur. J. 2002, 8, 3157–3161. [Google Scholar] [CrossRef]
- Hatfield, W.E. New magnetic and structural results for uniformly spaced, alternatingly spaced, and ladder-like copper (II) linear chain compounds. J. Appl. Phys. 1981, 52, 1985–1990. [Google Scholar] [CrossRef]
- Nudleman, L. Valence bond description of anti-ferromagnetic coupling in transition-metal dimers. J. Chem. Phys. 1981, 74, 5737–5743. [Google Scholar] [CrossRef]
- Nudleman, K.; Davidson, E.R. Ligand spin polarization and antiferromagnetic coupling in transition metal dimers. Chem. Phys. 1986, 109, 131–143. [Google Scholar] [CrossRef]
- Nudleman, L.; Case, D.A.; Mouesca, J.M. Orbital interactions, electron delocalization and spin coupling in iron-sulfur clusters. Coordin. Chem. Rev. 1995, 144, 199–244. [Google Scholar] [CrossRef]
- Nagao, H.; Nishino, M.; Shigeta, Y.; Soda, T.; Kitagawa, Y.; Onishi, T.; Yoshioka, Y.; Yamaguchi, K. Theoretical studies on effective spin interactions, spin alignments and macroscopic spin tunneling in polynuclear manganese and related complexes and their mesoscopic clusters. Coordin. Chem. Rev. 2000, 198, 265–295. [Google Scholar] [CrossRef]
- Deumal, M.; Bearpark, M.J.; Novoa, J.J; Robb, M.A. Magnetic properties of organic molecular crystals via an algebraic Heisenberg Hamiltonian. Applications to WILVIW, TOLKEK, and KAXHAS nitronyl nitroxide crystals. J. Phys. Chem. A 2002, 106, 1299–1315. [Google Scholar]
- Johnston, D.C. Normal-state magnetic properties of single-layer cuprate high-temperature superconductors and related materials compounds. In Handbook of Magnetic Materials; Buschow, K.H.J, Ed.; Elsevier: Amsterdam, The Netherlands, 1997. [Google Scholar]
- Nath, R.; Mahajan, A.V.; Buttgen, N.; Kegler, C.; Hemberger, J.; Loidl, A. 31P NMR study of Na2CuP2O7: An S = 1/2 two dimensional Heisenberg antiferromagnetic system. J. Phys. Cond. Mat. 2006, 18, 4285–4294. [Google Scholar]
- Rushbrooke, G.S.; Wood, P.J. On the high-temperature susceptibility for the Heisenberg model of a ferromagnet. Proc. Phys. Soc. Lond. A 1955, 68, 1161–1169. [Google Scholar] [CrossRef]
- Rushbrooke, G.S.; Wood, P.J. On the Curie points and high temperature susceptibilities of Heisenberg model ferromagnetics. Mol. Phys. 1958, 1, 257–283. [Google Scholar] [CrossRef]
- Thompson, L.K.; Waldmann, O.; Xu, Z. Polynuclear manganese grids and clusters—a magnetic perspective. Coordin. Chem. Rev. 2005, 249, 2677–2690. [Google Scholar] [CrossRef]
- Han, T.-H.; Helton, J.S.; Chu, S.; Rodriguez-Rivera, J.A.; Broholm, C.; Lee, Y.S. Fractionalized excitations in the spin-liquid state of a kagome-lattice antiferromagnet. Nature 2012, 492, 406–410. [Google Scholar] [CrossRef]
- Loth, S.; Baumann, S.; Lutz, C.P.; Eigler, D.M.; Heinrich, A.J. Bistability in atomic-scale antiferromagnets. Science 2012, 335, 196–198. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lonchakov, A.V.; Rakitin, O.A.; Gritsan, N.P.; Zibarev, A.V. Breathing Some New Life into an Old Topic: Chalcogen-Nitrogen π-Heterocycles as Electron Acceptors. Molecules 2013, 18, 9850-9900. https://doi.org/10.3390/molecules18089850
Lonchakov AV, Rakitin OA, Gritsan NP, Zibarev AV. Breathing Some New Life into an Old Topic: Chalcogen-Nitrogen π-Heterocycles as Electron Acceptors. Molecules. 2013; 18(8):9850-9900. https://doi.org/10.3390/molecules18089850
Chicago/Turabian StyleLonchakov, Anton V., Oleg A. Rakitin, Nina P. Gritsan, and Andrey V. Zibarev. 2013. "Breathing Some New Life into an Old Topic: Chalcogen-Nitrogen π-Heterocycles as Electron Acceptors" Molecules 18, no. 8: 9850-9900. https://doi.org/10.3390/molecules18089850