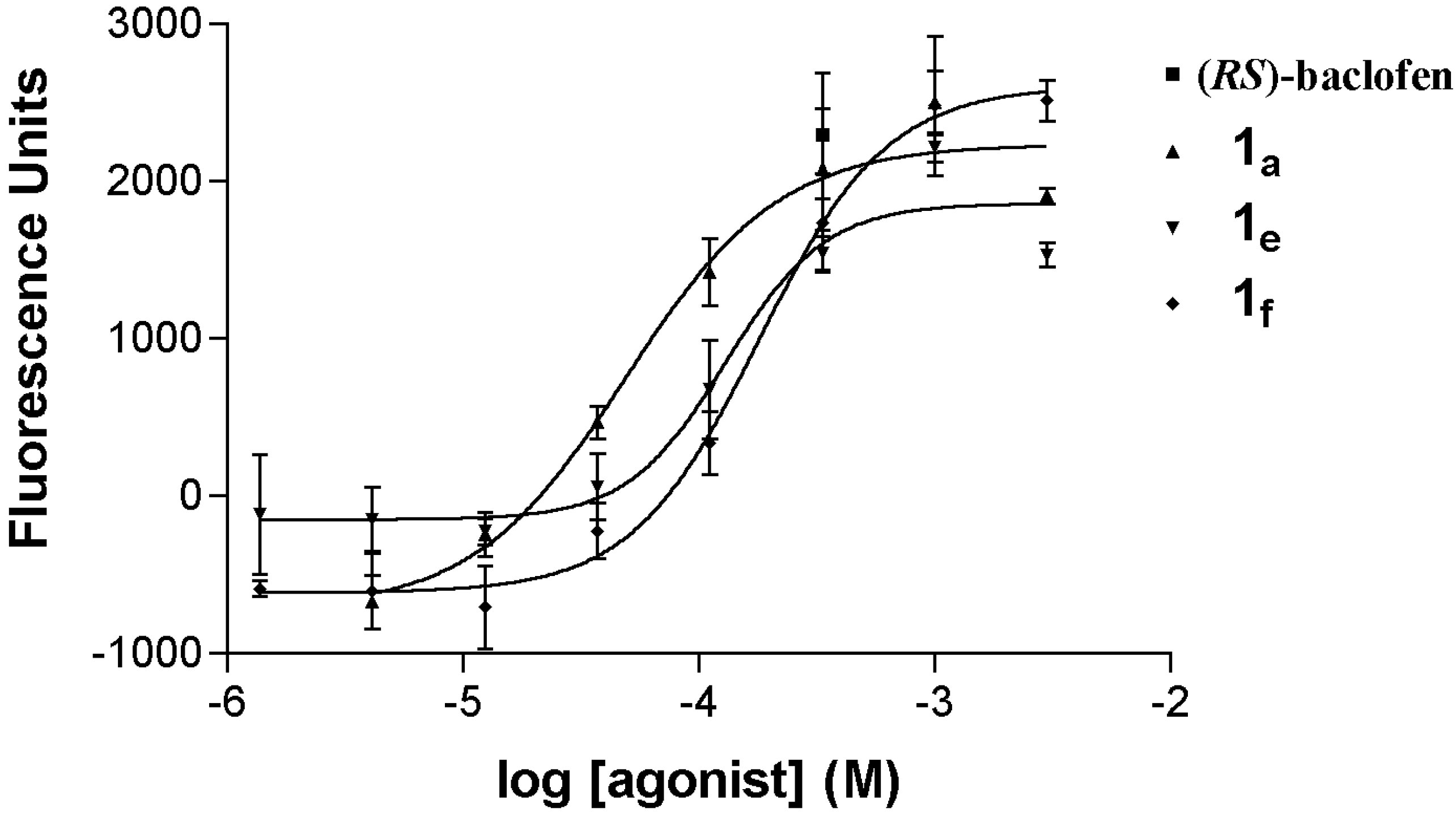
3.1. Chemistry
3.1.1. General
Melting points were determined using a capillary melting point apparatus (Gallenkamp, Sanyo) and are uncorrected. Infrared (IR) spectra were recorded as thin layer films (for oils) or as pellets (for solids) with BIO-RAD spectrometer and values are represented in cm−1. NMR (1H-NMR and 13C- NMR) spectra were recorded on a Bruker AC 250 spectrometer (at 250 MHz for 1H-NMR and 63 MHz for 13C-NMR) and chemical shift values were recorded in ppm on the δ scale. All samples were measured at room temperature. The 1H-NMR data are presented as follows: Chemical shifts, multiplicity, number of protons, assignment. Column chromatography was carried out on silica gel 60 (0.063–0.200 mm) obtained from Merck. Elemental analyses were performed by the microanalytical section of the Institute of Inorganic Chemistry, University of Würzburg, Würzburg, Germany.
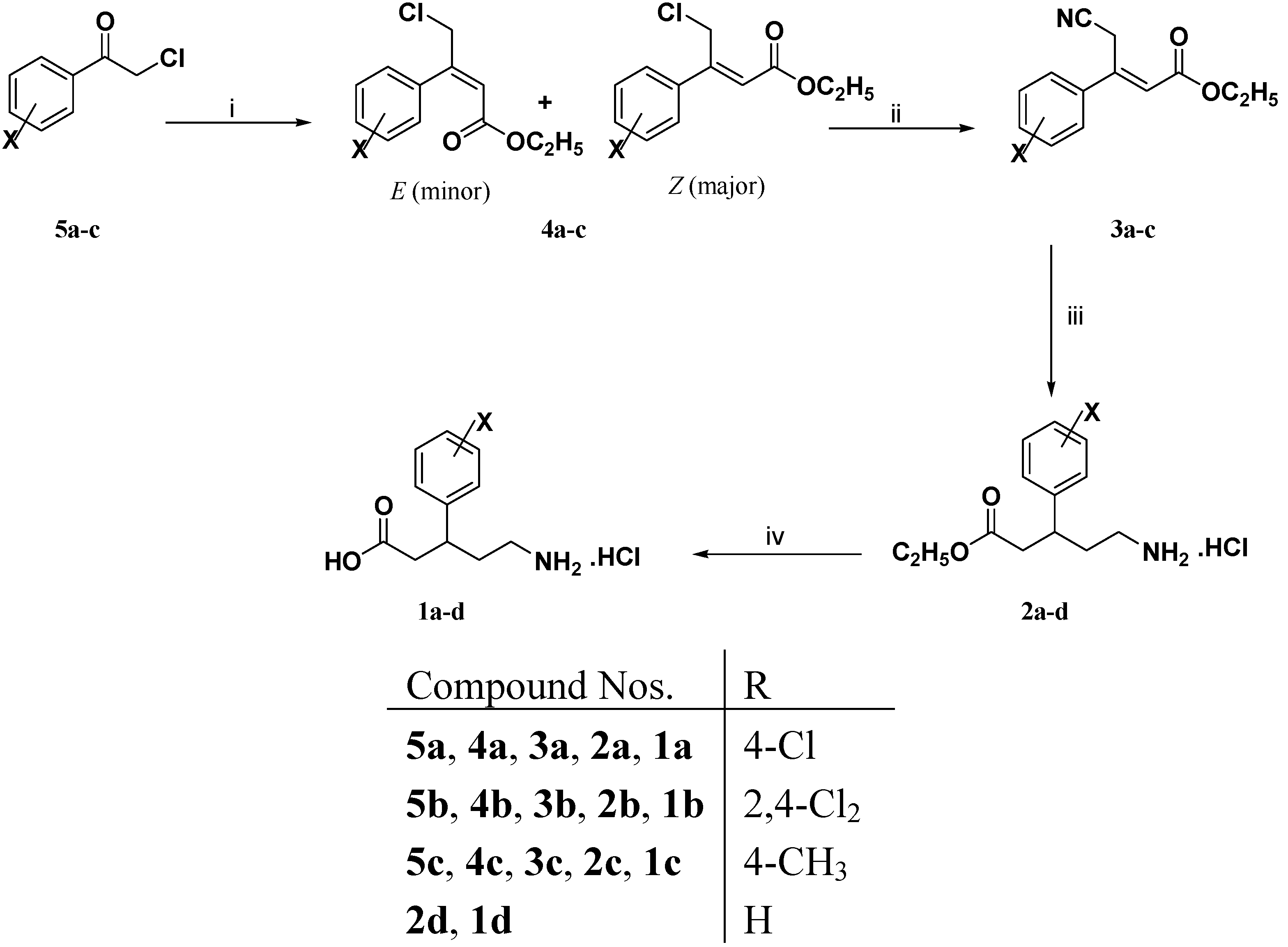
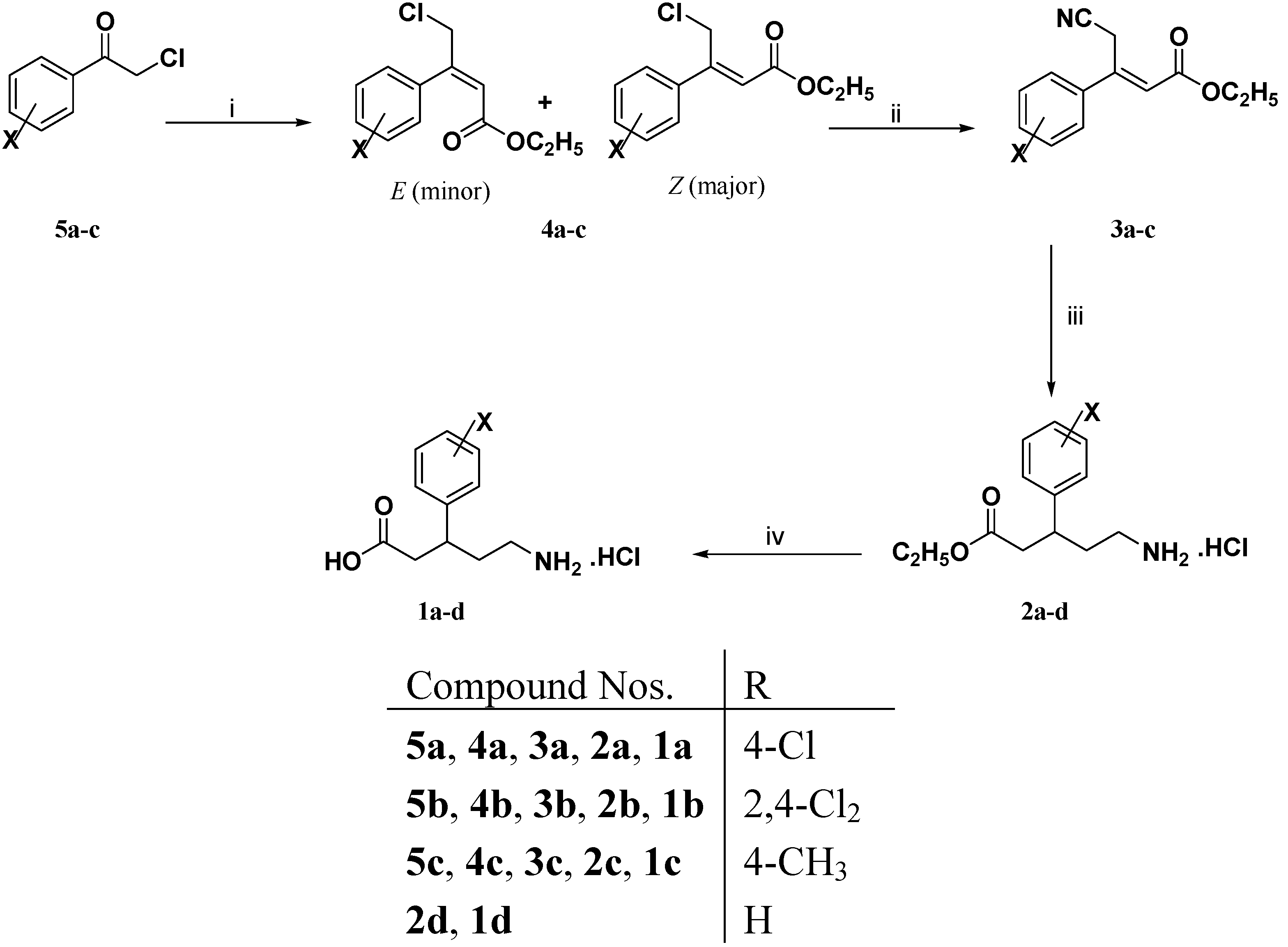
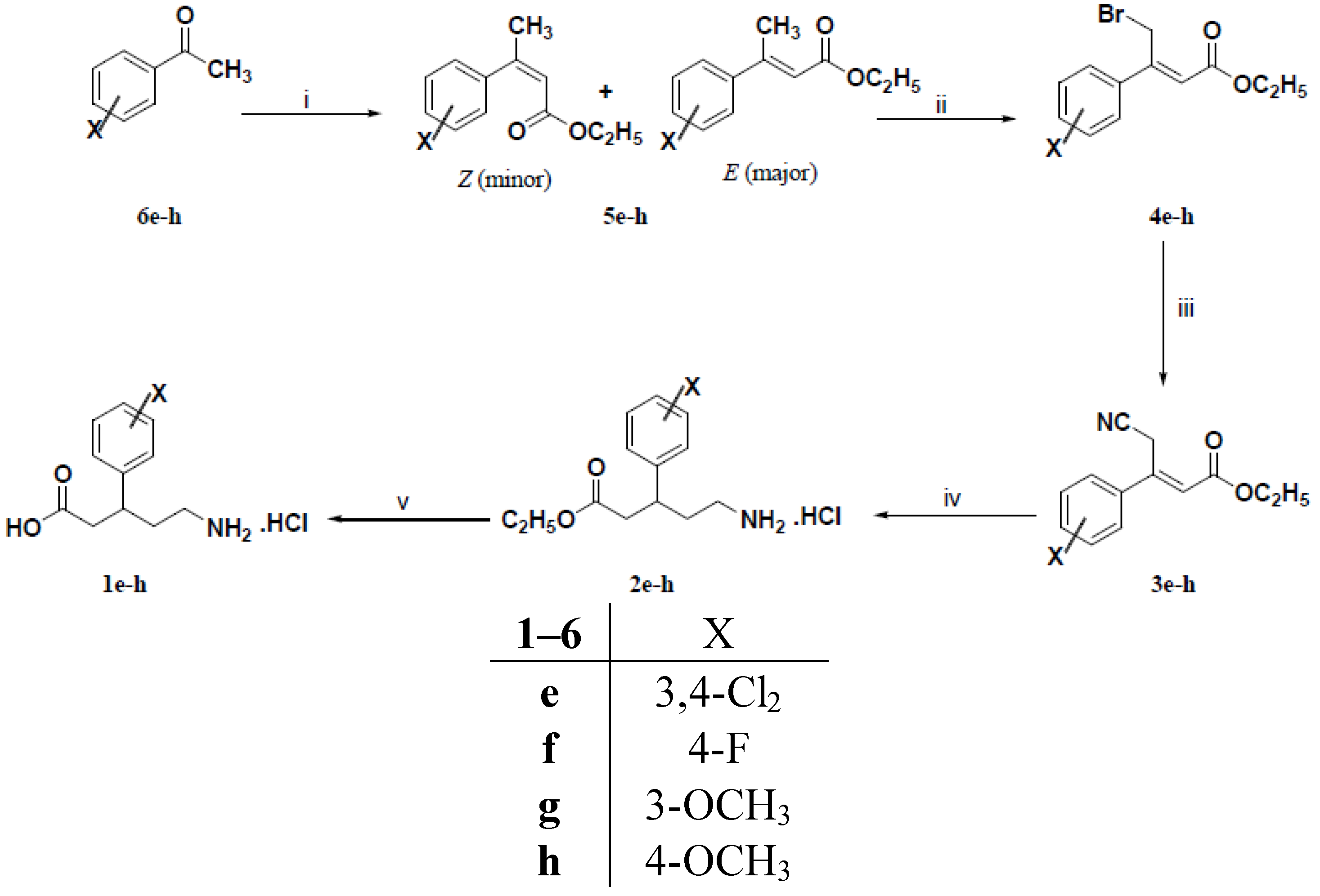
3.1.2. General Procedure for the Preparation of 3-Aryl-4-chloro-2-butenoic Acid Ethyl Esters 4a–c
Triethyl phosphonoacetate (2.92 g, 13 mmol) was added dropwise to a cold (5–10 °C) stirred slurry of 60% sodium hydride (0.52 g, 13 mmol) in dry 1,2 dimethoxyethane (20 mL). After complete addition, the reaction mixture was stirred at ambient temperature for 30 min or until gas evolution ceased. A solution of the appropriate ketone 5a–c (10 mmol) in dry 1,2 dimethoxyethane (10 mL) was then added dropwise to the resulting solution. The reaction mixture was heated under stirring at 50 °C for 18 h. The reaction mixture was cooled to room temperature, poured into water (100 mL) and extracted with diethyl ether (3 × 50 mL). The organic extract was dried (Na2SO4), filtered and evaporated under vacuum to afford viscous oils which were purified by column chromatography using petroleum ether (40–60 °C): Diethyl ether (9:1) to give compounds 4a–c in 40%–88% yields as pale yellow viscous oils.
(Z)-4-Chloro-3-(4-chlorophenyl)-2-butenoic acid ethyl ester [(Z)-4a]. Yield 80%; IR (neat): ν (cm−1) = 1711, 1628, 1492, 1176, 1160; 1H-NMR (CDCl3): δ (ppm) = 1.15 (t, J = 7.33 Hz, 3H, CH3–CH2–), 4.08 (q, J = 7.33 Hz, 2H, –CH2–CH3), 4.88 (s, 2H, 4-H), 6.03 (s, 1H, 2-H), 7.20 (d, JAB = 8.85 Hz, 2H, Harom.), 7.30 (d, JAB = 8.85 Hz, 2H, Harom.); 13C-NMR (CDCl3): δ (ppm) = 14.6 (CH3–CH2–), 39.4 (C-4), 61.1 (–CH2–CH3), 121.0 (C-2), 128.5, 129.4, 136.2, 137.0 (Carom.), 151.8 (C-3), 165.7 (C-1).
(E)-4-Chloro-3-(4-chlorophenyl)-2-butenoic acid ethyl ester [(E)-4a]. Yield 8%; IR (neat): ν (cm−1) = 1720, 1651, 1491, 1225, 1163; 1H-NMR (CDCl3): δ (ppm) = 1.16 (t, J = 7.03 Hz, 3H, CH3–CH2–), 4.07 (q, J = 7.03 Hz, 2H, –CH2–CH3), 4.31 (d, J = 1.23 Hz, 2H, 4-H), 6.28 (t, J = 1.23 Hz, 1H, 2-H), 7.21 (d, JAB = 8.55 Hz, 2H, Harom.), 7.39 (d, JAB = 8.55 Hz, 2H, Harom.); 13C-NMR (CDCl3): δ (ppm) = 14.3 (CH3–CH2–), 48.7 (C-4), 121.5 (C-2), 128.8, 129.5, 135.0, 135.7 (Carom.), 151.3 (C-3), 165.5 (C-1).
(Z)-4-Chloro-3-(2,4-dichlorophenyl)-2-butenoic acid ethyl ester [(Z)-4b]. Yield 48%; IR (neat): ν (cm−1) = 1707, 1641, 1581, 1436, 1341, 1186; 1H-NMR (CDCl3): δ (ppm) = 1.11 (t, J = 7.03 Hz, 3H, CH3–CH2–), 4.04 (q, J = 7.03 Hz, 2H, –CH2–CH3), 4.79 (s, 2H, 4-H), 5.70 (s, 1H, 2-H), 6.98–7.22 (m, 3H, Harom.); 13C-NMR (CDCl3): δ (ppm) = 14.6 (CH3–CH2–), 40.9 (C-4), 61.3 (–CH2–CH3), 124.6 (C-2), 127.6, 129.9, 130.0, 132.1, 135.7, 136.9, (Carom.), 151.9 (C-3), 165.2 (C-1).
(E)-4-Chloro-3-(2,4-dichlorophenyl)-2-butenoic acid ethyl ester [(E)-4b]. Yield 34%; IR (neat): ν (cm−1) = 1720, 1585, 1473, 1226, 1164; 1H-NMR (CDCl3): δ (ppm) = 1.14 (t, J = 7.03 Hz, 3H, CH3–CH2–), 4.06 (q, J = 7.03 Hz, 2H, –CH2–CH3), 4.31 (s, 2H, 4-H), 6.39 (t, J = 1.23 Hz, 1H, 2-H), 7.13–7.48 (m, 3H, Harom.); 13C-NMR (CDCl3): δ (ppm) = 14.3 (CH3–CH2–), 47.4 (C-4), 60.9 (–CH2–CH3), 123.4 (C-2), 127.4, 129.7, 130.9, 132.8, 134.8, 135.2, (Carom.), 149.3 (C-3), 164.8 (C-1).
(Z)-4-Chloro-3-(4-methylphenyl)-2-butenoic acid ethyl ester [(Z)-4c]. Yield 36%; IR (neat): ν (cm−1) = 1710, 1626, 1609, 1173, 1158; 1H-NMR (CDCl3): δ (ppm) = 1.37 (t, J = 7.03 Hz, 3H, CH3–CH2–), 2.42 (s, 3H, 4–CH3), 4.29 (q, J = 7.03 Hz, 2H, –CH2–CH3), 5.12 (s, 2H, 4-H), 6.27 (s, 1H, 2-H), 7.26 (d, JAB = 8.25 Hz, 2H, Harom.), 7.50 (d, JAB = 8.25 Hz, 2H, Harom.). 13C-NMR (CDCl3): δ (ppm) = 14.6 (CH3–CH2–), 21.7 (4–CH3), 39.5 (C-4), 60.9 (–CH2 –CH3), 119.7 (C-2), 127.0, 129.9, 135.6, 140.4, (Carom.), 153.0 (C-3), 166.0 (C-1).
(E)-4-Chloro-3-(4-methylphenyl)-2-butenoic acid ethyl ester [(E)-4c]. Yield 4%; IR (neat): ν (cm−1) = 1703, 1607, 1512, 1225, 1163; 1H-NMR (CDCl3): δ (ppm) = 1.15 (t, J = 7.03 Hz, 3H, CH3–CH2–), 2.40 (s, 3H, 4–CH3), 4.07 (q, J = 7.03 Hz, 2H, –CH2–CH3), 4.33 (d, J = 1.23 Hz, 2H, 4-H), 6.26 (t, J = 1.23 Hz, 1H, 2-H), 7.16 (d, JAB = 8.25 Hz, 2H, Harom.), 7.23 (d, JAB = 8.25 Hz, 2H, Harom.); 13C-NMR (CDCl3): δ (ppm) = 14.4 (CH3–CH2–), 21.8 (4–CH3), 48.9 (C-4), 120.5 (C-2 ), 127.9, 129.3, 134.2, 138.9 (Carom.), 152.6 (C-3), 165.9 (C-1).
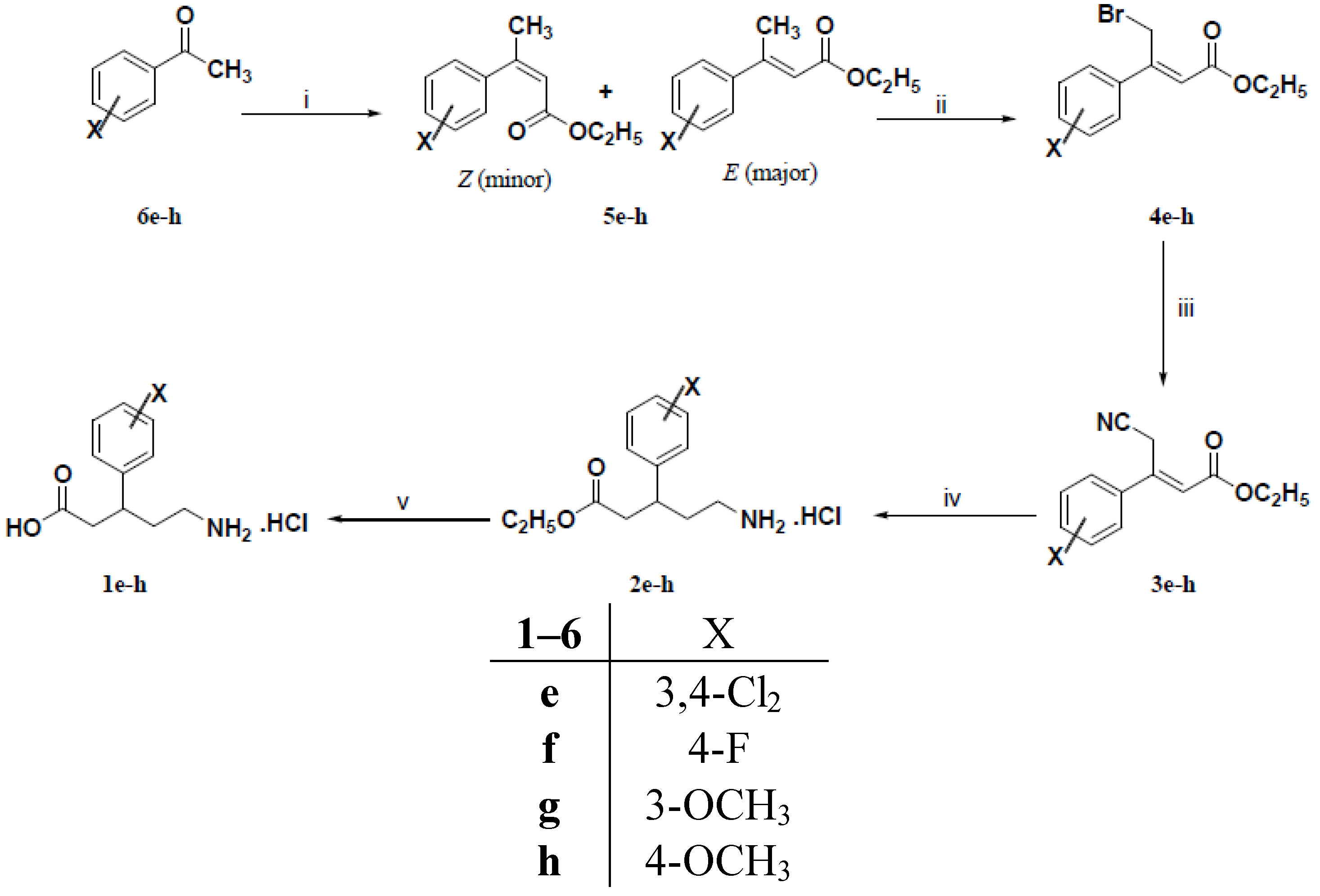
3.1.3. General Procedure for the Preparation of 3-Aryl-2-butenoic Acid Ethyl Esters 5e–h
To a cold (5–10 °C) solution of potassium t-butoxide (1.46 g, 13 mmol) in dry tetrahydrofuran (20 mL) was added dropwise triethyl phosphonoacetate (2.92 g, 13 mmol). The resulting solution was stirred at room temperature for 30 min. A solution of the appropriate ketone 6e–h (10 mmol) in dry tetrahydrofuran (10 mL) was added dropwise to the resulting solution. The reaction mixture was refluxed under stirring for 18 h. The reaction mixture was concentrated under vacuum, diluted with water (100 mL) and extracted with diethyl ether (3 × 50 mL). The combined organic extracts were dried (Na2SO4), filtered and evaporated under reduced pressure to give viscous oils which were purified by column chromatography using petroleum ether (40–60 °C): Diethyl ether (9:1) to afford compounds 5e–h in 75%–91% yields as pale yellow viscous oils.
(E)-3-(3,4-Dichlorophenyl)-2-butenoic acid ethyl ester [(E)-5e]. Yield 78%; IR (neat): ν (cm−1) = 1711, 1630, 1469, 1277, 1169; 1H-NMR (CDCl3): δ (ppm) = 1.21 (t, J = 7.03 Hz, 3H, CH3–CH2–), 2.42 (d, J = 1.23 Hz, 3H, 4-H), 4.12 (q, J = 7.03 Hz, 2H, –CH2–CH3), 5.99 (q, J = 1.23 Hz, 1H, 2-H), 7.10–7.44 (m, 3H, Harom.); 13C-NMR (CDCl3): δ (ppm) = 14.7 (CH3–CH2–), 18.1 (C-4), 60.5 (–CH2–CH3), 118.8 (C-2), 125.9, 128.7, 130.8, 133.2, 133.4, 142.5 (Carom.), 152.9 (C-3), 166.7 (C-1).
(Z)-3-(3,4-Dichlorophenyl)-2-butenoic acid ethyl ester [(Z)-5e]. Yield 6%; IR (neat): ν (cm−1) = 1717, 1644, 1472, 1229, 1165; 1H-NMR (CDCl3): δ (ppm) = 1.16 (t, J = 7.00 Hz, 3H, CH3–CH2–), 2.17 (d, J = 1.53 Hz, 3H, 4-H), 4.07 (q, J = 7.00 Hz, 2H, –CH2–CH3), 5.96 (q, J = 1.53 Hz, 1H, 2-H), 7.05–7.46 (m, 3H, Harom.). 13C-NMR (CDCl3): δ (ppm) = 14.4 (CH3–CH2–), 27.3 (C-4), 60.5 (CH2–CH3), 119.4 (C-2), 126.9, 129.3, 130.3, 132.1, 132.5, 141.1 (Carom.), 152.9 (C-3) 165.8 (C-1).
(E)-3-(4-Fluorophenyl)-2-butenoic acid ethyl ester [(
E)-
5f] [
27]. Yield 69%; IR (neat): ν (cm
−1) = 1710, 1631, 1602, 1508, 1233, 1157;
1H-NMR (CDCl
3): δ (ppm) = 1.32 (t,
J = 7.03 Hz, 3H, C
H3–CH
2–), 2.57 (d,
J = 1.23 Hz, 3H, 4-H), 4.22 (q,
J = 7.03 Hz, 2H, –C
H2–CH
3), 6.10 (q,
J = 1.23 Hz, 1H, 2-H), 7.02–7.11 (m, 2H, H
arom.), 7.43–7.49 (m, 2H, H
arom.).
13C-NMR (CDCl
3): δ (ppm) = 14.7 (
CH
3–CH
2–), 18.3 (C-4), 60.3 (
CH
2–CH
3), 115.8 (d,
JC-3`, F& C-5`, F = 21.99 Hz, C-3` and C-5`), 117.5 (C-2), 128.5 (d,
JC-2`, F& C-6`,F = 7.64 Hz, C-2` and C-6`), 138.6 (d,
JC-1`, F = 2.87 Hz, C-1`), 154.6 (C-3), 163.6 (d,
JC-4`, F = 249.45 Hz, C-4`), 167.1 (C-1).
(Z)-3-(4-Fluorophenyl)-2-butenoic acid ethyl ester [(Z)-5f]. Yield 10%; IR (neat): ν (cm−1) = 1718, 1638, 1603, 1509, 1226, 1153; 1H-NMR (CDCl3): δ (ppm) = 1.00 (t, J = 7.00 Hz, 3H, CH3–CH2–), 2.05 (d, J= 1.53 Hz, 3H, 4-H), 3.89 (q, J = 7.00 Hz, 2H, –CH2–CH3), 5.79 (q, J = 1.53 Hz, 1H, 2-H), 6.86–6.97 (m, 2H, Harom. ), 7.04–7.12 (m, 2H, Harom.). 13C-NMR (CDCl3): δ (ppm) = 14.4 (CH3–CH2–), 27.6 (C-4), 60.2 (–CH2–CH3), 115.3 (d, JC-3`, F& C-5`, F = 21.98 Hz, C-3`and C-5`), 118.5 (C-2), 129.2 (d, JC-2`, F& C-6`, F = 7.60 Hz, C-2` and C-6`), 137.0 (d, JC-1`, F = 3.82 Hz, C-1`), 154.7 (C-3), 162.8 (d, JC-4`, F = 247.41 Hz, C-4`), 166.2 (C-1).
(E)-3-(3-Methoxyphenyl)-2-butenoic acid ethyl ester [(
E)-
5g] [
28]. Yield 82%; IR (neat): ν (cm
−1) = 1709, 1627, 1578, 1216, 1156;
1H-NMR (CDCl
3): δ (ppm) = 1.35 (t,
J = 7.03 Hz, 3H, C
H3–CH
2–), 2.59 (d,
J = 1.23 Hz, 3H, 4-H), 3.85 (s, 3H, OCH
3), 4.25 (q,
J = 7.03 Hz, 2H, –C
H2–CH
3), 6.16 (q,
J = 1.23 Hz, 1H, 2-H), 6.19–7.34 (m, 4H, H
arom.).
13C-NMR (CDCl
3): δ (ppm) = 14.7 (
CH
3–CH
2–), 18.4 (C-4), 55.7 (OCH
3), 60.3 (–
CH
2–CH
3), 112.5 (C-2), 114.7, 117.7, 119.2, 129.9, 144.2 (C
arom.), 155.8 (C-3), 160.0 (C
arom. ), 167.2 (C-1).
(Z)-3-(3-Methoxyphenyl)-2-butenoic acid ethyl ester [(Z)-5g]. Yield 9%; IR (neat): ν (cm−1) = 1724, 1599, 1578, 1213, 1151; 1H-NMR (CDCl3): δ (ppm) = 1.13 (t, J = 7.00 Hz, 3H, CH3–CH2–), 2.20 (d, J = 1.53 Hz, 3H, 4-H), 3.83 (s, 3H, OCH3), 4.04 (q, J = 7.00 Hz, 2H, –CH2–CH3), 5.93 (q, J = 1.53 Hz, 1H, 2-H), 6.77–7.33 (m, 4H, Harom.). 13C-NMR (CDCl3): δ (ppm) = 14.4 (CH3–CH2–), 27.5 (C-4), 55.6 (OCH3), 60.2 (–CH2–CH3), 113.1 (C-2), 113.4, 118.3, 119.7, 129.4, 142.7 (Carom.), 155.3 (C-3), 159.6 (Carom.), 166.3 (C-1).
(E)-3-(4-Methoxyphenyl)-2-butenoic acid ethyl ester [(
E)-
5h] [
29]. Yield 71%; IR (neat): ν (cm
−1) = 1707, 1603, 1512, 1250, 1153;
1H-NMR (CDCl
3): δ (ppm) = 1.34 (t,
J = 7.03 Hz, 3H, C
H3–CH
2–), 2.59 (d,
J =1.23 Hz, 3H, 4-H), 3.84 (s, 3H, OCH
3), 4.23 (q,
J = 7.03 Hz, 2H, –C
H2–CH
3), 6.14 (q,
J = 1.23 Hz, 1H, 2-H), 6.91 (d,
JAB = 8.85 Hz, 2H, H
arom.), 7.48 (d,
JAB = 8.85 Hz, 2H, H
arom.).
13C-NMR (CDCl
3): δ (ppm) = 14.8 (
CH
3–CH
2–), 18.0 (C-4), 55.7 (OCH
3), 60.1 (–
CH
2–CH
3), 114.2 (C
arom.), 115.7 (C-2), 128.1, 134.7 (C
arom.), 155.2 (C-3), 160.8 (C
arom.), 167.5 (C-1).
(Z)-3-(4-Methoxyphenyl)-2-butenoic acid ethyl ester [(Z)-5h]. Yield 4%; IR (neat): ν (cm−1) = 1711, 1606, 1511, 1229, 1156; 1H-NMR (CDCl3): δ (ppm) = 1.17 (t, J = 7.00 Hz, 3H, CH3–CH2– ), 2.20 (d, J = 1.53 Hz, 3H, 4-H), 3.84 (s, 3H, OCH3), 4.07 (q, J = 7.00 Hz, 2H, –CH2–CH3), 5.91 (q, J = 1.53 Hz, 1H, 2-H), 6.91 (d, JAB = 8.85 Hz, 2H, Harom.), 7.23 (d, JAB = 8.85 Hz, 2H, Harom.); 13C-NMR (CDCl3): δ (ppm) = 14.5 (CH3–CH2–), 27.5 (C-4), 55.6 (OCH3), 60.1 (–CH2–CH3), 113.6 (Carom.), 117.5 (C-2), 128.9, 133.1 (Carom.), 155.3 (C-3), 159.8 (Carom.), 166.5 (C-1).
3.1.4. General Procedure for the Preparation of (Z)-3-Aryl-4-bromo-2-butenoic Acid Ethyl Esters 4e–h
A mixture of 3-aryl-2-butenoic acid ethyl esters 5e–h (9 mmol) and N-bromosuccinimide (1.69 g, 10 mmol) was refluxed with stirring. Benzoyl peroxide (0.02 g) was added to the reaction mixture and refluxing was continued for further 24 h. The reaction mixture was chilled and the solid succinimide was filtered off. The filtrate was dried (Na2SO4), filtered and evaporated under reduced pressure to give viscous oils which were purified by column chromatography using petroleum ether (40–60 °C): Diethyl ether (9:1) to yield mainly (Z)-3-aryl-4-bromo-2-butenoic acid ethyl esters 4e–h in 59%–71% yields as light brown viscous oils.
(Z)-4-Bromo-3-(3,4-dichlorophenyl)-2-butenoic acid ethyl ester [(Z)-4e]. Yield 59% as light brown viscous oil; IR (neat): ν (cm−1) = 1711, 1626, 1474, 1290, 1178; 1H-NMR (CDCl3): δ (ppm) = 1.36 (t, J = 7.03 Hz, 3H, CH3–CH2–), 4.29 (q, J = 7.03 Hz, 2H, –CH2–CH3), 4.93 (s, 2H, 4-H), 6.19 (s, 1H, 2-H), 7.38–7.65 (m, 3H, Harom.); 13C-NMR (CDCl3): δ (ppm) = 14.6 (CH3–CH2–), 26.3 (C-4), 61.2 (–CH2–CH3), 121.4 (C-2), 126.3, 129.0, 131.2, 133.6, 134.3, 138.9 (Carom.), 151.1 (C-3), 165.5 (C-1).
(Z)-4-Bromo-3-(4-fluorophenyl)-2-butenoic acid ethyl ester [(
Z)-
4f] [
27]. Yield 67% as light brown viscous oil; IR (neat): ν (cm
−1) = 1709, 1626, 1610, 1510, 1234, 1162;
1H-NMR (CDCl
3): δ (ppm) = 1.36 (t,
J = 7.03 Hz, 3H, C
H3–CH
2–), 4.29 (q,
J = 7.03 Hz, 2H, –C
H2–CH
3), 4.98 (s, 2H, 4-H), 6.19 (s, 1H, 2-H), 7.04–7.17 (m, 2H, H
arom.), 7.52–7.60 (m, 2H, H
arom.);
13C-NMR (CDCl
3): δ (ppm) = 14.6 (
CH
3–CH
2–), 26.9 (C-4), 61.0 (–
CH
2–CH
3), 116.3 (d,
JC-3`, F& C-5`, F = 21.57 Hz, C-3`and C-5`), 120.1 (C-2), 129.0 (d,
JC-2`, F& C-6`, F = 8.30 Hz, C-2` and C-6`), 134.9 (d,
JC-1`, F = 3.43 Hz, C-1`), 152.5 (C-3), 163.9 (d,
JC-4`, F = 239.36 Hz, C-4`), 165.9 (C-1).
(Z)-4-Bromo-3-(3-methoxyphenyl)-2-butenoic acid ethyl ester [(
Z)-
4g] [
30]. Yield 73% as light brown viscous oil; IR (neat): ν (cm
−1) = 1709, 1625, 1579, 1224, 1161;
1H-NMR (CDCl
3): δ (ppm) = 1.37 (t,
J = 7.00 Hz, 3H, C
H3–CH
2– ), 3.87 (s, 3H, OCH
3), 4.27 (q,
J = 7.00 Hz, 2H, –C
H2–CH
3), 4.98 (s, 2H, 4-H), 6.23 (s, 1H, 2-H), 6.96–7.39 (m, 4H, H
arom.).
13C-NMR (CDCl
3): δ (ppm) = 14.6 (
CH
3–CH
2–), 27.1 (C-4), 55.8 (OCH
3), 60.9 (–
CH
2–CH
3), 112.9 (C-2), 115.5, 119.4, 120.4, 130.2, 140.4 (C
arom.), 153.5 (C-3), 160.2 (C
arom. ), 165.9 (C-1).
(Z)-4-Bromo-3-(4-methoxyphenyl)-2-butenoic acid ethyl ester [(
Z)-
4h] [
31]. Yield 71% as pale yellow solid m.p. 80–82 °C; IR (neat): ν (cm
−1) = 1701, 1603, 1512, 1250, 1169;
1H NMR (CDCl
3): δ (ppm) = 1.36 (t,
J = 7.03 Hz, 3H, C
H3–CH
2–), 3.86 (s, 3H, OCH
3), 4.28 (q,
J = 7.03 Hz, 2H, –C
H2–CH
3), 5.01 (s, 2H, 4-H), 6.21 (s, 1H, 2-H), 6.96 (d,
JAB = 9.15 Hz, 2H, H
arom.), 7.55 (d,
JAB = 9.15 Hz, 2H, H
arom.).
13C-NMR (CDCl
3): δ (ppm) = 14.7 (
CH
3–CH
2–), 26.8 (C-4), 55.8 (OCH
3), 60.8 (–
CH
2–CH
3), 118.1 (C-2), 114.6, 128.4, 130.8, 161.4 (C
arom.), 152.3 (C-3), 166.2 (C-1).
3.1.5. General Procedure for the Preparation of (E)-3-Aryl-4-cyano-2-butenoic Acid Ethyl Esters 3a–c and 3e–h
A solution of tetraethylammonium cyanide (0.78 g, 5 mmol) in acetonitrile (5 mL) was added dropwise to a stirred solution of 3-aryl-4-chloro-2-butenoic acid ethyl esters 4a–c and/or (Z)-3- aryl-4-bromo-2-butenoic acid ethyl esters 4e–h (5 mmol) in acetonitrile (10 mL) under nitrogen atmosphere. After complete addition, the reaction mixture was heated at 50 °C for 18 h. The reaction mixture was cooled, diluted with diethyl ether (30 mL) and washed with water (3 × 20 mL). The organic layer was dried (Na2SO4) and evaporated under reduced pressure to give dark red viscous oils which were purified by column chromatography using petroleum ether (40–60 °C): Diethyl ether (8:2) to afford mainly (E)–3-aryl-4-cyano-2-butenoic acid ethyl esters 3a–c and/or 3e–h as pale yellow viscous oils in 42%–66% yields
(E)-4-Cyano-3-(4-chlorophenyl)-2-buenoic acid ethyl ester [(
E)-
3a] [
32]. Yield 42%; IR (neat): ν (cm
−1) = 2217, 1731, 1591, 1493, 1176, 1162;
1H-NMR (CDCl
3): δ (ppm) = 1.21 (t,
J = 7.03 Hz, 3H, C
H3–CH
2–), 3.88 ( s, 2H, 4-H), 4.15 (q.
J = 7.03 Hz, 2H, –C
H2–CH
3), 5.79 (s, 1H, 2-H), 7.39 (s, 4H, H
arom.).
13C-NMR (CDCl
3): δ (ppm) = 14.4 (
CH
3–CH
2–), 39.7 (C-4), 62.0 (
CH
2–CH
3), 99.9 (C-2), 116.9 (C≡N), 127.9, 129.9, 135.7, 137.1 (C
arom.), 154.9 (C-3), 168.8 (C-1).
(E)-4-Cyano-3-(2,4-dichloro-phenyl)-2-butenoic acid ethyl ester [(E)-3b]. Yield 46%; IR (neat): ν (cm−1) = 2223, 1733, 1585, 1472, 1180; 1H-NMR (CDCl3): δ (ppm) = 1.26 (t, J = 7.03 Hz, 3H, CH3–CH2–), 3.93 (s, 2H, 4-H), 4.16 (q, J = 7.03 Hz, 2H, –CH2–CH3), 5.62 (s, 1H, 2-H), 7.26–7.49 (m, 3H, Harom.); 13C-NMR (CDCl3): δ (ppm) = 14.4 (CH3–CH2–), 40.6 (C-4), 61.9 (–CH2–CH3), 105.2 (C-2), 115.8 (C≡N), 127.9, 130.3, 131.8, 132.8, 134.4, 136.1 (Carom.), 155.4 (C-3), 168.4 (C-1).
(E)-4-Cyano-3-(4-methyl-phenyl)-2-butenoic acid ethyl ester [(E)-3c]. Yield 66%; IR (neat): ν (cm−1) = 2214, 1733, 1603, 1314, 1175, 1159; 1H-NMR (CDCl3): δ (ppm) = 1.22 (t, J = 7.03 Hz, 3H, CH3–CH2–), 2.40 (s, 3H, 4`-CH3), 3.90 (s, 2H, 4-H), 4.15 (q, J = 7.03 Hz, 2H, –CH2–CH3), 5.78 (s, 1H, 2-H), 7.23 (d, JAB = 8.23 Hz, 2H, Harom.), 7.38 (d, JAB = 8.23 Hz, 2H, Harom.). 13C-NMR (CDCl3): δ (ppm) = 14.4 (CH3–CH2–), 21.7 (4`-CH3), 39.7 (C-4), 61.9 (–CH2–CH3), 98.3 (C-2), 117.5 (C≡N), 126.4, 130.1, 134.3, 141.4 (Carom.), 155.9 (C-3), 169.1 (C-1).
(E)-4-Cyano-3-(3,4-dichlorophenyl)-2-butenoic acid ethyl ester [(E)-3e]. Yield 44%; IR (neat): ν (cm−1) = 2219, 1732, 1550, 1472, 1179; 1H-NMR (CDCl3): δ (ppm) = 1.05 (t, J = 7.03 Hz, 3H, CH3–CH2–), 3.68 (s, 2H, 4-H), 3.98 (q, J = 7.03 Hz, 2H, –CH2–CH3), 5.61 (s, 1H, 2-H), 7.08–7.37 (m, 3H, Harom.); 13C-NMR (CDCl3): δ (ppm) = 14.4 (CH3–CH2–), 39.6 (C-4), 62.2 (–CH2–CH3), 101.1 (C-2), 116.5 (C≡N), 125.8, 128.5, 131.4, 133.9, 135.2, 137.3, (Carom.),153.9 (C-3), 168.5 (C-1).
(E)-4-Cyano-3-(4-fluorophenyl)-2-butenoic acid ethyl ester [(E)-3f]. Yield 48%; IR (neat): ν (cm−1) = 2217, 1732, 1601, 1511, 1237, 1162; 1H-NMR (CDCl3): δ (ppm) = 1.22 (t, J = 7.00 Hz, 3H, CH3–CH2–), 3.89 (s, 2H, 4-H), 4.16 (q, J = 7.00 Hz, 2H, –CH2–CH3), 5.76 (s, 1H, 2-H), 7.07–7.16 (m, 2H, Harom.), 7.43–7.51 (m, 2H, Harom.); 13C-NMR (CDCl3): δ (ppm) = 14.4 (CH3–CH2–), 39.8 (C-4), 62.0 (–CH2–CH3), 99.4 (C-2), 116.5 (d, JC-3`, F& C-5`, F = 21.95 Hz, C-3` and C-5`), 117.1 (C≡N), 128.6 (d, JC-2`, F& C-6`, F = 8.57 Hz, C-2` and C-6`), 133.5 (d, JC-1`, F = 3.82 Hz, C-1`), 155.1 (C-3), 164.4 (d, JC-4`, F = 252.23 Hz, C-4`), 168.9 (C-1).
(E)-4-Cyano-3-(3-methoxy-phenyl)-2-butenoic acid ethyl ester [(E)-3g]. Yield 53%; IR (neat): ν (cm−1) = 2216, 1733, 1599, 1577, 1229, 1177; 1H-NMR (CDCl3): δ (ppm) = 1.22 (t, J = 7.03 Hz, 3H, CH3–CH2–), 3.84 (s, 3H, OCH3), 3.89 (s, 2H, 4-H), 4.16 (q, J = 7.03 Hz, 2H, –CH2–CH3), 5.79 (s, 1H, 2-H), 6.97–7.37 (m, 4H, Harom.); 13C-NMR (CDCl3): δ (ppm) = 14.4 (CH3–CH2–), 39.8 (C-4), 55.8 (OCH3), 61.9 (–CH2–CH3), 99.7 (C-2), 112.4, 116.2 (Carom.), 117.2 (C≡N), 118.9, 130.5, 138.7 (Carom.), 156.2 (C-3), 160.3 (Carom.), 168.9 (C-1).
(E)-4-Cyano-3-(4-methoxy-phenyl)-2-butenoic acid ethyl ester [(
E)-
3h] [
32]. Yield 45%; IR (neat): ν (cm
−1) = 2213, 1732, 1599, 1514, 1251, 1179;
1H-NMR (CDCl
3): δ (ppm) = 1.21 (t,
J = 7.03 Hz, 3H, C
H3–CH
2–), 3.84 (s, 3H, OCH
3), 3.88 (s, 2H, 4-H), 4.15 (q,
J = 7.03 Hz, 2H, –C
H2–CH
3), 5.72 (s, 1H, 2-H), 6.92 (d,
JAB = 8.85 Hz, 2H, H
arom.), 7.43 (d,
JAB = 8.85 Hz, 2H, H
arom.).
13C-NMR (CDCl
3): δ (ppm) = 14.4 (
CH
3–CH
2–), 39.6 (C-4), 55.8 (OCH
3), 61.9 (–
CH
2–CH
3), 96.9 (C-2), 117.7 (C≡N), 155.3 (C-3), 114.8, 128.1, 129.4, 161.9 (C
arom.), 169.2 (C-1).
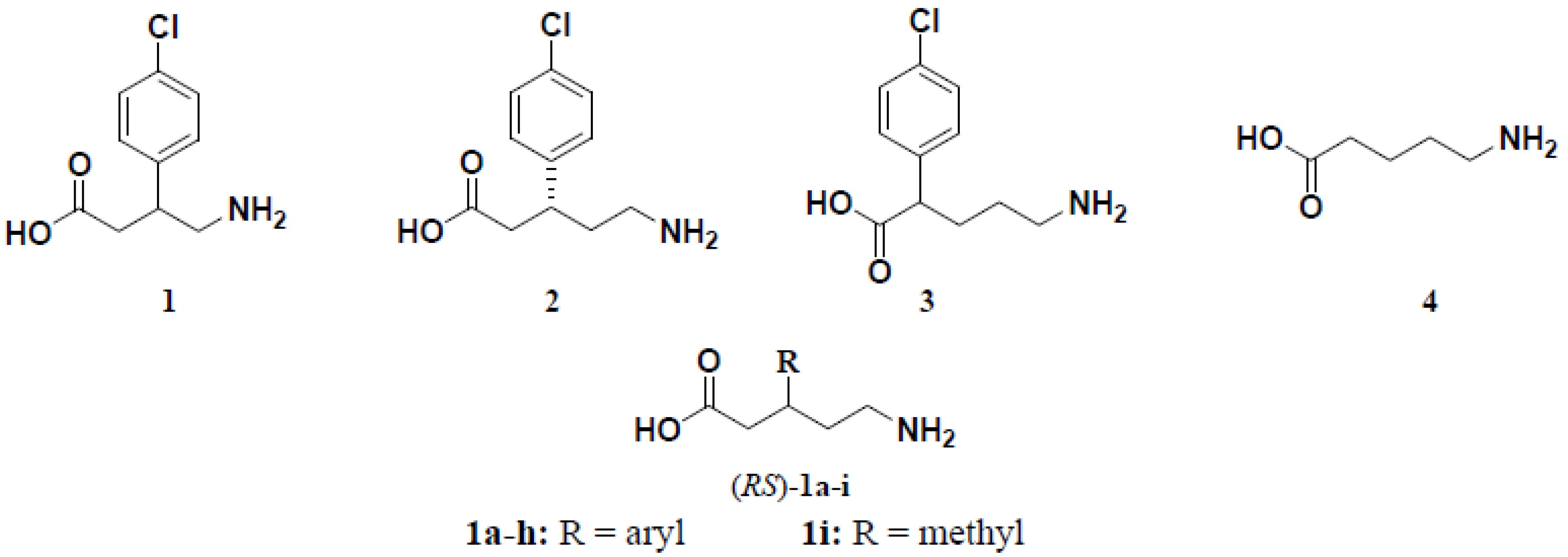
3.1.6. General Procedure for the Preparation of (R,S)-5-Amino-3-arylpentanoic Acid Hydrochlorides 1a–h
To a solution of (E)–3-aryl-4-cyano-2-butenoic acid ethyl esters 3a–c and/or 3e–h (2 mmol) in 95% ethanol (10 mL) and concentrated hydrochloric acid (1 mL) was added PtO2 (0.05 g) for compounds 3a, 3b, 3e and 3f or 10% Pd/C (0.10 g) for compounds 3b, 3c, 3g and 3h. The reaction mixture was hydrogenated on a Parr shaker apparatus under 4 bar of H2 for 18 h at room temperature. The catalyst was removed by filtration and the solvent was evaporated under reduced pressure to give (RS)-5-amino-3-arylpentanoic acid ethyl ester hydrochlorides 2a–h which were dissolved in 5 N hydrochloric acid (15 mL) and washed with diethyl ether (2 × 10 mL). Without further purification, the aqueous layer was refluxed with stirring for 4 h. The reaction mixture was evaporated under vacuum to give (RS)-5-amino-3-aryl-pentanoic acid hydrochlorides 1a–h which were recrystallized from the isopropanol.
(R,S)-5-Amino-3-(4-chlorophenyl)pentanoic acid hydrochloride (1a). Yield 76% as white solid m.p. 201–203 °C; IR (neat): ν (cm−1) = 3200–2727 and 1726; 1H-NMR (D2O): δ (ppm) = 1.81–2.05 (m, 2H, 4-H), 2.50–2.87 (m, 4H, 2-H and 5-H), 2.99-3.12 (m, 1H, 3-H), 7.17 (d, JAB = 8.55 Hz, 2H, Harom.), 7.26 (d, JAB = 8.55 Hz, 2H, Harom.); 13C-NMR (D2O): δ (ppm) = 33.2 (C-4), 38.0 (C-2), 39.1 (C-3), 41.1 (C-5), 129.2, 129.4, 132.7, 140.9 (Carom.), 176.6 (C-1); MS (EI), m/z (%): 209 (100), 181 (30), 138 (64), 97 (56), 43 (43); MS (CI), m/z (%): 227 [(100), M+].: Anal. Calcd. for C11H15Cl2NO2: C 50.02, H 5.72, N 5.30; found C 49.93, H 5.72, N 5.36.
(R,S)-5-Amino-3-(2,4-chlorophenyl)pentanoic acid hydrochloride (1b). Yield 70% as white solid m.p. 215–217 °C; IR (neat): ν (cm−1) = 3200–2700 and 1728; 1H-NMR (D2O): δ (ppm) = 1.87–2.12 (m, 2H, 4-H), 2.57–2.98 (m, 4H, 2-H and 5-H), 3.58–3.70 (m, 1H, 3-H), 7.20–7.32 (m, 3H, Harom.); 13C-NMR (D2O): δ (ppm) = 32.6 (C-4), 35.0 (C-2), 37.8 (C-3), 39.7 (C-5), 128.3, 129.3, 129.7, 133.1, 134.6, 138.5 (Carom.), 176.3 (C-1); MS (EI), m/z (%): 243 (37), 208 (72), 172 (49), 97 (100), 43 (46); MS (CI), m/z (%): 261 [(100), M+ -1]; Anal. Calcd. for C11H14Cl3NO2: C 44.25, H 4.73, N 4.69; found C 44.10, H 4.76, N 4.79.
(R,S)-5-Amino-3-(4-methylphenyl)pentanoic acid hydrochloride (1c). Yield 78% as white solid m.p. 204–206 °C; IR (neat): ν (cm−1) = 3200-2720 and 1726; 1H-NMR (D2O): δ (ppm) = 1.80–2.03 (m, 2H, 4-H), 2.19 (s, 3H, 4`-CH3), 2.50–2.86 (m, 4H, 2-H and 4-H), 2.96–3.08 (m, 1H, 3-H), 7.12 (s, 4H, Harom.); 13C-NMR (D2O): δ (ppm) = 20.5 (4`-CH3), 33.3 (C-4), 38.1 (C-2), 39.3 (C-3), 41.3 (C-5), 127.8, 129.9, 137.8, 139.2 (Carom.), 176.9 (C-1); MS (CI), m/z (%): 207 [(100), M+]; Anal. Calcd. for C12H18ClNO2: C 59.14, H 7.44, N 5.75; found C 58.75, H 7.39, N 5.76.
(R,S)-5-Amino-3-phenylpentanoic acid hydrochloride (1d). Yield 69% as white solid m.p. 195–196 °C; IR (neat): ν (cm−1) = 3200–2690 and 1724; 1H-NMR (D2O): δ (ppm) = 1.83–2.06 (m, 2H, 4-H), 2.54–2.86 (m, 4H, 2-H and 5-H), 3.00–3.12 (m, 1H, 3-H), 7.18–7.33 (m, 5H, Harom.); 13C-NMR (D2O): δ (ppm) = 33.3 (C-4), 38.1 (C-2), 39.7 (C-3), 41.2 (C-5), 127.8, 127.9, 129.4, 142.3 (Carom.), 176.9 (C-1); MS (EI), m/z (%): 194 [(10) M++ 1], 175 (95), 104 (100), 91 (41), 43 (42); Anal. Calcd. for C11H16ClNO2: C 57.52, H 7.02, N 6.09; found C 57.12, H 7.13, N 5.99.
(R,S)-5-Amino-3-(3,4-chlorophenyl)pentanoic acid hydrochloride (1e). Yield 80% as white solid m.p. 201–203 °C; IR (neat): ν (cm−1) = 3200–2700 and 1715; 1H-NMR (D2O): δ (ppm) = 1.81–2.05 (m, 2H, 4-H), 2.51–2.95 (m, 4H, 2-H and 5-H), 3.00–3.12 (m, 1H, 3-H), 7.09–7.39 (m, 3H, Harom.); 13C-NMR (D2O): δ (ppm) = 32.9 (C-4), 37.9 (C-2), 38.9 (C-3), 40.9 (C-5),127.7, 129.8, 130.7, 131.1, 132.4, 142.9 (Carom.), 176.4 (C-1); MS (CI), m/z (%): 261 [(100), M+ -1]; Anal. Calcd. for C11H14Cl3NO2: C 44.25, H 4.73, N 4.69; found C 44.04, H 4.99, N 4.72.
(R,S)-5-Amino-3-(4-fluorophenyl)pentanoic acid hydrochloride (1f). Yield 81% as white solid m.p. 208–210 °C; IR (neat): ν (cm−1) = 3200–2700 and 1724. 1H-NMR (D2O): δ (ppm) = 1.81–2.05 (m, 2H, 4-H), 2.49–2.87 (m, 4H, 2-H and 5-H), 3.00–3.12 (m, 1H, 3-H), 6.96–7.03 (m, 2H, Harom.), 7.17–7.23 (m, 2H, Harom.); 13C-NMR (D2O): δ (ppm) = 33.3 (C-4), 38.0 (C-2), 38.9 (C-3), 41.3 (C-5), 115.9 (d, JC-3`, F& C5`, F = 21.38 Hz, C-3` and C-5`), 129.5 (d, JC-2`, F& C-6`, F = 8.17 Hz, C-2` and C-6`), 137.9 (d, JC-1`, F = 3.02 Hz, C-1`), 162.0 (d, JC-4`, F = 242.87 Hz, C-4`), 176.8 (C-1); MS (CI), m/z (%): 211 [(100), M+]; Anal. Calcd. for C11H15ClFNO2: C 53.34, H 6.10, N 5.66; found C 53.17, H 6.34, N 5.66.
(R,S)-5-Amino-3-(3-methoxyphenyl)pentanoic acid hydrochloride (1g). Yield 85% as pale yellow solid m.p. 182–184 °C; IR (neat): ν (cm−1) = 3200–2700 and 1722; 1H-NMR (D2O): δ (ppm) = 1.82–2.04 (m, 2H, 4-H), 2.52–2.87 (m, 4H, 2-H and 5-H), 2.98–3.10 (m, 1H, 3-H), 3.69 (s, 3H, OCH3), 6.77–7.25 (m, 4H, Harom.); 13C-NMR (D2O): δ (ppm) = 33.2 (C-4), 38.1 (C-2), 39.7 (C-3), 41.1 (C-5), 55.7 (OCH3), 113.1, 113.6, 120.6, 130.6, 144.2, 159.6 (Carom.), 176.8 (C-1); MS (CI), m/z (%): 223 [(100), M+]; Anal. Calcd. for C12H18ClNO3: C 55.49, H 6.99, N 5.39; found C 55.20, H 7.01, N 5.33.
(R,S)-5-Amino-3-(4-methoxyphenyl)pentanoic acid hydrochloride (1h). Yield 76% as pale yellow solid m.p. 194–195 °C; (neat): ν (cm−1) = 3200–2721 and 1724; 1H-NMR (D2O): δ (ppm) = 1.79–2.03 (m, 2H, 4-H), 2.49–2.86 (m, 4H, 2-H and 5-H), 2.96–3.08 (m, 1H, 3-H), 3.68 (s, 3H, OCH3), 6.86 (d, JAB = 8.85 Hz, 2H, Harom.), 7.15 (d, JAB = 8.85 Hz, 2H, Harom.). 13C-NMR (D2O): δ (ppm) = 33.4 (C-4), 38.1 (C-2), 38.9 (C-3), 41.4 (C-5), 55.8 (OCH3), 114.7, 129.0, 134.8, 158.2 (Carom.), 176.9 (C-1); MS (CI), m/z (%): 223 [(100), M+]; Anal. Calcd. for C12H18ClNO3: C 55.49, H 6.99, N 5.39; found C 55.23, H 7.07, N 5.35.
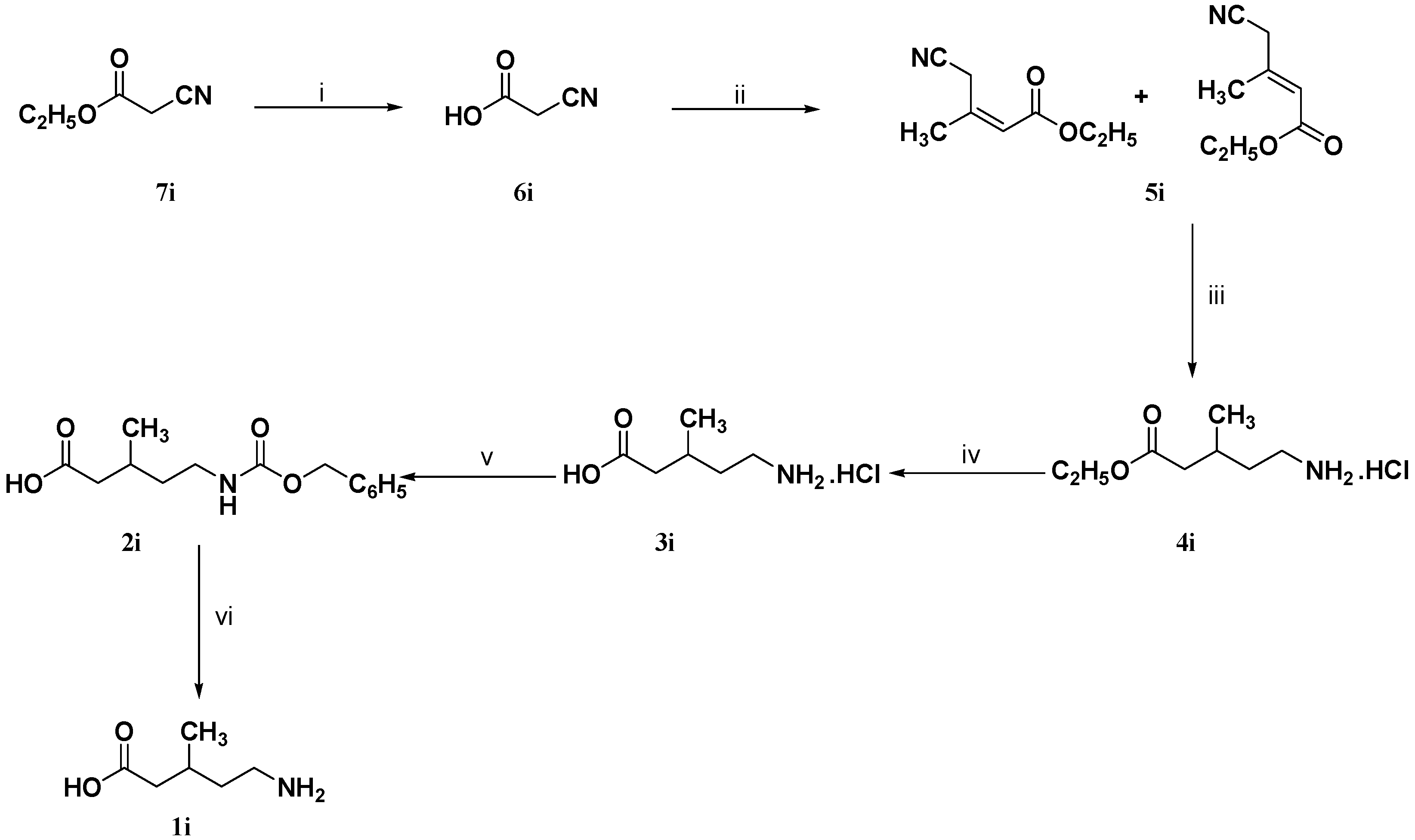
3.1.7. Synthesis of Cyanoacetic Acid (6i)
A mixture of ethyl cyanoacetate (7i, 10 g, 88 mmol) and 1 N hydrochloric acid (35 mL) was heated at 100 °C for 1.5 h. The reaction mixture was evaporated under reduced pressure to give 7.5 g (100%) of 6i as a colorless crystals m.p. 63–65 °C which was pure enough to be used in the next step without further purification. IR (neat): ν (cm−1) = 3300–2973, 2269, 1725, 1388, 1183; 1H-NMR (DMSO-d6): δ (ppm) = 3.28 (s, 2H, 2-H), 8.1–8.7 (br.s, 1H, COOH); 13C-NMR (DMSO-d6): δ (ppm) = 25.5 (C-2), 116.3 (C≡N), 166.5 (C-1).
3.1.8. Synthesis of 4-Cyano-3-methyl-2-butenoic Acid Ethyl Ester (5i)
A mixture of cyanoacetic acid (
6i, 4.51 g, 53 mmol), ethyl acetoacetate (6.51 g, 50 mmol), ammonium acetate (0.77 g, 10 mmol) and acetic acid (1.58 g, 1.5 mL, 26.3 mmol) in benzene (15 mL) was refluxed for 8 h using a Dean-Stark apparatus. The reaction mixture was evaporated under reduced pressure, water (10 mL) was added to the residue and extracted with diethyl ether (3 × 15 mL). The organic layer was separated, dried (Na
2SO
4) and evaporated under vacuum. The residue was distilled under vacuum to yield 5.2 g (68%) of
5i as a colorless oil b.p. 100–102 °C/5 mm (lit. [
19] 130 °C/20 mm) with
E/
Z ratio = 1.7 as detected by
1H-NMR. IR (neat): ν (cm
−1) = 2221, 1733, 1636, 1175, 1161;
1H-NMR (CDCl
3): δ (ppm) = 1.24–1.31 (2 x t, 3H, C
H3–CH
2–), 2.01 [d,
J = 1.53 Hz, 3H, (
Z)-3-CH
3], 2.13 [d,
J = 0.93 Hz, 3H, (
E)-3-CH
3], 3.18 [d,
J = 0.90 Hz, 2H, (
E)-4-H], 3.42 [s, 2H, (
Z)-4-H], 4.12–4.22 (2 × q, 2H, –C
H2–CH
3), 5.29–5.32 (m, 1H, 2-H);
13C-NMR (CDCl
3): δ (ppm) = 14.5 (
CH
3–CH
2–), 21.7 [(
E)-3-CH
3], 23.8 [(
Z)-3-CH
3], 41.6 [(
Z)-C-4], 43.9 [(
E)-C-4], 61.8 (–CH
2–CH
3), 99.7 [(
E)-C-2], 99.8 [(
Z)-C-2], 116.6 [(
Z)-C≡N], 116.7 [(
E)-C≡N], 157.1 [(
Z)-C-3], 157.2 [(
E)-C-3], 168.9 [(
Z)-C-1], 169.2 [(
E)-C-1].
3.1.9. Synthesis of (R,S)-5-Benzyloxycarbonylamino-3-methylpentanoic Acid (2i)
To a solution of 4-cyano-3-methyl-2-butenoic acid ethyl ester (5i, 0.77 g, 5 mmol) in 95% ethanol (25 mL) was added concentrated hydrochloric acid (1 mL) and 10% Pd/C (0.26 g). The reaction mixture was hydrogenated on a Parr shaker apparatus under 4 bar of H2 for 18 h at room temperature. The catalyst was removed by filtration and the solvent was evaporated under vacuum to give (RS)-5-amino-3-methyl-pentanoic acid ethyl ester hydrochloride (4i) which was dissolved in 5 N hydrochloric acid (10 mL) and extracted with diethyl ether (3 × 10 mL). Without further purification the aqueous layer was refluxed under stirring for 4 h. The reaction mixture containing (RS)-5-amino-3-methyl-pentanoic acid hydrochloride (3i) was cooled (0–5 °C) and basified using 4 N sodium hydroxide solution (14 mL). To this basic solution was added simultaneously in portions and under cooling (0 °C) benzyl chloroformate (0.85 g, 5 mmol) and 4 N sodium hydroxide solution (1.25 mL) during 30 min. The reaction mixture was extracted with diethyl ether (3 × 10 mL), the aqueous layer was cooled (0–5 °C) and acidified using concentrated hydrochloric acid. The reaction mixture was extracted with diethyl ether (3 × 10 mL), dried (Na2SO4) and evaporated under reduced pressure to give 0.86 g (65%) of 2i as a viscous pale yellow oil which was used in the next step without further purification. IR (neat): ν (cm−1) = 3066–2588, 1699, 1528, 1454, 1523; 1H-NMR (CDCl3): δ (ppm) = 1.02 (d, J = 6.1 Hz, 3H, 3-CH3), 1.39–1.50 (m, 1H, 4-Ha), 1.53–1.67 (m, 1H, 4-Hb), 1.98–2.15 (m, 1H, 3-H), 2.21–2.55 (m, 2H, 2-H), 3.25 (m, 2H, 5-H), 5.03 (br.s 1H, N–H), 5.13 (s, 2H, –CH2–C6H5), 7.37 (s, 5H, Harom.), 10.27 (br.s, 1H, COOH); 13C-NMR (CDCl3): δ (ppm) = 19.9 (3-CH3), 27.9 (C-3), 36.8 (C-4), 39.3 (C-5), 41.7 (C-2), 67.2 (–CH2–C6H5), 127.5, 128.6, 128.9, 136.9 (Carom.), 157 (O=C–N–H), 178.8 (C-1).
3.1.10. Synthesis of (R,S)-5-Amino-3-methylpentanoic Acid (1i)
To a solution of (
R,S)-5-benzyloxycarbonylamino-3-methyl-pentanoic acid (
2i, 0.53 g, 2 mmol) in 50% 2-propanol (10 mL) was added 10% Pd/C (0.85 g). The reaction mixture was hydrogenated on a Parr shaker apparatus under 4 bar of H
2 for 18 h at room temperature. The catalyst was removed by filtration and the solvent was evaporated under vacuum. The residue was recrystallized (2-propanol/water) to give 0.18 g (69%) of
1i as a white powder m.p. 164–165 °C (lit. [
21] 133–135 °C). IR (neat): ν (cm
−1) = 3019–2659, 1630, 1528, 1460, 1398;
1H-NMR (D
2O): δ (ppm) = 0.78 (d,
J = 6.73 Hz, 3H, 3-CH
3), 1.31–1.58 (m, 2H, 4-H), 1.71–1.85 (m, 1H, 3-H), 1.87–1.96 (m, 1H, 2-H
a), 2.02–2.10 (m, 1H, 2-H
b), 2.77–2.95 (m, 2H, 5-H);
13C-NMR (CDCl
3): δ (ppm) = 19.1 (3-CH
3), 28.5 (C-3), 33.9 (C-4), 37.9 (C-5), 44.7 (C-2), 181.9 (C-1); MS (CI), m/z (%): 149.1 [(100), M
++18]; Anal. Calcd. for C
6H
13NO
2: C 54.94, H 9.99, N 10.68; found C 54.64, H 10.11, N 10.60.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}