Enantio- and Periselective Nitroalkene Diels-Alder Reactions Catalyzed by Helical-Chiral Hydrogen Bond Donor Catalysts

Abstract
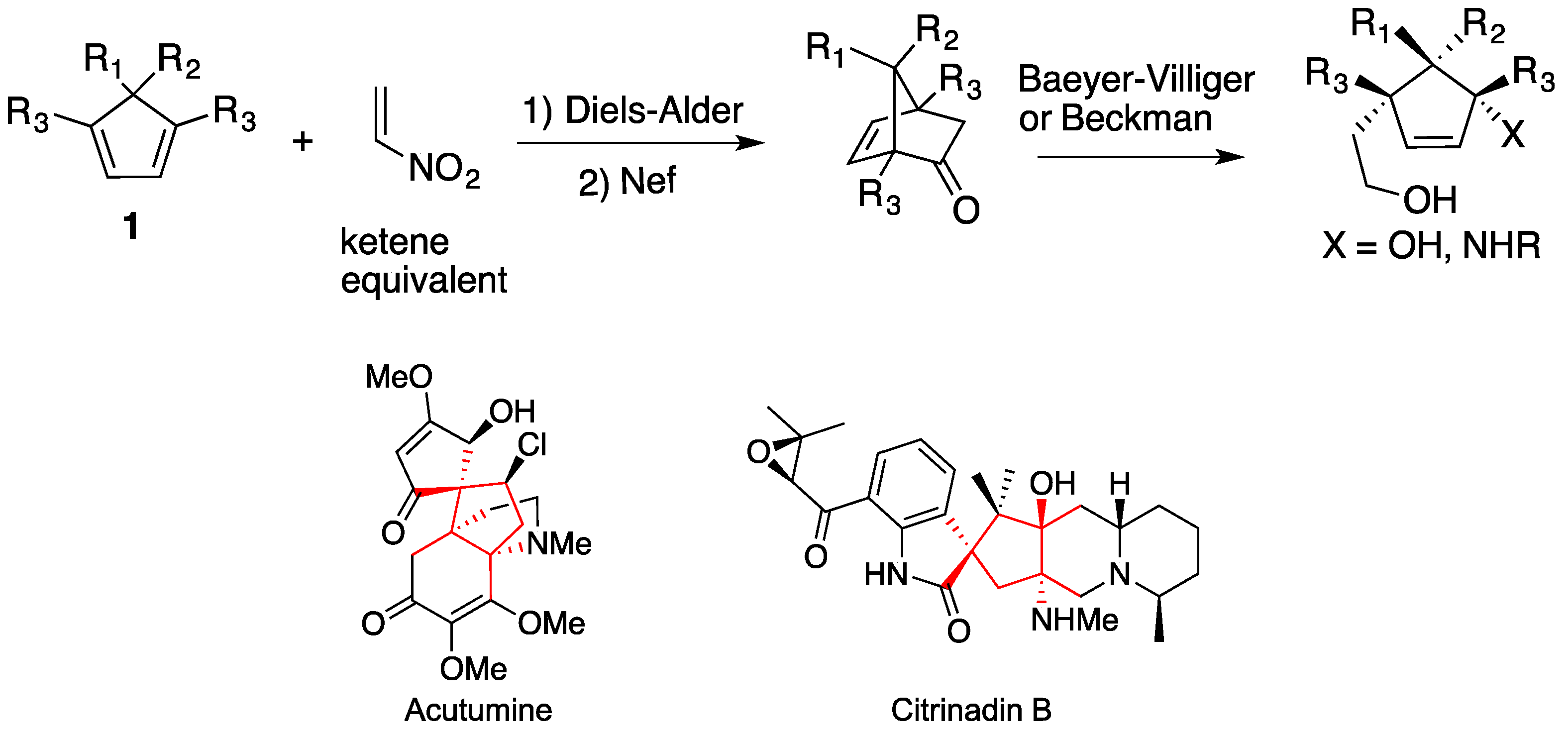
:1. Introduction
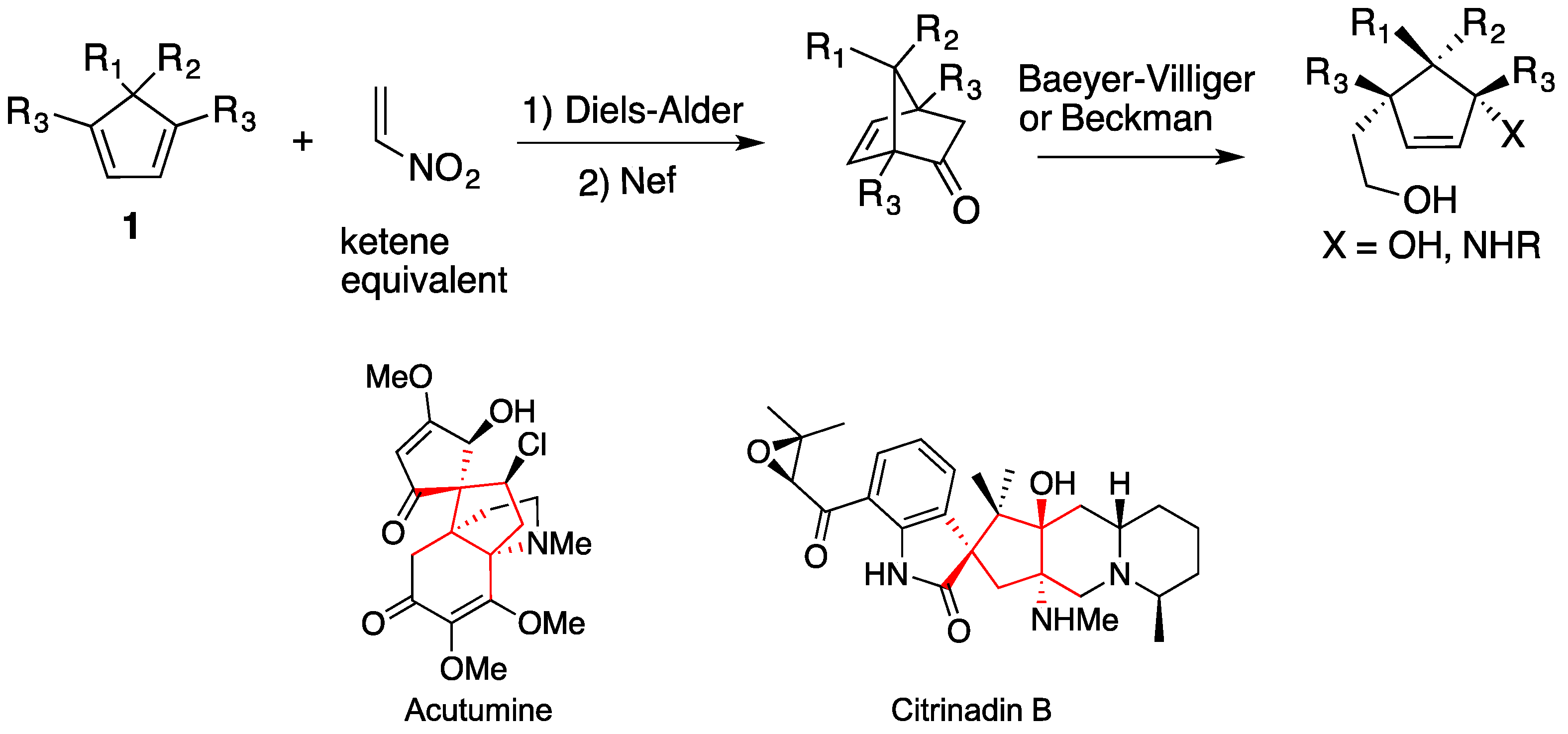
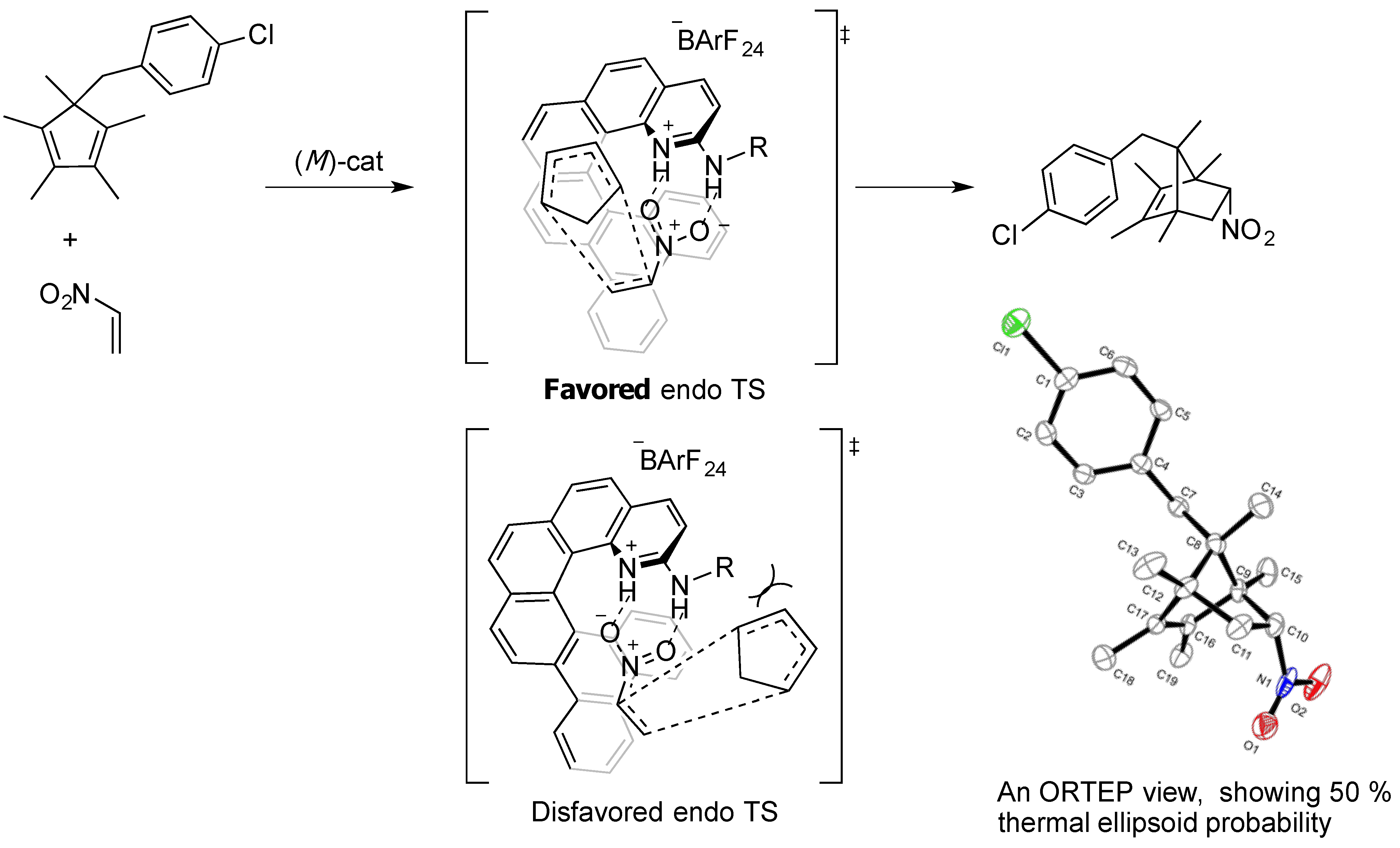
2. Results and Discussion
2.1. Evaluation of Catalysts
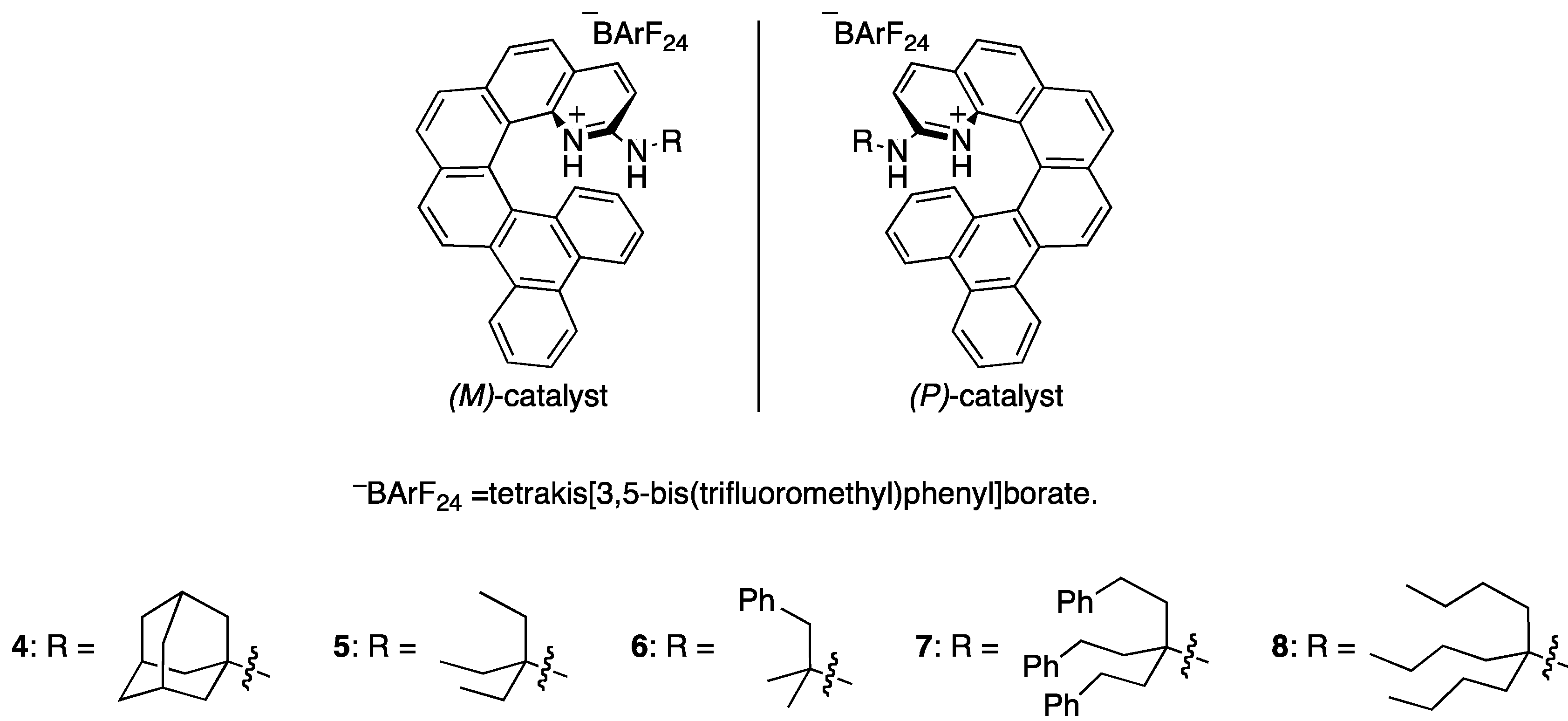


| Entry | Catalyst | Yield c (%) | er d |
|---|---|---|---|
| 1 | - | <5 | - |
| 2 | (M)-4 | 70 | 64:36 |
| 3 | (P)-5 | 84 | 38:62 |
| 4 | (M)-6 | 70 | 59.5:40.5 |
| 5 | (M)-7 | 70 | 67:33 |
| 6 | (M)-8 | 77 | 70:30 |
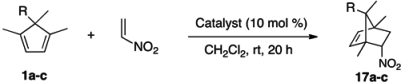
2.2. Evaluation of Dienes

| Entry | Diene | Catalyst | Ar | Yield d (%) | er e |
|---|---|---|---|---|---|
| 1 | 2 | (M)-8 | Ph | 77 | 70:30 |
| 2 | 9a | (M)-8 | 4-F-Ph | 80 | 65:35 |
| 3 | 9b | (M)-8 | 4-Cl-Ph | 38 | 66:34 |
| 4 | 9b | (M)-4 | 4-Cl-Ph | 80 | 64:36 |
| 5 | 9c | (M)-8 | 3-Cl-Ph | 67 | 69:31 |
| 6 | 9d | (P)-4 | 2-Cl-Ph | 55 | 32:68 |
| 7 | 9e | (P)-4 | 2-I-Ph | 65 | 40:60 |
| 8 | 9f | (P)-4 | 4-Me-Ph | 85 | 35:65 |
| 9 | 9g | (P)-4 | 4-CF3-Ph | 75 | 36:64 |
| 10 | 9h | (P)-4 | 4-tert-Butyl-Ph | 80 | 30:70 |
| 11 | 9i | (P)-4 | 2-naphthyl | 83 | 31:69 |
| 12 | 9j | (P)-4 | 3-OMe-Ph | 80 | 34:66 |
| 13 | 9k | (P)-4 | 4-SMe-Ph | 89 | 33:67 |
| 14 f | 9l | (P)-4 | 4-NO2-Ph | 0 | - |
| 15 | 9m | (P)-4 |  | 79 | 37:63 |
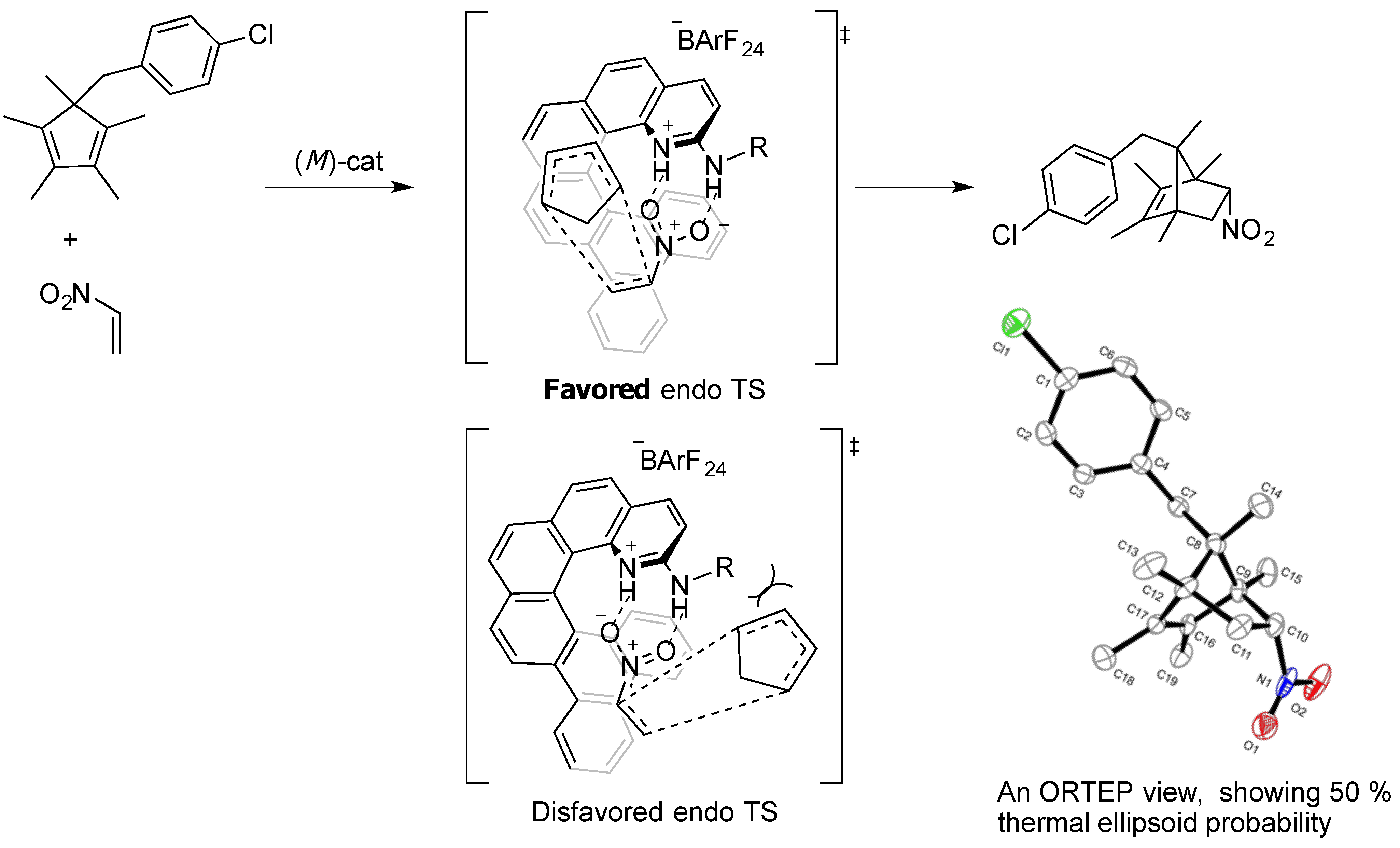
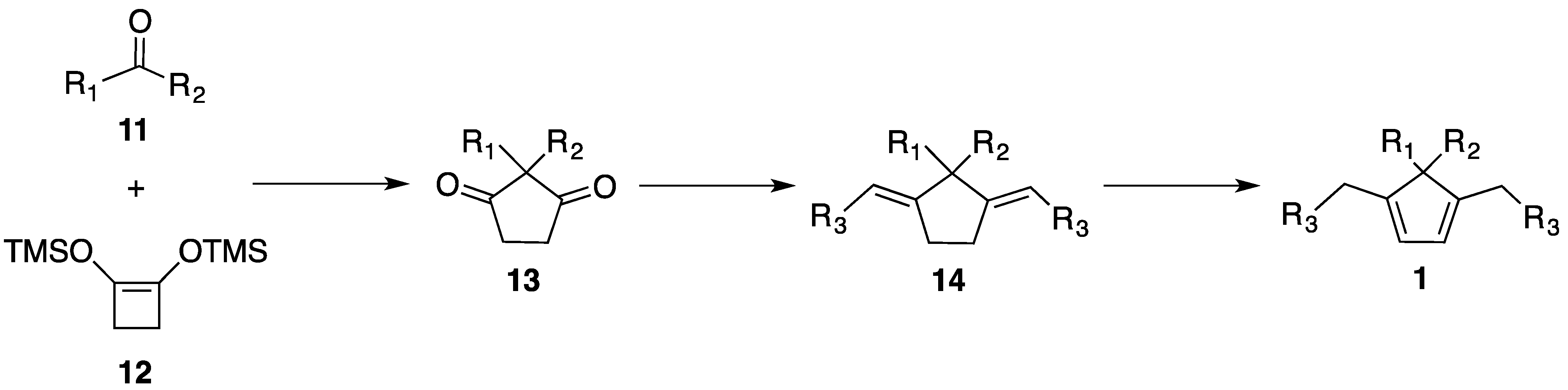
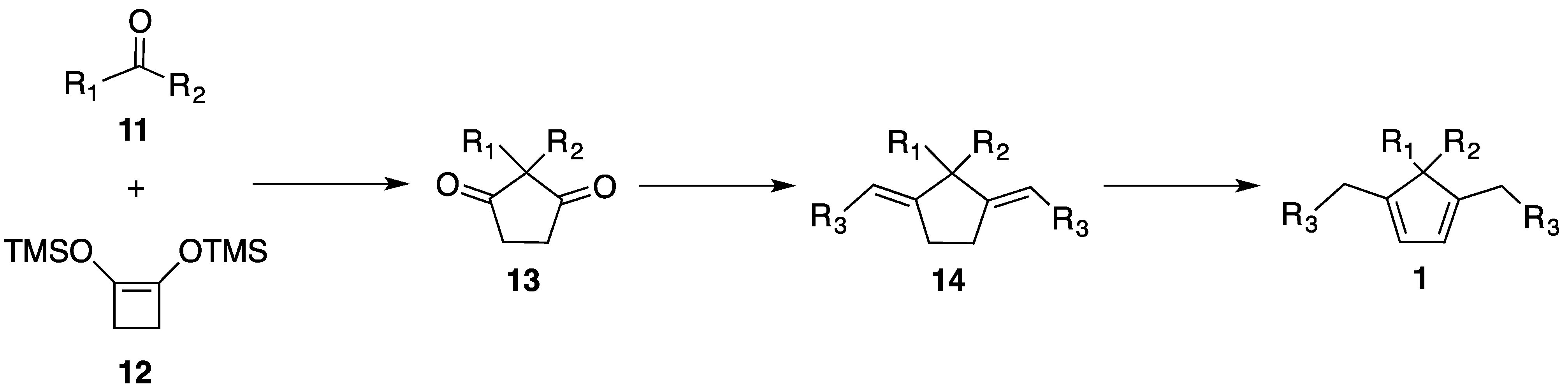
2.3. Synthesis of New Dienes

| Entry | Protocols [Ref.] | Reagents | Yield a (%) |
|---|---|---|---|
| 1 | Wittig [41] | MeP+Ph3I− | 0 |
| 2 | Lombardo [43] | CH2Br2-Zn-TiCl4 | 7 |
| 3 | Curran [38] | CH2Br2-Zn-TiCl4 | 13 |
| 4 | Takai [48] | CH2I2-Zn-TiCl4 | trace |
| 5 | Yan [49] | CH2Cl2-Mg-TiCl4-THF | 50% b |
2.4. Evaluation of New Dienes
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Yield a (%) | |
|---|---|---|---|
| 1 | 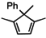 | none | 0 |
| 2 |  | none | 25 |
| 3 | NaBArF24∙2.5H2O | 67 b | |
| 4 |  | none | 11 |
| 5 | (P)-4 | 14 c | |
| 6 | NaBArF24∙2.5H2O | 42 b |
3. Experimental
3.1. General Information
3.2. Preparation of 5-Substituted Pentamethylcyclopentadienes
3.3. General Procedure for Asymmetric Nitroalkene Diels-Alder Reaction
3.4. New Dienes Synthesis
3.4.1. Synthesis of Diones 13
3.4.2. General Procedure for the Synthesis of Dienes 1
3.5. General Procedure for NaBArF24∙2.5H2O Catalyzed Nitroalkene Diels-Alder Reaction
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Payette, J.N.; Yamamoto, H. Cationic-oxazaborolidine-catalyzed enantioselective Diels-Alder reaction of α,β-unsaturated acetylenic ketones. Angew. Chem. Int. Ed. 2009, 48, 8060–8062. [Google Scholar] [CrossRef]
- Balskus, E.P.; Jacobsen, E.N. Asymmetric catalysis of the transannular Diels-Alder reaction. Science 2007, 317, 1736–1740. [Google Scholar] [CrossRef]
- Snyder, S.A.; Corey, E.J. Concise total syntheses of palominol, dolabellatrienone, β-araneosene, and isoedunol via an enantioselective Diels-Alder macrobicyclization. J. Am. Chem. Soc. 2006, 128, 740–742. [Google Scholar] [CrossRef]
- Wilson, R.M.; Jen, W.S.; MacMillan, D.W.C. Enantioselective organocatalytic intramolecular Diels-Alder reactions. The asymmetric synthesis of solanapyrone D. J. Am. Chem. Soc. 2005, 127, 11616–11617. [Google Scholar] [CrossRef]
- Corey, E.J.; Shibata, T.; Lee, T.W. Asymmetric Diels-Alder reactions catalyzed by a triflic acid activated chiral oxazaborolidine. J. Am. Chem. Soc. 2002, 124, 3808–3809. [Google Scholar] [CrossRef]
- Erkkila, A.; Majander, I.; Pihko, P.M. Iminium catalysis. Chem. Rev. 2007, 107, 5416–5470. [Google Scholar] [CrossRef]
- Corey, E.J. Catalytic enantioselective Diels-Alder reactions: Methods, mechanistic fundamentals, pathways, and applications. Angew. Chem. Int. Ed. 2002, 41, 1650–1667. [Google Scholar] [CrossRef]
- Ҫelebi-Ölçüm, N.; Ess, D.H.; Aviyente, V.; Houk, K.N. Lewis acid catalysis alters the shapes and products of bis-pericyclic Diels-Alder transition states. J. Am. Chem. Soc. 2007, 129, 4528–4529. [Google Scholar] [CrossRef]
- Denmark, S.E.; Kesler, B.S.; Moon, Y.C. Intermolecular and intramolecular [4 + 2] cycloadditions of nitroalkenes with olefins. 2-nitrostyrenes. J. Org. Chem. 1992, 57, 4912–4924. [Google Scholar] [CrossRef]
- Takenaka, N.; Sarangthem, R.S.; Seerla, S.K. 2-aminopyridinium ions activate nitroalkenes through hydrogen bonding. Org. Lett. 2007, 9, 2819–2822. [Google Scholar] [CrossRef]
- Thayumanavan, R.; Dhevalapally, B.; Sakthivel, K.; Tanaka, F.; Barbas, C.F. Amine-catalyzed direct Diels-Alder reactions of α,β-unsaturated ketones with nitro olefins. Tetrahedron Lett. 2002, 43, 3817–3820. [Google Scholar] [CrossRef]
- Wu, L.Y.; Bencivenni, G.; Mancinelli, M.; Mazzanti, A.; Bartoli, G.; Melchiorre, P. Organocascade reactions of enones catalyzed by a chiral primary amine. Angew. Chem. Int. Ed. 2009, 48, 7196–7199. [Google Scholar] [CrossRef]
- Xu, D.Q.; Xia, A.B.; Luo, S.P.; Tang, J.; Zhang, S.; Jiang, J.R.; Xu, Z.Y. In situ enamine activation in aqueous salt solutions: Highly efficient asymmetric organocatalytic Diels-Alder reaction of cyclohexenones with nitroolefins. Angew. Chem. Int. Ed. 2009, 48, 3821–3824. [Google Scholar] [CrossRef]
- Sunden, H.; Rios, R.; Xu, Y.; Eriksson, L.; Cordova, A. Direct enantioselective synthesis of bicyclic Diels-Alder products. Adv. Synth. Catal. 2007, 349, 2549–2555. [Google Scholar] [CrossRef]
- Bartelson, K.J.; Singh, R.P.; Foxman, B.M.; Deng, L. Catalytic asymmetric [4 + 2] additions with aliphatic nitroalkenes. Chem. Sci. 2011, 2, 1940–1944. [Google Scholar] [CrossRef]
- King, S.M.; Calandra, N.A.; Herzon, S.B. Total syntheses of (-)-acutumine and (-)-dechloroacutumine. Angew. Chem. Int. Ed. 2013, 52, 3642–3645. [Google Scholar] [CrossRef]
- Li, F.; Tartakoff, S.S.; Castle, S.L. Total synthesis of (-)-acutumine. J. Am. Chem. Soc. 2009, 131, 6674–6675. [Google Scholar]
- Mugishima, T.; Tsuda, M.; Kasai, Y.; Ishiyama, H.; Fukushi, E.; Kawabata, J.; Watanabe, M.; Akao, K.; Kobayashi, J. Absolute stereochemistry of citrinadins A and B from marine-derived fungus. J. Org. Chem. 2005, 70, 9430–9435. [Google Scholar] [CrossRef]
- Ranganat, S.; Ranganat, D.; Mehrotra, A.K. Nitroethylene as a versatile ketene equivalent. Novel one-step preparation of prostaglandin intermediates by reduction and abnormal nef reaction. J. Am. Chem. Soc. 1974, 96, 5261–5262. [Google Scholar] [CrossRef]
- Corey, E.J.; Weinshenker, N.M.; Schaaf, T.K.; Huber, W. Stereo-controlled synthesis of prostaglandins F2α and E2 (dl). J. Am. Chem. Soc. 1969, 91, 5675–5677. [Google Scholar]
- Fujiwara, Y.; Fu, G.C. Application of a new chiral phosphepine to the catalytic asymmetric synthesis of highly functionalized cyclopentenes that bear an array of heteroatom-substituted quaternary stereocenters. J. Am. Chem. Soc. 2011, 133, 12293–12297. [Google Scholar] [CrossRef]
- Han, X.; Wang, Y.; Zhong, F.; Lu, Y. Enantioselective [3 + 2] cycloaddition of allenes to acrylates catalyzed by dipeptide-derived phosphines: Facile creation of functionalized cyclopentenes containing quaternary stereogenic centers. J. Am. Chem. Soc. 2011, 133, 1726–1729. [Google Scholar] [CrossRef]
- Xiao, H.; Chai, Z.; Zheng, C.W.; Yang, Y.Q.; Liu, W.; Zhang, J.K.; Zhao, G. Asymmetric [3 + 2] cycloadditions of allenoates and dual activated olefins catalyzed by simple bifunctional N-acyl aminophosphines. Angew. Chem. Int. Ed. 2010, 49, 4467–4470. [Google Scholar] [CrossRef]
- Cowen, B.J.; Miller, S.J. Enantioselective [3 + 2]-cycloadditions catalyzed by a protected, multifunctional phosphine-containing α-amino acid. J. Am. Chem. Soc. 2007, 129, 10988–10989. [Google Scholar] [CrossRef]
- Chiang, P.C.; Kaeobamrung, J.; Bode, J.W. Enantioselective, cyclopentene-forming annulations via NHC-catalyzed benzoin-oxy-cope reactions. J. Am. Chem. Soc. 2007, 129, 3520–3521. [Google Scholar] [CrossRef]
- Wilson, J.E.; Fu, G.C. Synthesis of functionalized cyclopentenes through catalytic asymmetric [3 + 2] cycloadditions of allenes with enones. Angew. Chem. Int. Ed. 2006, 45, 1426–1429. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Xiang, B.P.; Kong, N.; Stafford, D.G. Catalytic asymmetric synthesis of highly functionalized cyclopentenes by a [3 + 2] cycloaddition. J. Am. Chem. Soc. 2001, 123, 7461–7462. [Google Scholar]
- Yu, X.; Wang, W. Hydrogen-bond-mediated asymmetric catalysis. Chem. Asian J. 2008, 3, 516–532. [Google Scholar] [CrossRef]
- Doyle, A.G.; Jacobsen, E.N. Small-molecule H-bond donors in asymmetric catalysis. Chem. Rev. 2007, 107, 5713–5743. [Google Scholar] [CrossRef]
- Taylor, M.S.; Jacobsen, E.N. Asymmetric catalysis by chiral hydrogen-bond donors. Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef]
- Takemoto, Y. Recognition and activation by ureas and thioureas: Stereoselective reactions using ureas and thioureas as hydrogen-bonding donors. Org. Biomol. Chem. 2005, 3, 4299–4306. [Google Scholar] [CrossRef]
- Peng, Z.; Takenaka, N. Applications of helical-chiral pyridines as organocatalysts in asymmetric synthesis. Chem. Rec. 2013, 13, 28–42. [Google Scholar] [CrossRef]
- Takenaka, N.; Chen, J.; Captain, B.; Sarangthem, R.S.; Chandrakumar, A. Helical chiral 2-aminopyridinium ions: A new class of hydrogen bond donor catalysts. J. Am. Chem. Soc. 2010, 132, 4536–4537. [Google Scholar] [CrossRef]
- Narcis, M.J.; Sprague, D.J.; Captain, B.; Takenaka, N. Enantio- and periselective nitroalkene Diels-Alder reaction. Org. Biomol. Chem. 2012, 10, 9134–9136. [Google Scholar] [CrossRef]
- Ben Romdhane, H.; Baklouti, M.; Chaabouni, M.R.; Grenier-Loustalot, M.F.; Delolme, F.; Sillion, B. Polypentamethylnadimides obtained by Diels-Alder reaction. Polymer 2002, 43, 255–268. [Google Scholar] [CrossRef]
- Adam, W.; Jacob, U.; Prein, M. Importance of steric effects in the [4 + 2] cycloaddition of 5-substituted pentamethylcyclopentadienes. J. Chem. Soc., Chem. Commun. 1995, 839–840. [Google Scholar] [CrossRef]
- Baranovsky, A.V.; Bolibrukh, D.A.; Khripach, V.A.; Schneider, B. Diels-Alder reaction of androsta-14,16-dien-17-yl acetates with nitroethylene: Product distribution and selected adduct transformations. Steroids 2013, 78, 282–287. [Google Scholar] [CrossRef]
- Schwartz, C.E.; Curran, D.P. New tandem radical cyclizations directed toward the synthesis of Crinipellin A. J. Am. Chem. Soc. 1990, 112, 9272–9284. [Google Scholar] [CrossRef]
- Crane, S.N.; Burnell, D.J. The BF3∙Et2O catalyzed reaction of 1,2-bis (trimethylsilyl)oxy cyclobutene and analogues with aromatic ketones. J. Org. Chem. 1998, 63, 1352–1355. [Google Scholar] [CrossRef]
- Jenkins, T.J.; Burnell, D.J. Lewis-acid catalyzed geminal acylation reaction of ketones with 1,2-bis((trimethylsilyl)oxy)cyclobutene: Direct formation of 2,2-disubstituted 1,3-cyclopentanediones. J. Org. Chem. 1994, 59, 1485–1491. [Google Scholar] [CrossRef]
- Leiris, S.J.; Khdour, O.M.; Segerman, Z.J.; Tsosie, K.S.; Chapuis, J.C.; Hecht, S.M. Synthesis and evaluation of verticipyrone analogues as mitochondrial complex I inhibitors. Biorg. Med. Chem. 2010, 18, 3481–3493. [Google Scholar] [CrossRef]
- Petasis, N.A.; Staszewski, J.P. Dibromomethane-Zinc-Titanium (IV) Chloride. In Encyclopedia of reagents for organic synthesis, 2nd ed.; Paquette, L.A., Crich, D., Fuchs, P.L., Molander, G.A., Eds.; Wiley: Chichester, UK, 2009; Volume 4, pp. 3132–3135. [Google Scholar]
- Lombardo, L. Methylenation of carbonyl compounds: (+)-3- methylene-cis-p-menthane. Org. Synth. 1987, 65, 81–85. [Google Scholar]
- Lombardo, L. Methylenation of carbonyl-compounds with Zn-CH2Br2-TiCl4. Applications to gibberellins. Tetrahedron Lett. 1982, 23, 4293–4296. [Google Scholar] [CrossRef]
- Peng, F.; Danishefsky, S.J. Total synthesis of (+/-)-maoecrystal V. J. Am. Chem. Soc. 2012, 134, 18860–18867. [Google Scholar] [CrossRef]
- Skepper, C.K.; Quach, T.; Molinski, T.F. Total synthesis of enigmazole A from cinachyrella enigmatica. Bidirectional bond constructions with an ambident 2,4-disubstituted oxazole synthon. J. Am. Chem. Soc. 2010, 132, 10286–10292. [Google Scholar] [CrossRef]
- Takai, K.; Hotta, Y.; Oshima, K.; Nozaki, H. Effective methods of carbonyl methylenation using CH2I2-Zn-Me3Al and CH2Br2-Zn-TiCl4 system. Tetrahedron Lett. 1978, 2417–2420. [Google Scholar]
- Hibino, J.; Okazoe, T.; Takai, K.; Nozaki, H. Carbonyl methylenation of easily enolizable ketones. Tetrahedron Lett. 1985, 26, 5579–5580. [Google Scholar] [CrossRef]
- Yan, T.H.; Tsai, C.C.; Chien, C.T.; Cho, C.C.; Huang, P.C. Dichloromethane activation. Direct methylenation of ketones and aldehydes with CH2Cl2 promoted by Mg/TiCl4/THF. Org. Lett. 2004, 6, 4961–4963. [Google Scholar] [CrossRef]
- Aguado, A.; Takenaka, N. Intramolecular nitroalkene Diels-Alder reaction catalyzed by Brønsted acids. Synlett 2011, 1259–1261. [Google Scholar] [CrossRef]
- Ranganathan, D.; Rao, C.B.; Ranganathan, S.; Mehrotra, A.K.; Iyengar, R. Nitroethylene: A stable, clean, and reactive agent for organic synthesis. J. Org. Chem. 1980, 45, 1185–1189. [Google Scholar] [CrossRef]
- Wu, Y.J.; Strickland, D.W.; Jenkins, T.J.; Liu, P.Y.; Burnell, D.J. Formation of 2,2-disubstituted 1,3-cyclopentanediones from ketals with 1,2-bis(trimethylsilyloxy)cyclobutene. Can. J. Chem. 1993, 71, 1311–1318. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Peng, Z.; Narcis, M.J.; Takenaka, N. Enantio- and Periselective Nitroalkene Diels-Alder Reactions Catalyzed by Helical-Chiral Hydrogen Bond Donor Catalysts. Molecules 2013, 18, 9982-9998. https://doi.org/10.3390/molecules18089982
Peng Z, Narcis MJ, Takenaka N. Enantio- and Periselective Nitroalkene Diels-Alder Reactions Catalyzed by Helical-Chiral Hydrogen Bond Donor Catalysts. Molecules. 2013; 18(8):9982-9998. https://doi.org/10.3390/molecules18089982
Chicago/Turabian StylePeng, Zhili, Maurice J. Narcis, and Norito Takenaka. 2013. "Enantio- and Periselective Nitroalkene Diels-Alder Reactions Catalyzed by Helical-Chiral Hydrogen Bond Donor Catalysts" Molecules 18, no. 8: 9982-9998. https://doi.org/10.3390/molecules18089982