Palladium-Catalyzed Synthesis of Natural and Unnatural 2-, 5-, and 7-Oxygenated Carbazole Alkaloids from N-Arylcyclohexane Enaminones

Abstract
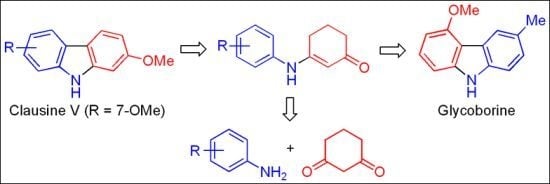
:
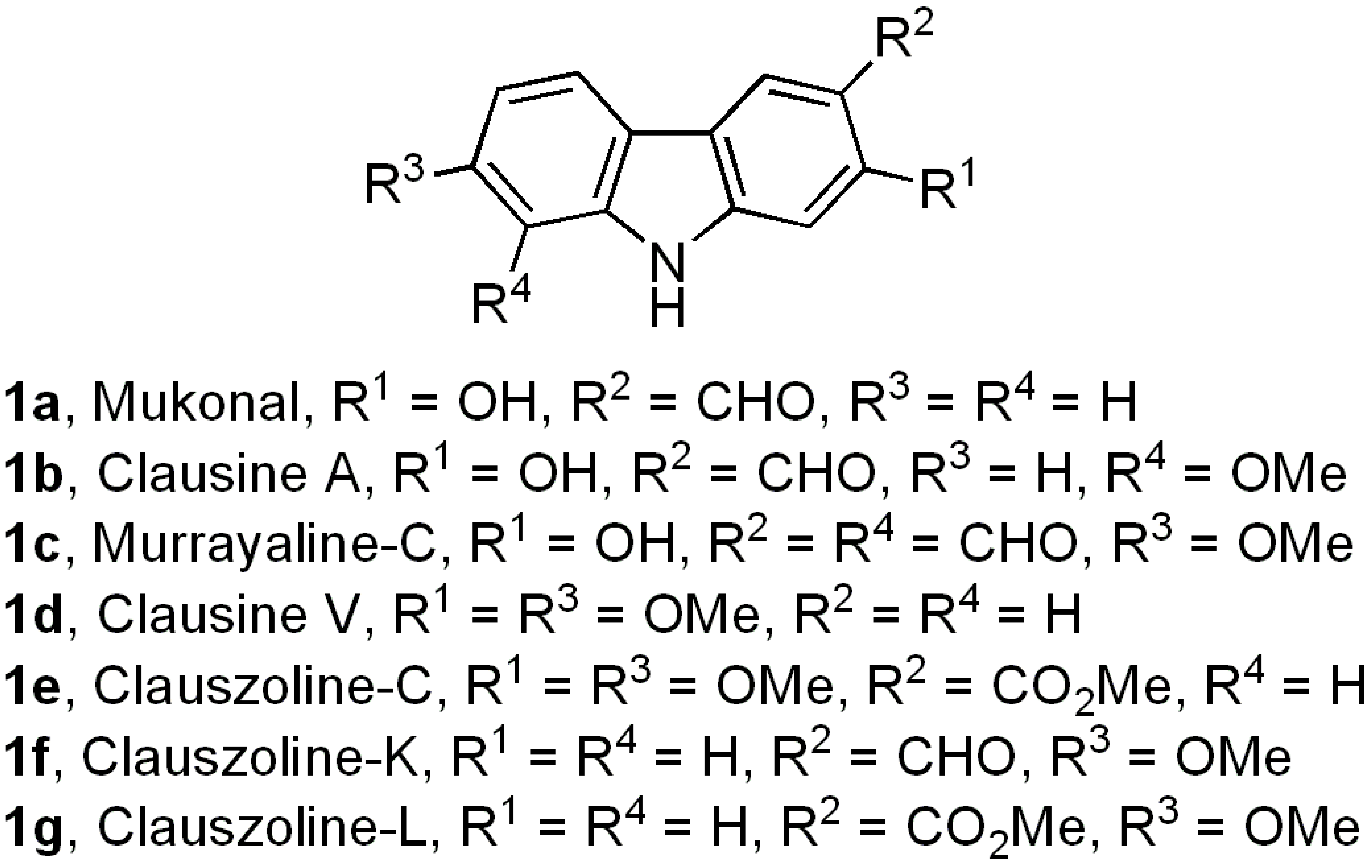
1. Introduction

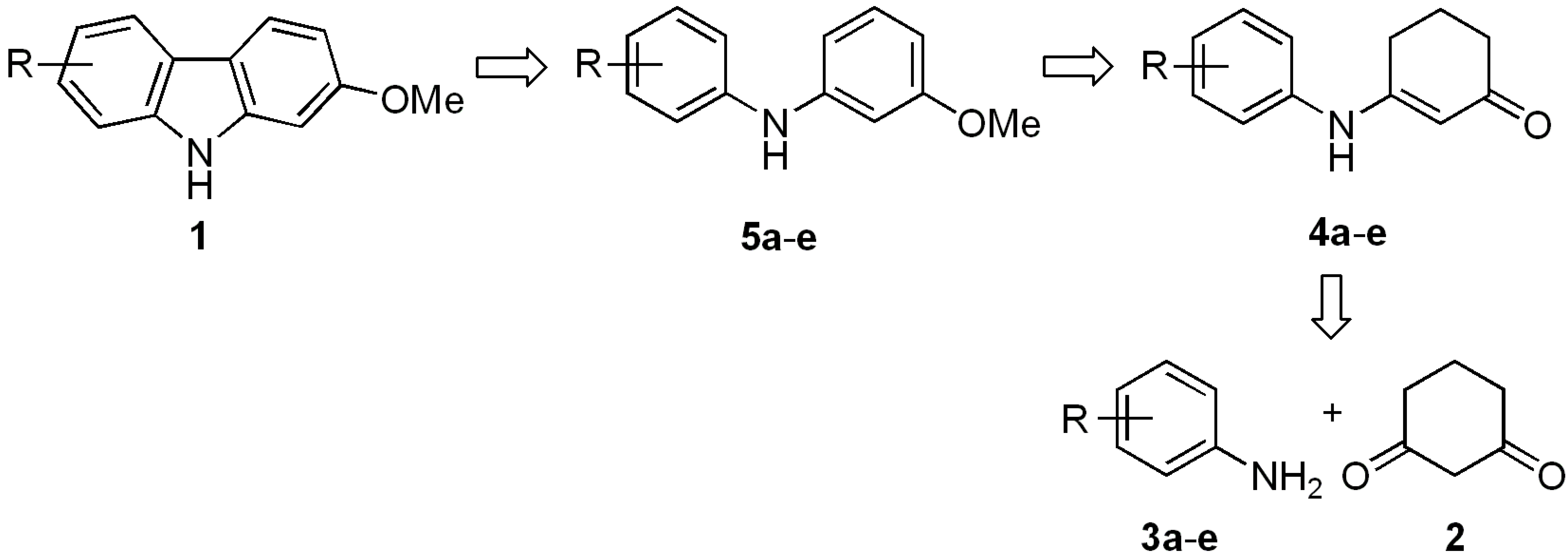
2. Results and Discussion
2.1. Synthesis of Diarylamines


| Entry | 3 (Ar) | 4 (%) b |
|---|---|---|
| 1 | 3a (C6H4-4-Me) | 4a (92) |
| 2 | 3b (C6H4-4-OMe) | 4b (95) |
| 3 | 3c (C6H4-3-Me) | 4c (90) |
| 4 | 3d (C6H4-3-OMe) | 4d (93) |
| 5 | 3e (C6H3-3,5-(OMe)2 | 4e (96) |

{kind=link}
{kind=link}
{kind=link}
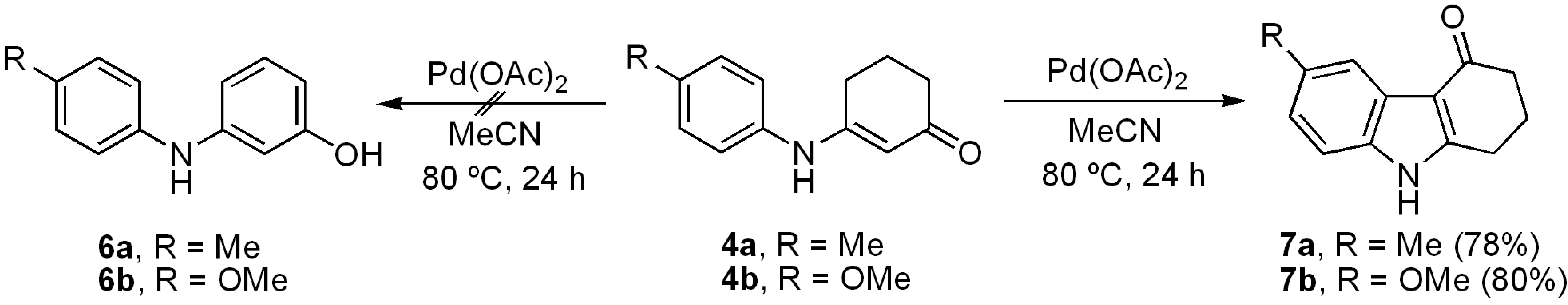{kind=link}
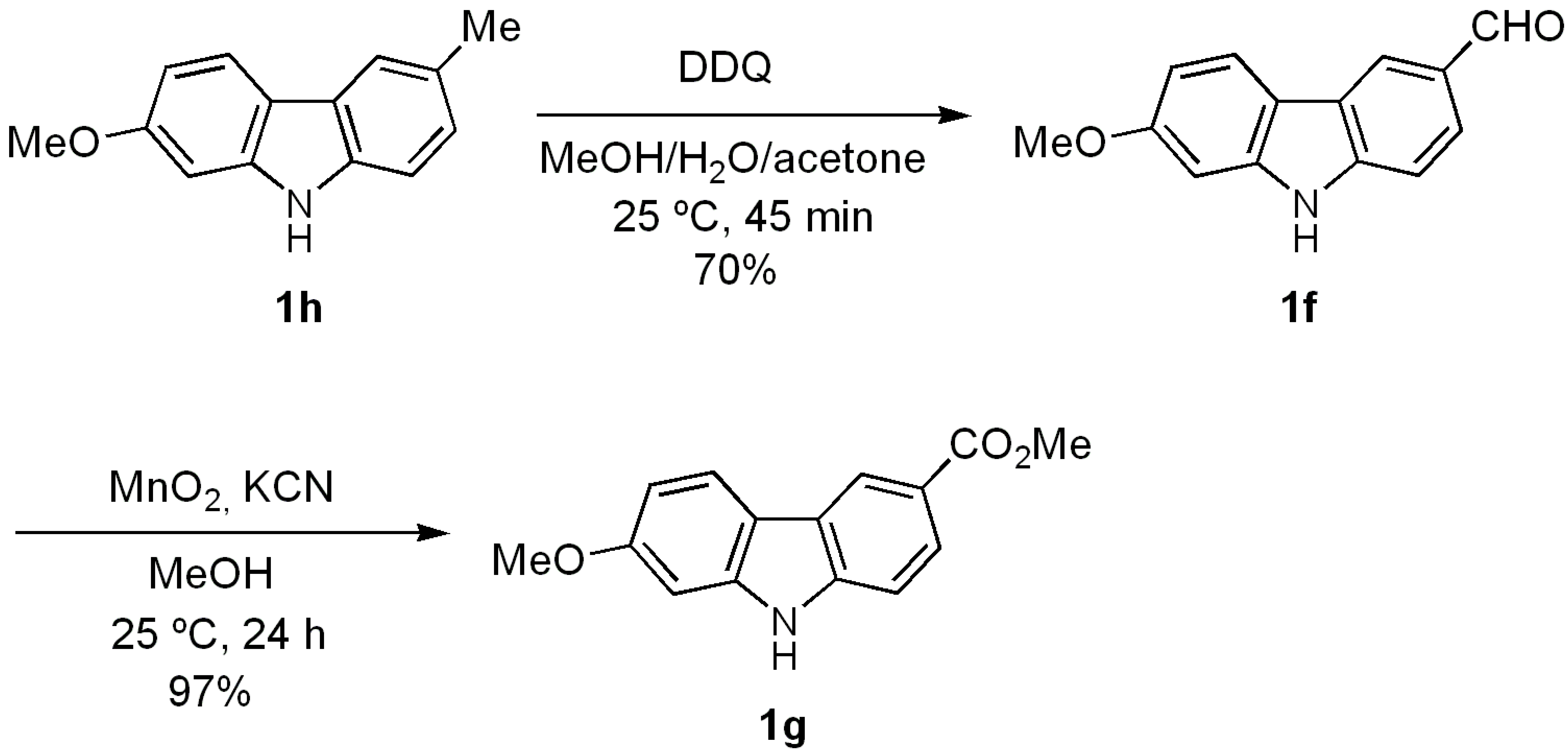{kind=link}
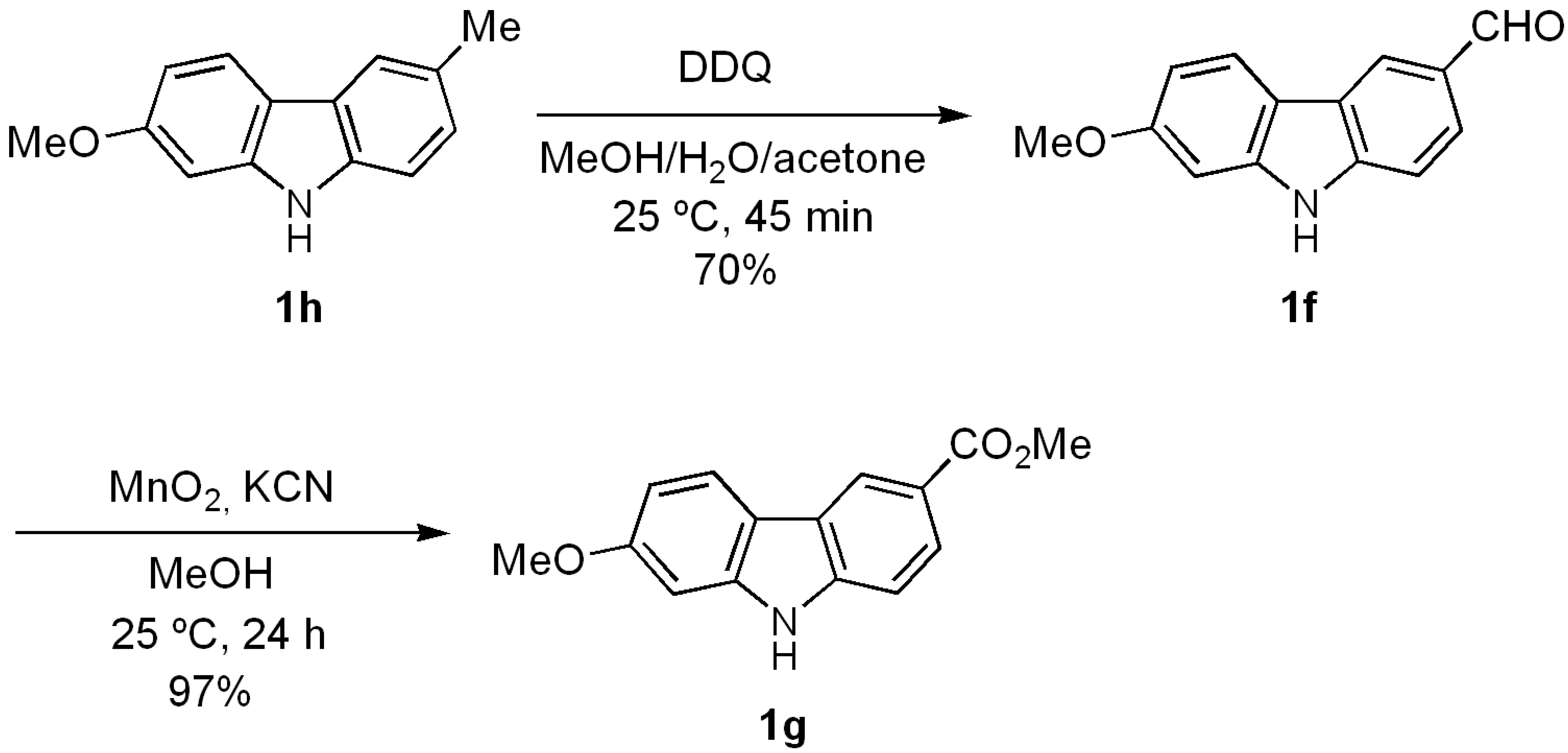
{kind=link}
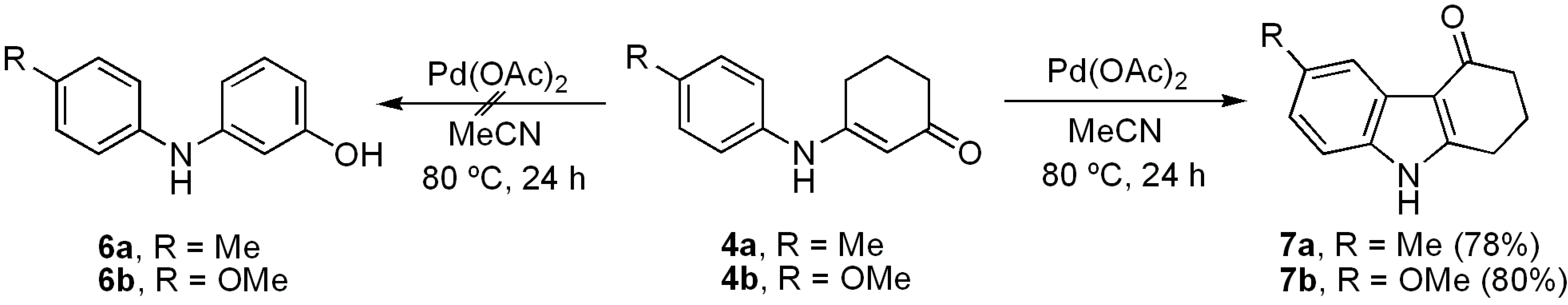
| Entry | 4 (R) | Pd/C (10%) (mol%) b | 6 (%) c | 5 (%) d |
|---|---|---|---|---|
| 1 e | 4b (4-OMe) | 1.9 | 6b (65) | ---- |
| 2 f | 4b (4-OMe) | 3.8 | 6b (75) | ---- |
| 3 | 4b (4-OMe) | 5.7 | 6b (87) | 5b (85) |
| 4 | 4a (4-Me) | 5.7 | 6a (85) | 5a (83) |
| 5 | 4c (3-Me) | 5.7 | (g) | 5c (87) |
| 6 | 4d (3-OMe) | 5.7 | 6d (84) | 5d (81) |
| 7 | 4e (3,5-(OMe)2 | 5.7 | 6e (88) | 5e (86) |
2.2. Synthesis of Carbazoles

| Entry | 5 (R) | 1 | Isolated yield (%) b |
|---|---|---|---|
| 1 | 5a (4-Me) |  | 80 |
| 2 | 5b (4-OMe) |  | 87 |
| 3 | 5c (3-Me) |  | 82 |
| 4 | 5d (3-OMe) |  | 90 |
| 5 | 5e (3,5-(OMe)2 |  | 92 |
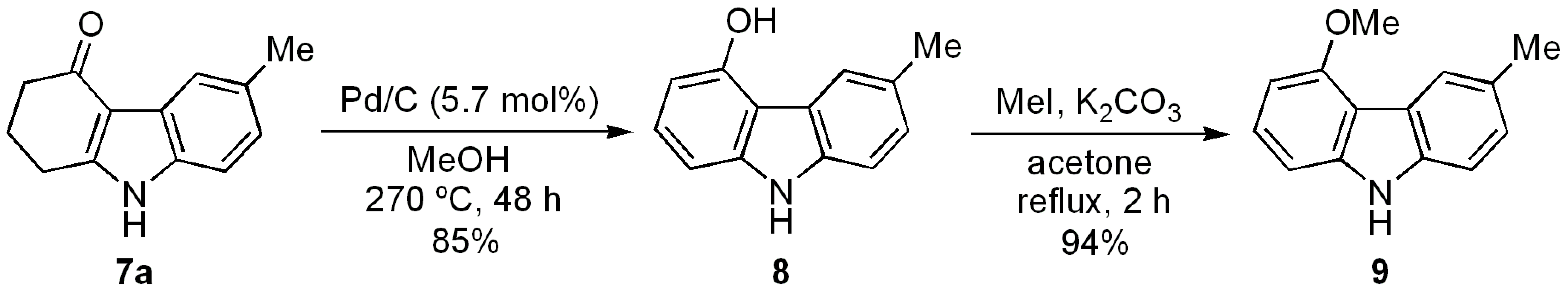
2.3. Total Synthesis of Glycoborine (Glycrophylamine, 9)
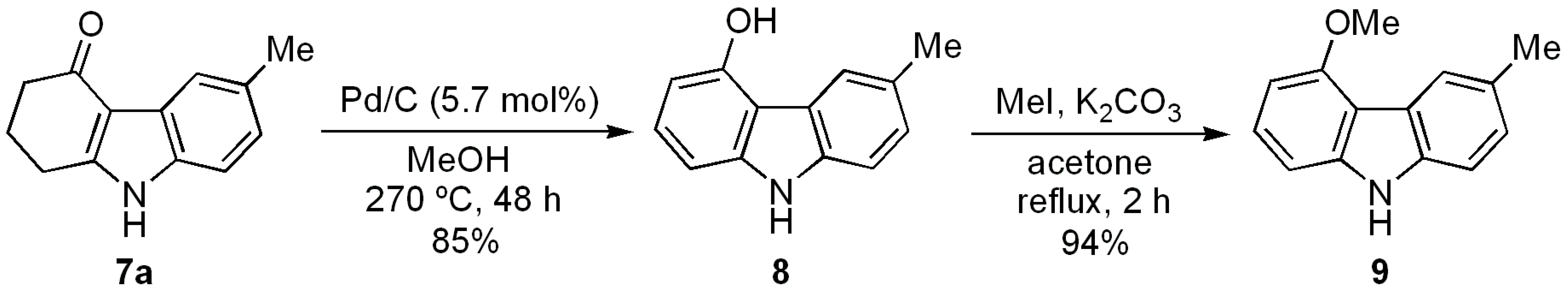
3. Experimental
3.1. General
3.2. Synthesis and Characterization
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Knölker, H.J.; Reddy, K.R. Isolation and synthesis of biologically active carbazole alkaloids. Chem. Rev. 2002, 102, 4303–4427. [Google Scholar] [CrossRef]
- Knölker, H.J.; Reddy, K.R. Chemistry and Biology of Carbazole Alkaloids. In The Alkaloids Chemistry and Biology; Cordell, G.A., Ed.; Academic Press: Amsterdam, The Netherlands, 2008; Volume 65. [Google Scholar]
- Schmidt, A.W.; Reddy, K.R.; Knölker, H.J. Occurrence, biogenesis, and synthesis of biologically active carbazole alkaloids. Chem. Rev. 2012, 112, 3193–3328. [Google Scholar] [CrossRef]
- Knölker, H.J. Occurrence, biological activity, and convergent organometallic synthesis of carbazole alkaloids. Top. Curr. Chem. 2005, 244, 115–148. [Google Scholar]
- Knölker, H.J. Synthesis of biologically active carbazole alkaloids using selective transition-metal-catalyzed coupling reactions. Chem. Lett. 2009, 38, 8–13. [Google Scholar] [CrossRef]
- Chakraborty, D.P. Progress in the Chemistry of Organic Natural Products; Herz, W., Grisebach, H., Kirby, G.W., Eds.; Springer-Verlag: Wien, Austria, 1977; Volume 34. [Google Scholar]
- Chaichantipyuth, C.; Pummangura, S.; Naowsaran, K.; Thanyavuthi, D. Two new bioactive carbazole alkaloids from the rootbark of clausena harmandiana. J. Nat. Prod. 1988, 51, 1285–1288. [Google Scholar] [CrossRef]
- Wu, T.S.; Huang, S.C.; Wu, P.L.; Teng, C.M. Carbazole alkaloids from Clausena excavata and their biological activity. Phytochemistry 1996, 43, 133–140. [Google Scholar]
- Sunthitikawinsakul, A.; Kongkathip, N.; Kongkathip, B.; Phonnakhu, S.; Daly, J.W.; Spande, T.F.; Nimit, Y.; Rochanaruangrai, S. Coumarins and carbazoles from Clausena excavata exhibited antimycobacterial and antifungal activities. Planta Med. 2003, 69, 155–157. [Google Scholar] [CrossRef]
- Ma, C.; Case, R.J.; Wang, Y.; Zhang, H.J.; Tan, G.T.; Hung, N.V.; Cuong, N.M.; Franzblau, S.G.; Soejarto, D.D.; Fong, H.H.S.; Pauli, G.F. Anti-tuberculosis constituents from the stem bark of Micromelum hirsutum. Planta Med. 2005, 71, 261–267. [Google Scholar] [CrossRef]
- Meragelman, K.M.; McKee, T.C.; Boyd, M.R. Siamenol, a new carbazole alkaloid from Murraya siamensis. J. Nat. Prod. 2000, 63, 427–428. [Google Scholar] [CrossRef]
- Kongkathip, B.; Kongkathip, N.; Sunthitikawinsakul, A.; Napaswat, C.; Yoosook, C. Anti-HIV-1 constituents from Clausena excavata: Part II. Carbazoles and a pyranocoumarin. Phytother. Res. 2005, 19, 557–731. [Google Scholar] [CrossRef]
- Wang, J.; Zheng, Y.; Efferth, T.; Wang, R.; Shen, Y.; Hao, X. Indole and carbazole alkaloids from Glycosmis montana with weak anti-HIV and cytotoxic activities. Phytochemistry 2005, 66, 697–701. [Google Scholar] [CrossRef]
- Yenjai, C.; Sripontan, S.; Sriprajun, P.; Kittakoop, P.; Jintasirikul, A.; Tanticharoen, M.; Thebtaranonth, Y. Coumarins and carbazoles with antiplasmodial activity from Clausena harmandiana. Planta Med. 2000, 66, 277–279. [Google Scholar] [CrossRef]
- Shoeb, A.; Anwer, F.; Kapil, R.S.; Popli, S.P. N-Alkylaminocarbazoles as potential anticonvulsant and diuretic agents. J. Med. Chem. 1973, 16, 425–427. [Google Scholar] [CrossRef]
- Ferris, R.M.; White, H.L.; Tang, F.L.M.; Russell, A.; Harfenist, M. Rimcazole (BW 234U), a novel antipsychotic agent whose mechanism of action cannot be explained by a direct blockade of postsynaptic dopaminergic receptors in brain. Drug Dev. Res. 1986, 9, 171–188. [Google Scholar] [CrossRef]
- Gilmore, D.L.; Liu, Y.; Matsumoto, R.R. Review of the pharmacological and clinical profile of rimcazole. CNS Drug Rev. 2004, 10, 1–22. [Google Scholar] [CrossRef]
- Bhattacharyya, P.; Chakraborty, A. Mukonal, a probable biogenetic intermediate of pyranocarbazole alkaloids from Murraya koenigii. Phytochemistry 1984, 23, 471–472. [Google Scholar] [CrossRef]
- Chakraborty, D.P.; Bhattacharyya, P.; Islam, A.; Roy, M.S. Structure of murrayacinine: A new carbazole alkaloid from Murraya koenigii spreng. Chem. Ind. 1974, 1974, 165–166. [Google Scholar]
- Wu, T.S.; Huang, S.C.; Wu, P.L. Carbazole alkaloids from stem bark of Clausena excavata. Phytochemistry 1996, 43, 1427–1429. [Google Scholar] [CrossRef]
- Ito, C.; Nakagawa, M.; Wu, T.-S.; Furukawa, H. New carbazole alkaloids from Murraya euchrestifolia. Chem. Pharm. Bull. 1991, 39, 2525–2528. [Google Scholar] [CrossRef]
- Wu, T.S.; Huang, S.C.; Wu, P.L.; Kuoh, C.S. Alkaloidal and other constituents from the root bark of Clausena excavata. Phytochemistry 1999, 52, 523–527. [Google Scholar]
- Ito, C.; Katsuno, S.; Ohta, H.; Omura, M.; Kajiura, I.; Furukawa, H. Constituents of Clausena excavate. Isolation and structural elucidation of new carbazole alkaloids. Chem. Pharm. Bull. 1997, 45, 48–52. [Google Scholar] [CrossRef]
- Knölker, H.J.; Wolpert, M. Transition metal complexes in organic synthesis. Part 68: Iron-mediated total synthesis of mukonine and mukonidine by oxidative cyclization with air as the oxidizing agent. Tetrahedron 2003, 59, 5317–5322. [Google Scholar] [CrossRef]
- St. Jean, D.J., Jr.; Poon, S.F.; Schwarzbach, J.L. A tandem cross-coupling/SNAr approach to functionalized carbazoles. Org. Lett. 2007, 9, 4893–4896. [Google Scholar] [CrossRef]
- Forke, R.; Krahl, M.P.; Däbritz, F.; Jäger, A.; Knölker, H.J. Transition metals in organic synthesis, Part 87: An efficient palladium-catalyzed route to 2-oxygenated and 2,7-dioxygenated carbazole alkaloids—Total synthesis of 2-methoxy-3-methylcarbazole, glycosinine, clausine L, mukonidine, and clausine V. Synlett 2008, 2008, 1870–1876. [Google Scholar] [CrossRef]
- Forke, R.; Jäger, A.; Knölker, H.J. First total synthesis of clausine L and pityriazole, a metabolite of the human pathogenic yeast Malassezia furfur. Org. Biomol. Chem. 2008, 6, 2481–2483. [Google Scholar] [CrossRef]
- Watanabe, T.; Oishi, S.; Fujii, N.; Ohno, H. Palladium-catalyzed direct synthesis of carbazoles via one-pot N-arylation and oxidative biaryl coupling: Synthesis and mechanistic study. J. Org. Chem. 2009, 74, 4720–4726. [Google Scholar] [CrossRef]
- Sridharan, V.; Martín, M.A.; Menéndez, J.C. Acid-free synthesis of carbazoles and carbazolequinones by intramolecular Pd-catalyzed, microwave-assisted oxidative biaryl coupling reactions—Efficient syntheses of murrayafoline A, 2-methoxy-3-methylcarbazole, and glycozolidine. Eur. J. Org. Chem. 2009, 4614–4621. [Google Scholar] [CrossRef]
- Tsang, W.C.P.; Munday, R.H.; Brasche, G.; Zheng, N.; Buchwald, S.L. Palladium-catalyzed method for the synthesis of carbazoles via tandem C-H functionalization and C-N bond formation. J. Org. Chem. 2008, 73, 7603–7610. [Google Scholar] [CrossRef]
- Witulski, B.; Alayrac, C. A highly efficient and flexible synthesis of substituted carbazoles by rhodium-catalyzed inter- and intramolecular alkyne cyclotrimerizations. Angew. Chem. Int. Ed. 2002, 41, 3281–3284. [Google Scholar] [CrossRef]
- Krahl, M.P.; Jäger, A.; Krause, T.; Knölker, H.J. First total synthesis of the 7-oxygenated carbazole alkaloid clauszoline-K, 3-formyl-7-hydroxycarbazole, clausine M, clausine N and the anti-HIV active siamenol using a highly efficient palladium-catalyzed approach. Org. Biomol. Chem. 2006, 4, 3215–3219. [Google Scholar] [CrossRef]
- Kuethe, J.T.; Childers, K.G. Suzuki-miyaura cross-coupling of 2-nitroarenediazonium tetrafluoroborates: Synthesis of unsymmetrical 2-nitrobiphenyls and highly functionalized carbazoles. Adv. Synth. Catal. 2008, 350, 1577–1586. [Google Scholar] [CrossRef]
- Kataeva, O.; Krahl, M.P.; Knölker, H.J. First total synthesis of the biologically active 2,7-dioxygenated tricyclic carbazole alkaloids 7-methoxy-O-methylmukonal, clausine H (clauszoline-C), clausine K (clauszoline-J) and clausine O. Org. Biomol. Chem. 2005, 3, 3099–3101. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Alonso, J.M.; Fernández, I. Carbocyclization versus oxycyclization on the metal-catalyzed reactions of oxyallenyl C3-linked índoles. J. Org. Chem. 2013, 78, 6688–6701. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Alonso, J.M.; Cembellín, S.; Fernández, I.; Martínez del Campo, T.; Torres, M.R. Iodine recycling via 1,3-migration in iodoindoles under metal catalysis. Chem. Commun. 2013, 49, 7779–7781. [Google Scholar] [CrossRef]
- Bautista, R.; Jerezano, A.V.; Tamariz, J. Synthetic approach for constructing the 1-oxygenated carbazole core and its application to the preparation of natural alkaloids. Synthesis 2012, 44, 3327–3336. [Google Scholar] [CrossRef]
- Knölker, H.J.; Fröhner, W. Palladium-catalyzed total synthesis of the antibiotic carbazole alkaloids carbazomycin G and H. J. Chem. Soc. Perkin Trans. 1 1998, 1998, 173–175. [Google Scholar]
- Knölker, H.J.; Reddy, K.R. Indoloquinones, Part 6. First palladium-mediated oxidative cyclization of arylamino-1,2-benzoquinones to carbazole-3,4-quinones—Application to the total synthesis of carbazoquinocin C and (±)-carquinostatin A. Synlett 1999, 1999, 596–598. [Google Scholar] [CrossRef]
- Knölker, H.J.; O’Sullivan, N. Palladium-promoted synthesis of hydroxy-substituted 5-cyano-5H-benzo[b]carbazole-6,11-diones. Tetrahedron 1994, 50, 10893–10908. [Google Scholar] [CrossRef]
- Hagelin, H.; Oslob, J.D.; Åkermark, B. Oxygen as oxidant in palladium-catalyzed inter- and intramolecular coupling reactions. Chem. Eur. J. 1999, 5, 2413–2416. [Google Scholar] [CrossRef]
- Sissouma, D.; Maingot, L.; Collet, S.; Guingant, A. Concise and efficient synthesis of calothrixin B. J. Org. Chem. 2006, 71, 8384–8389. [Google Scholar] [CrossRef]
- Iida, H.; Yuasa, Y.; Kibayashi, C. A convenient synthesis of m-aminophenols by mercury(II) acetate oxidation of 3-amino-2-cyclohexenones. Synthesis 1982, 1982, 471–472. [Google Scholar] [CrossRef]
- Bautista, R.; Bernal, P.; Montiel, L.E.; Delgado, F.; Tamariz, J. Total synthesis of the natural carbazoles glycozolicine, mukoline, and mukolidine, starting from 4,5-dimethyleneoxazolidin-2-ones. Synthesis 2011, 2011, 929–933. [Google Scholar] [CrossRef]
- Mithani, S.; Weeratunga, G.; Taylor, N.J.; Dmitrienko, G.I. The kinamycins are diazofluorenes and not cyanocarbazoles. J. Am. Chem. Soc. 1994, 116, 2209–2210. [Google Scholar] [CrossRef]
- Yogo, M.; Ito, C.; Furukawa, H. Synthesis of some carbazolequinone alkaloids and their analogues. Facile palladium-assisted intramolecular ring closure of arylamino-1,4-benzoquinones to carbazole-1,4-quinones. Chem. Pharm. Bull. 1991, 39, 328–334. [Google Scholar] [CrossRef]
- Saha, C.; Chowdhury, B.K. Carbazoloquinones from Murraya koenigii. Phytochemistry 1998, 48, 363–366. [Google Scholar] [CrossRef]
- Lin, G.; Zhang, A. The first synthesis of optically pure biscarbazoles and determination of their absolute configurations. Tetrahedron Lett. 1999, 40, 341–344. [Google Scholar] [CrossRef]
- Molina, P.; Fresneda, P.M.; Almendros, P. Fused carbazoles by tandem aza Wittig/electrocyclic ring closure, preparation of 6H-pyrido[4,3-b]carbazole, 11H-pyrido[4,3-a]carbazole and 11H-pyrido[3,4-a]carbazole derivatives. Tetrahedron 1993, 49, 1223–1236. [Google Scholar] [CrossRef]
- Chakraborty, D.P.; Chowdhury, B.K. Synthesis of murrayanine. J. Org. Chem. 1968, 33, 1265–1268. [Google Scholar] [CrossRef]
- Murakami, Y.; Yokoo, H.; Watanabe, T. New syntheses of murrayaquinone—A via Fischer indolization of 2-sulfonyloxyphenylhydrazone (Fischer indolization and its related compounds. Part 29). Heterocycles 1998, 49, 127–132. [Google Scholar] [CrossRef]
- Chakraborty, D.P.; Islam, A.; Bhattacharyya, P. Synthesis of murrayanine. J. Org. Chem. 1973, 38, 2728–2729. [Google Scholar] [CrossRef]
- Murphy, W.S.; Bertrand, M. Bromoquinone-enaminone annulations: Syntheses of murrayaquinone-A and (±)-bismurrayaquinone-A. J. Chem. Soc. Perkin Trans. 1 1998, 1998, 4115–4119. [Google Scholar] [CrossRef]
- Crum, J.D.; Sprague, P.W. The synthesis of murrayanine. Chem. Commun. 1966, 1966, 417–418. [Google Scholar]
- Sissouma, D.; Collet, S.C.; Guingant, A.Y. A synthesis of calothrixin B. Synlett 2004, 2004, 2612–2614. [Google Scholar]
- Forke, R.; Krahl, M.P.; Krause, T.; Schlechtingen, G.; Knölker, H.J. Transition metals in organic synthesis, Part 82. First total synthesis of methyl 6-methoxycarbazole-3-carboxylate, glycomaurrol, the anti-TB active micromeline, and the furo[2,3-c]carbazole alkaloid eustifoline-D. Synlett 2007, 2007, 268–272. [Google Scholar] [CrossRef]
- Schmidt, M.; Knölker, H.J. Transition metals in organic synthesis, Part 91: Palladium-catalyzed approach to 2,6-dioxygenated carbazole alkaloids—First total synthesis of the phytoalexin carbalexin C. Synlett 2009, 2009, 2421–2424. [Google Scholar] [CrossRef]
- Knölker, H.J.; Goesmann, H.; Hofmann, C. Transition metal complexes in organic synthesis, Part 31. A novel molybdenum-mediated synthesis of carbazole derivatives: Application to the total synthesis of mukonal and 1,1′-bis(2-hydroxy-3-methylcarbazole). Synlett 1996, 1996, 737–740. [Google Scholar] [CrossRef]
- Roy, S.; Bhattacharyya, L.; Chakraborty, D.P. Structure and synthesis of mukoline and mukolidine, two new carbazole alkaloids from Murraya koenigii spreng. J. Indian Chem. Soc. 1982, 59, 1369–1371. [Google Scholar]
- Cheenpracha, S.; Laphookhieo, S. Alkaloids and amides from Glycosmis macrophylla. Phytochem. Lett. 2011, 4, 187–189. [Google Scholar] [CrossRef]
- Chakravarty, A.K.; Sarkar, T.; Masuda, K.; Takey, T.; Doi, H.; Kotani, E.; Shiojima, K. Structure and synthesis of glycoborine, a new carbazole alkaloid from the roots of Glycosmis arborea: A note on the structure of glycozolicine. Indian J. Chem. 2001, 40B, 484–489. [Google Scholar]
- Jash, S.S.; Biswas, G.K.; Bhattacharyya, S.K.; Bhattacharyya, P.; Chakraborty, A.; Chowdhury, B.K. Carbazole alkaloids from Glycosmis pentaphylla. Phytochemistry 1992, 31, 2503–2505. [Google Scholar] [CrossRef]
- Makawana, J.A.; Patel, M.P.; Patel, R.G. Synthesis and in vitro antimicrobial activity of N-arylquinoline derivatives bearing 2-morpholinoquinoline moiety. Chinese Chem. Lett. 2012, 23, 427–430. [Google Scholar] [CrossRef]
- Iida, H.; Yuasa, Y.; Kibayashi, C. Intramolecular cyclization of enaminones involving arylpalladium complexes. Synthesis of carbazoles. J. Org. Chem. 1980, 45, 2938–2942. [Google Scholar] [CrossRef]
- Andrade, C.K.Z.; Barreto, A.F.S.; Wender, A.S. Microwave assisted solvent-, support- and catalyst-free synthesis of enaminones. Arkivoc 2008, xii, 226–232. [Google Scholar]
- Maiti, D.; Buchwald, S.L. Orthogonal Cu- and Pd-based catalyst systems for the O- and N-arylation of aminophenols. J. Am. Chem. Soc. 2009, 131, 17423–17429. [Google Scholar] [CrossRef]
- Urgaonkar, S.; Xu, J.H.; Verkade, J.G. Application of a new bicyclic triaminophosphine ligand in Pd-catalyzed Buchwald-Hartwig amination reactions of aryl chlorides, bromides, and iodines. J. Org. Chem. 2003, 68, 8416–8423. [Google Scholar] [CrossRef]
- Bhatia, S.K.; Kapil, R.S. Synthesis of O-methylcycloclausanitin. Indian J. Chem. Soc. 1984, 23B, 296–299. [Google Scholar]
- Oikawa, Y.; Yonemitsu, O. Selective oxidation of the side chain at C-3 of indoles. J. Org. Chem. 1977, 42, 1213–1216. [Google Scholar] [CrossRef]
- Weng, B.; Liu, R.; Li, J.-H. An improved method for the synthesis of carbazolones by palladium/copper-catalyzed intramolecular annulation of N-arylenaminones. Synthesis 2010, 2010, 2926–2930. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 4a–e, 5a, 5d, 5e, 1d, 1f–g, 1h–i, and 1k are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bautista, R.; Montoya, P.A.; Rebollar, A.; Burgueño, E.; Tamariz, J. Palladium-Catalyzed Synthesis of Natural and Unnatural 2-, 5-, and 7-Oxygenated Carbazole Alkaloids from N-Arylcyclohexane Enaminones. Molecules 2013, 18, 10334-10351. https://doi.org/10.3390/molecules180910334
Bautista R, Montoya PA, Rebollar A, Burgueño E, Tamariz J. Palladium-Catalyzed Synthesis of Natural and Unnatural 2-, 5-, and 7-Oxygenated Carbazole Alkaloids from N-Arylcyclohexane Enaminones. Molecules. 2013; 18(9):10334-10351. https://doi.org/10.3390/molecules180910334
Chicago/Turabian StyleBautista, Rafael, Pablo A. Montoya, Araceli Rebollar, Eleuterio Burgueño, and Joaquín Tamariz. 2013. "Palladium-Catalyzed Synthesis of Natural and Unnatural 2-, 5-, and 7-Oxygenated Carbazole Alkaloids from N-Arylcyclohexane Enaminones" Molecules 18, no. 9: 10334-10351. https://doi.org/10.3390/molecules180910334