A Macrocyclic Peptide that Serves as a Cocrystallization Ligand and Inhibits the Function of a MATE Family Transporter

Abstract
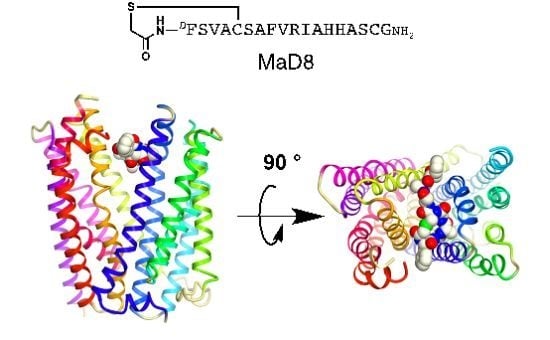
:
1. Introduction
2. Results and Discussion
2.1. RaPID Selection of PfMATE-Binding Macrocyclic Peptides
2.1.1. RaPID-Displayed Cyclic Peptide Library
2.1.2. Initial Selection with a Binding Step at 4 °C
2.1.3. Increasing Stringency with a Binding Step at 37 °C

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anti-MATE Selection Clone Sequences | |||||
|---|---|---|---|---|---|
| Name | Sequence | Round 7 | Round 6 | c-Round 6 | Frequency of appearance |
| MaL1 | AcFSTFCYFPTELLLLAC | 1/9 | 1/32 | ||
| MaL2 | AcFTDCLHARWIFPRVRQRQRQLGRGAEKSLVNSRP | 1/11 | 1/32 | ||
| MaL3 | AcFVYSAVCLYVGSLYPC | 2/11 | 4/12 | 6/32 | |
| MaL4 | AcFTFRDVWIFYGSLLSRC | 1/11 | 1/32 | ||
| MaL5 | AcFLYNAYCLWLAYCVNSC | 2/32 | |||
| MaL6 | AcFTFRYSPSLYTWFLFPC | 4/9 | 7/11 | 3/12 | 14/32 |
| MaL7 | AcFWTVASWGLVALDFVAC | 1/9 | 1/32 | ||
| MaL8 | AcFTHPIFCYPSADLC | 1/9 | 1/32 | ||
| MaL9 | AcFTDCLHARWIFPRVC | 1/9 | 1/32 | ||
| MaL10 | AcFTYSAFCYAIANIAYC | 3/12 | 3/32 | ||
| MaL11 | AcFAYECMWLTLPASWPPC | 1/9 | 1/32 | ||
| MaD1 | AcfQWQCHIFTNLALTC | 6/11 | 1/9 | 7/27 | |
| MaD2 | AcfHPVNCTNLWAAIRLAC | 1/7 | 1/27 | ||
| MaD3 | AcfVYSAVCLYVGSLYSC | 1/7 | 2/11 | 3/9 | 6/27 |
| MaD4 | AcfLYNAYCLWLAYCVNSC | 1/7 | 1/27 | ||
| MaD5 | AcfVYSAVCYSIAAAAAAARTGGGKITS | 1/11 | 2/9 | 3/27 | |
| MaD6 | AcfVDASACSFVNLWLTC | 1/7 | 1/9 | 2/27 | |
| MaD7 | AcfIECQTLVYLSLIPHNC | 1/7 | 1/27 | ||
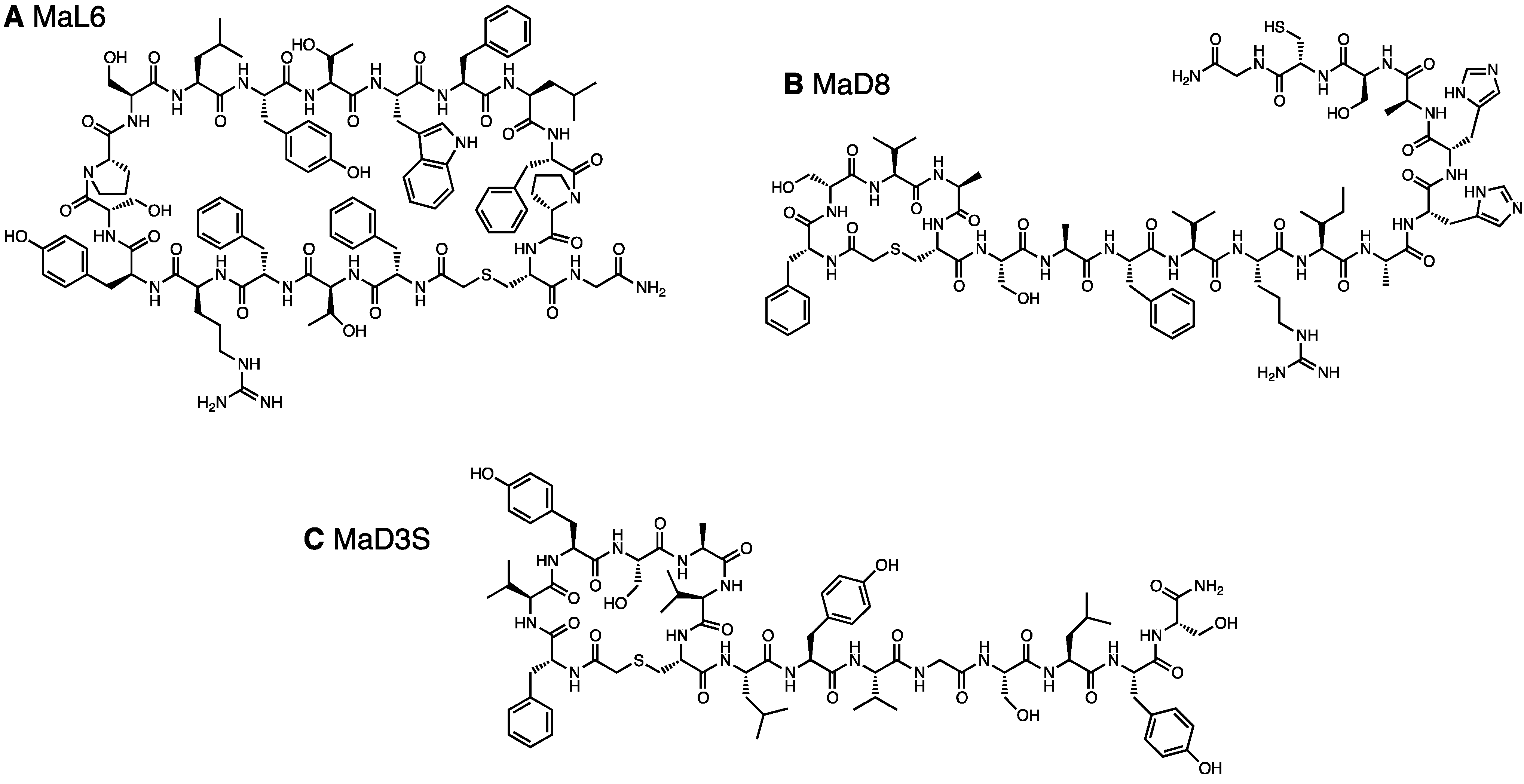
| MaD8 | AcfSVACSAFVRIAHHASC | 1/7 | 1/27 | ||
| MaD9 | AcfTTYSAFCYAIANIAYC | 1/7 | 1/27 | ||
| MaD10 | AcfTYSAFCYAIANIAYC | 1/11 | 2/9 | 3/27 | |
| MaD11 | AcfVNTSVCLFACWVNSC | 1/11 | 1/27 | ||

2.1.4. A Chemically Synthesized, in Vitro Selected Competitor
2.2. Co-Crystallization of the MaD8-PfMATE Complex
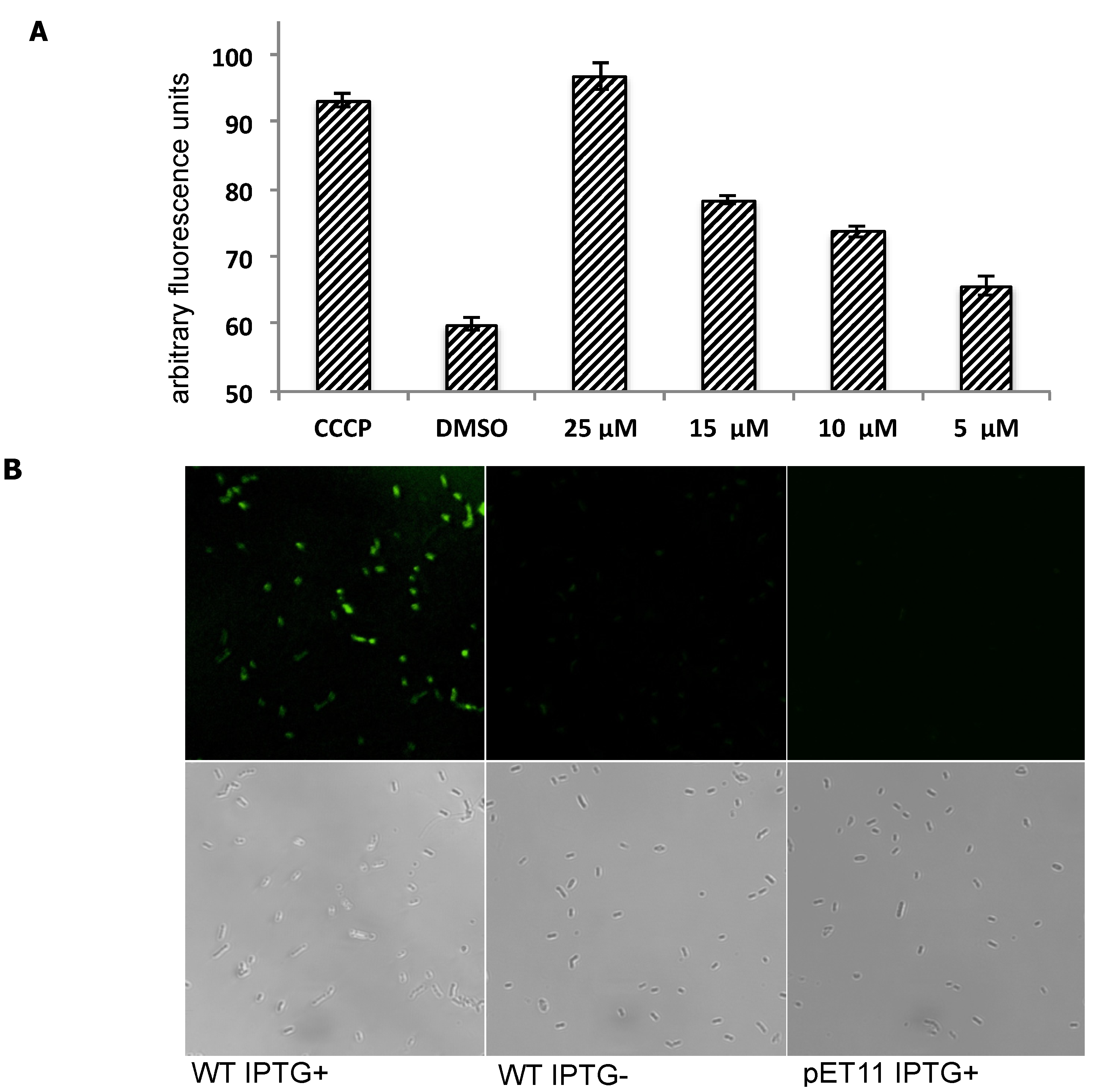
2.3. Inhibition of Transporter Activity


3. Experimental
3.1. Preparation of N-ClAc-l-phenylalanine-tRNAfMetCAU and N-ClAc-d-phenylalanine-tRNAfMetCAU
3.2. Library Construction
3.3. Protein Immobilization
3.4. Production of the mRNA-Puromycin Conjugates for Round 1
3.5. Production of the mRNA-Puromycin Conjugates for Round 2+
3.6. Selection with the Binding Step at 4 °C
3.7. Selection with the Binding Step at 37 °C
3.8. Selection Round Using a Chemically Synthesized Competitor
3.9. Single-Clone Assays
3.10. Peptide Chemical Synthesis
3.11. Inhibition Assay
3.12. Staining E. coli Cells with MaD8F
3.13. Crystallization, Data Collection, and Structure Determination
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Stahl, S.J.; Watts, N.R.; Rader, C.; DiMattia, M.A.; Mage, R.G.; Palmer, I.; Kaufman, J.D.; Grimes, J.M.; Stuart, D.I.; Steven, A.C.; et al. Generation and characterization of a chimeric rabbit/human Fab for co-crystallization of HIV-1 Rev. J. Mol. Biol. 2010, 397, 697–708. [Google Scholar] [CrossRef]
- McPherson, A.; Cudney, B. Searching for silver bullets: An alternative strategy for crystallizing macromolecules. J. Struct. Biol. 2006, 156, 387–406. [Google Scholar] [CrossRef]
- Paduch, M.; Koide, A.; Uysal, S.; Rizk, S.S.; Koide, S.; Kossiakoff, A.A. Generating conformation-specific synthetic antibodies to trap proteins in selected functional states. Methods 2013, 60, 3–14. [Google Scholar] [CrossRef]
- Lu, M.; Symersky, J.; Radchenko, M.; Koide, A.; Guo, Y.; Nie, R.; Koide, S. Structures of a Na+-coupled, substrate-bound MATE multidrug transporter. Proc. Natl. Acad. Sci. USA 2013, 110, 2099–2104. [Google Scholar] [CrossRef]
- Warke, A.; Momany, C. Addressing the protein crystallization bottleneck by cocrystallization. Cryst. Growth Des. 2007, 7, 2219–2225. [Google Scholar] [CrossRef]
- Hunte, C.; Koepke, J.; Lange, C.; Robmanith, T.; Michel, H. Structure at 2.3 Å resolution of the cytochrome bc1 complex from the yeast Saccharomyces cerevisiae co-crystallized with an antibody Fv fragment. Structure 2000, 8, 669–684. [Google Scholar] [CrossRef]
- Zhou, Y.; Morais-Cabral, J.H.; Kaufman, A.; MacKinnon, R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature 2001, 414, 43–48. [Google Scholar] [CrossRef]
- Huber, T.; Steiner, D.; Röthlisberger, D.; Plückthun, A. In vitro selection and characterization of DARPins and Fab fragments for the co-crystallization of membrane proteins: The Na+-citrate symporter CitS as an example. J. Struct. Biol. 2007, 159, 206–221. [Google Scholar] [CrossRef]
- Mittal, A.; Böhm, S.; Grütter, M.G.; Bordignon, E.; Seeger, M.A. Asymmetry in the homodimeric ABC transporter MsbA recognized by a DARPin. J. Biol. Chem. 2012, 287, 20395–20406. [Google Scholar]
- Smith, G. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar]
- Hanes, J.; Plückthun, A. In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl. Acad. Sci. USA 1997, 94, 4937–4942. [Google Scholar] [CrossRef]
- Koellhoffer, J.F.; Chen, G.; Sandesara, R.G.; Bale, S.; Saphire, E.O.; Chandran, K.; Sidhu, S.S.; Lai, J.R. Two synthetic antibodies that recognize and neutralize distinct proteolytic forms of the ebola virus envelope glycoprotein. Chembiochem 2012, 13, 2549–2557. [Google Scholar] [CrossRef]
- Nemoto, N.; Miyamoto-sato, E.; Husumi, Y.; Yanagawa, H. In vitro virus: Bonding of mRNA bearing puromycin at the 3'-terminal end to the C-terminal end of its encoded protein on the ribosome in vitro. FEBS Lett. 1997, 414, 405–408. [Google Scholar] [CrossRef]
- Roberts, R.; Szostak, J. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 12297–12302. [Google Scholar] [CrossRef]
- Yamagishi, Y.; Shoji, I.; Miyagawa, S.; Kawakami, T.; Katoh, T.; Goto, Y.; Suga, H. Natural product-like macrocyclic N-methyl-peptide inhibitors against a ubiquitin ligase uncovered from a ribosome-expressed de novo library. Chem. Biol. 2011, 18, 1562–1570. [Google Scholar] [CrossRef]
- Murakami, H.; Kourouklis, D.; Suga, H. Using a solid-phase ribozyme aminoacylation system to reprogram the genetic code. Chem. Biol. 2003, 10, 1077–1084. [Google Scholar]
- Morimoto, J.; Hayashi, Y.; Iwasaki, K.; Suga, H. Flexizymes: Their evolutionary history and the origin of catalytic function. Acc. Chem. Res. 2011, 44, 1359–1368. [Google Scholar] [CrossRef]
- Murakami, H.; Ohta, A.; Ashigai, H.; Suga, H. A highly flexible tRNA acylation method for non-natural peptide synthesis. Nat. Methods 2006, 3, 357–359. [Google Scholar] [CrossRef]
- Ohuchi, M.; Murakami, H.; Suga, H. The flexizyme system: A highly flexible tRNA aminoacylation tool for the translation apparatus. Curr. Opin. Chem. Biol. 2007, 11, 537–542. [Google Scholar] [CrossRef]
- Goto, Y.; Katoh, T.; Suga, H. Flexizymes for genetic code reprogramming. Nat. Protoc. 2011, 6, 779–790. [Google Scholar] [CrossRef]
- Ohta, A.; Yamagishi, Y.; Suga, H. Synthesis of biopolymers using genetic code reprogramming. Curr. Opin. Chem. Biol. 2008, 12, 159–167. [Google Scholar] [CrossRef]
- Kang, T.J.; Suga, H. Ribosomal Synthesis of nonstandard peptides. Biochem. Cell. Biol. 2008, 86, 92–99. [Google Scholar] [CrossRef]
- Hipolito, C.; Suga, H. Ribosomal Production and in vitro selection of natural product-like peptidomimetics: the FIT and RaPID systems. Curr. Opin. Chem. Biol. 2012, 16, 196–203. [Google Scholar] [CrossRef]
- Morimoto, J.; Hayashi, Y.; Suga, H. Discovery of macrocyclic peptides armed with a Mechanism-based warhead: Isoform-selective inhibition of human deacetylase SIRT2. Angew. Chem. Int. Ed. 2012, 51, 3423–3427. [Google Scholar] [CrossRef]
- Hayashi, Y.; Morimoto, J.; Suga, H. In vitro selection of anti-Akt2 thioether-macrocyclic peptides leading to isoform-selective inhibitors. Chem. Biol. 2012, 7, 607–613. [Google Scholar]
- He, X.; Szewczyk, P.; Karyakin, A.; Evin, M.; Hong, X.; Zhang, Q.; Chang, G. Structure of a cation-bound multidrug and toxic compound extrusion transporter. Nature 2010, 467, 991–994. [Google Scholar] [CrossRef]
- Otsuka, M.; Matsumoto, T.; Morimoto, R.; Arioka, S.; Omote, H.; Moriyama, Y. A human transporter protein that mediates the final excretion step for toxic organic cations. Proc. Natl. Acad. Sci. USA 2005, 102, 17923–17928. [Google Scholar] [CrossRef] [Green Version]
- Masuda, S.; Terada, T.; Yonezawa, A.; Tanihara, Y.; Kishimoto, K.; Katsura, T.; Ogawa, O.; Inui, K. Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J. Am. Soc. Nephrol. 2006, 17, 2127–2135. [Google Scholar] [CrossRef]
- Tanaka, Y.; Hipolito, C.J.; Maturana, A.D.; Ito, K.; Kuroda, T.; Higuchi, T.; Katoh, T.; Kato, H.E.; Hattori, M.; Kumazaki, K.; et al. Structural basis for the drug extrusion mechanism by a MATE multidrug transporter. Nature 2013, 496, 247–251. [Google Scholar] [CrossRef]
- Goto, Y.; Ohta, A.; Sako, Y.; Yamagishi, Y.; Murakami, H.; Suga, H. Reprogramming the translation initiation for the synthesis of physiologically stable cyclic peptides. ACS Chem. Biol. 2008, 3, 120–129. [Google Scholar] [CrossRef]
- Iwasaki, K.; Goto, Y.; Katoh, T.; Suga, H. Selective thioether macrocyclization of peptides having the N-terminal 2-chloroacetyl group and competing two or three cysteine residues in translation. Org. Biomol. Chem. 2012, 10, 5783–5786. [Google Scholar] [CrossRef]
- Cherezov, V. Lipidic cubic phase technologies for membrane protein structural studies. Curr. Opin. Struct. Biol. 2011, 21, 559–566. [Google Scholar] [CrossRef]
- Li, X.; Poole, K.; Nikaido, H. Contributions of MExAB-OprM and an EmrE homolog to intrinsic resistance of Pseudomonas aeruginosa to aminoglycosides and dyes. Antimicrob. Agents Chemother. 2003, 47, 27–33. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hipolito, C.J.; Tanaka, Y.; Katoh, T.; Nureki, O.; Suga, H. A Macrocyclic Peptide that Serves as a Cocrystallization Ligand and Inhibits the Function of a MATE Family Transporter. Molecules 2013, 18, 10514-10530. https://doi.org/10.3390/molecules180910514
Hipolito CJ, Tanaka Y, Katoh T, Nureki O, Suga H. A Macrocyclic Peptide that Serves as a Cocrystallization Ligand and Inhibits the Function of a MATE Family Transporter. Molecules. 2013; 18(9):10514-10530. https://doi.org/10.3390/molecules180910514
Chicago/Turabian StyleHipolito, Christopher J., Yoshiki Tanaka, Takayuki Katoh, Osamu Nureki, and Hiroaki Suga. 2013. "A Macrocyclic Peptide that Serves as a Cocrystallization Ligand and Inhibits the Function of a MATE Family Transporter" Molecules 18, no. 9: 10514-10530. https://doi.org/10.3390/molecules180910514