Ensemble-Based Interpretations of NMR Structural Data to Describe Protein Internal Dynamics

Abstract
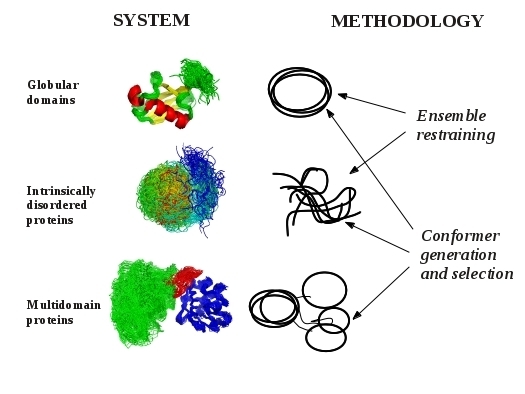
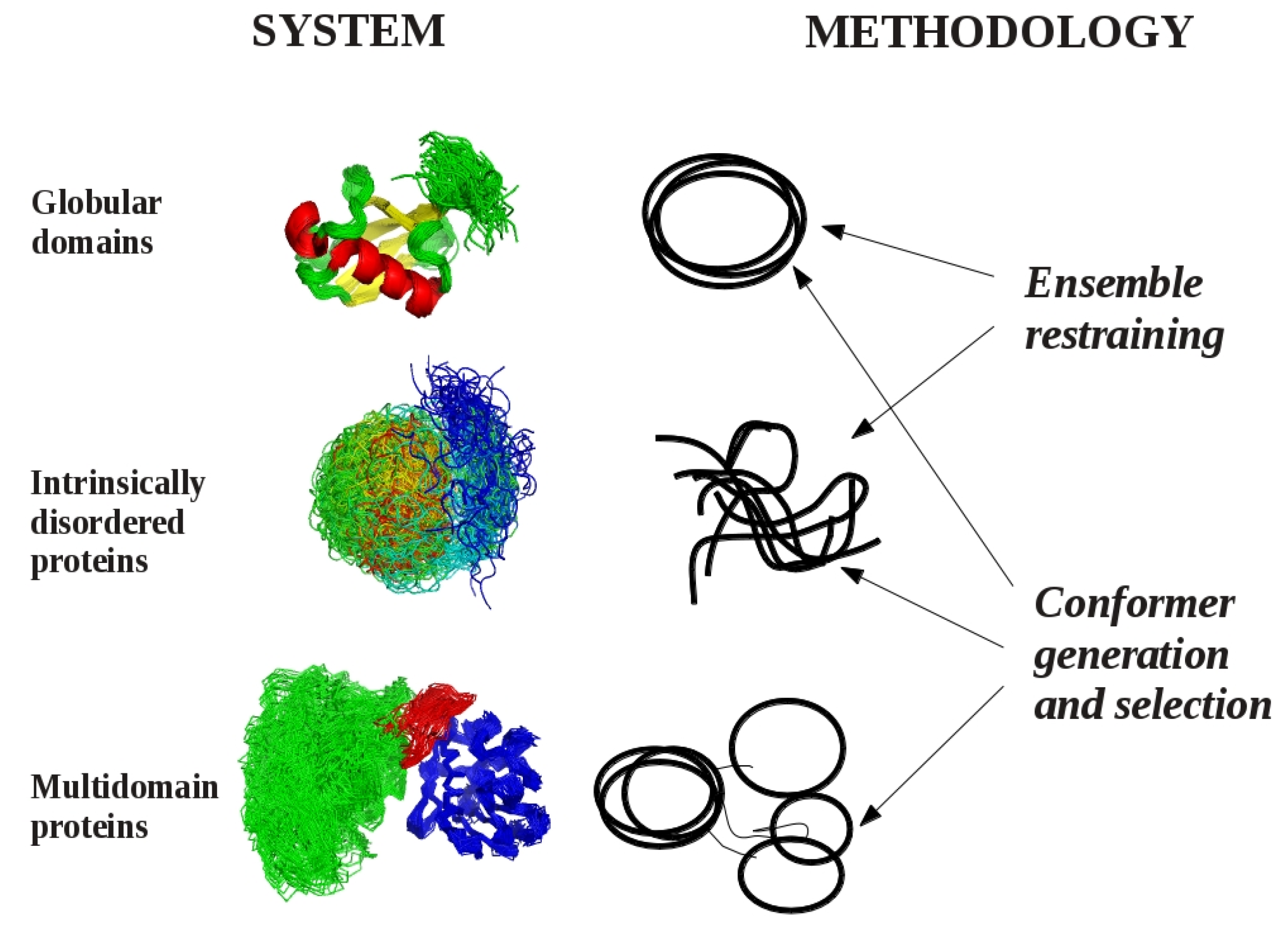
:
1. Introduction
2. Models of Protein Structures
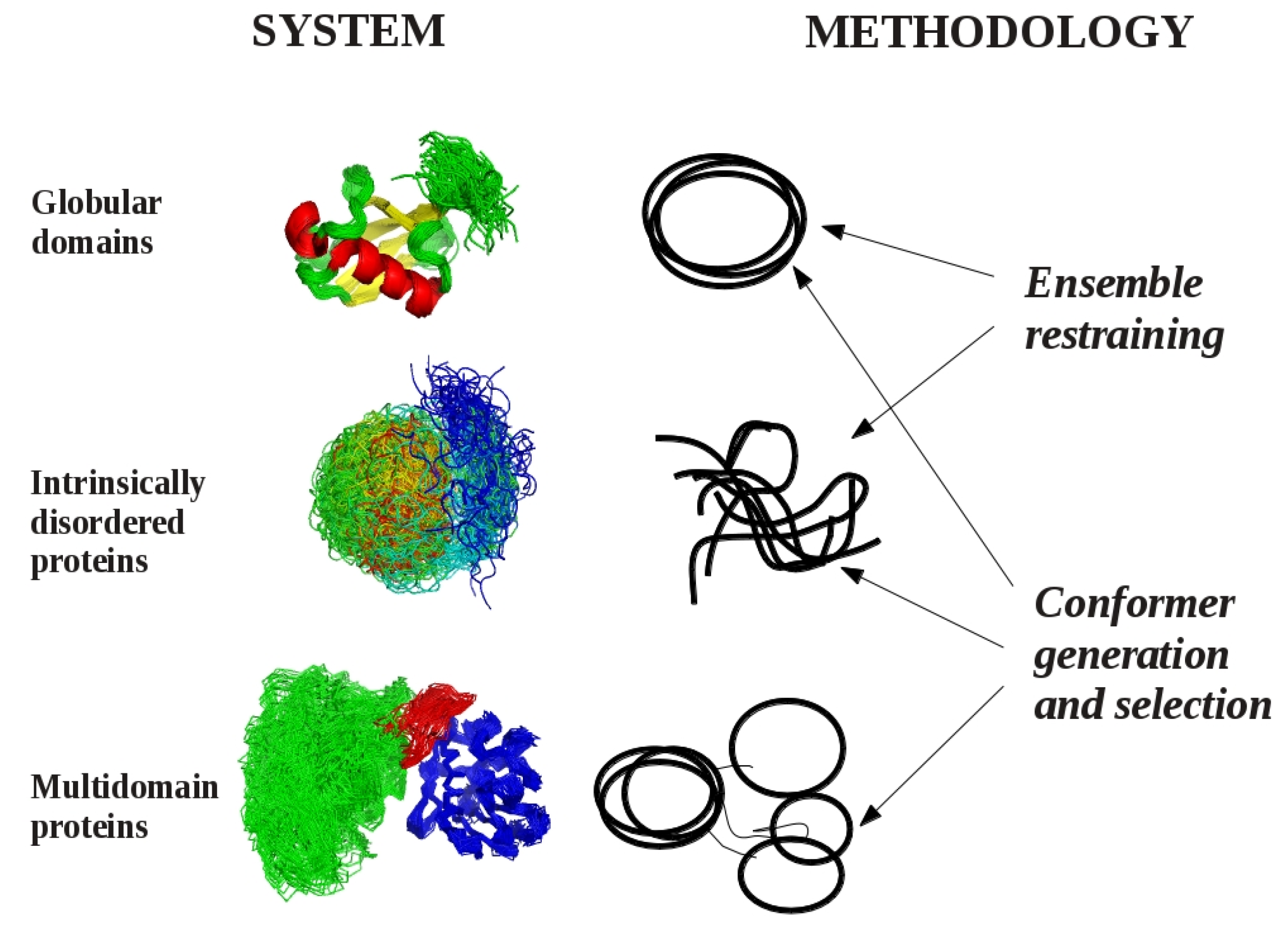
3. Methodological Aspects of Ensemble Generation
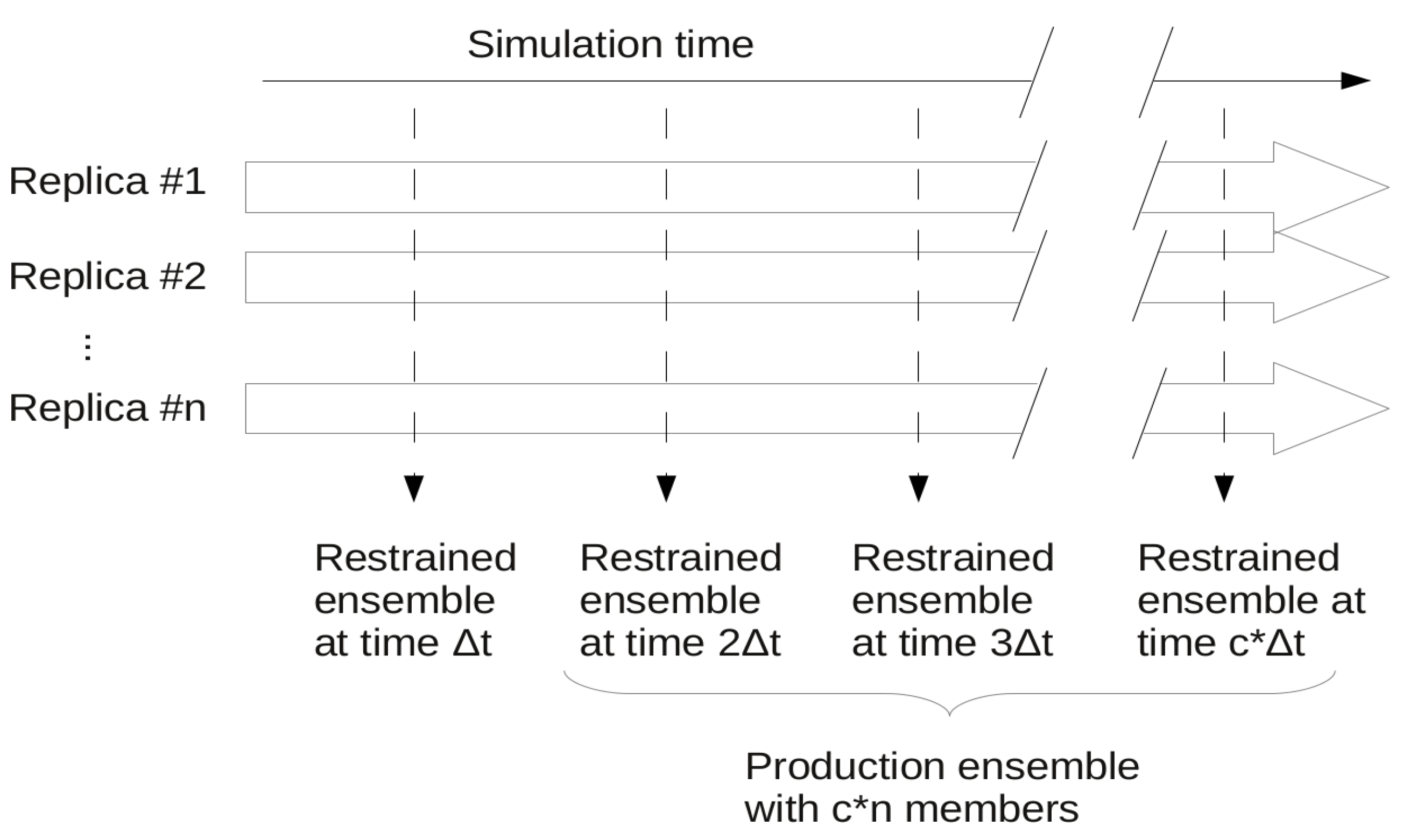
3.1. Ensemble Restraining Approaches


3.1.1. S2 General Order Parameters
3.1.2. NOE Distances
3.1.3. Residual Dipolar Couplings
3.1.4. Chemical Shifts
3.1.5. Other Types of Parameters
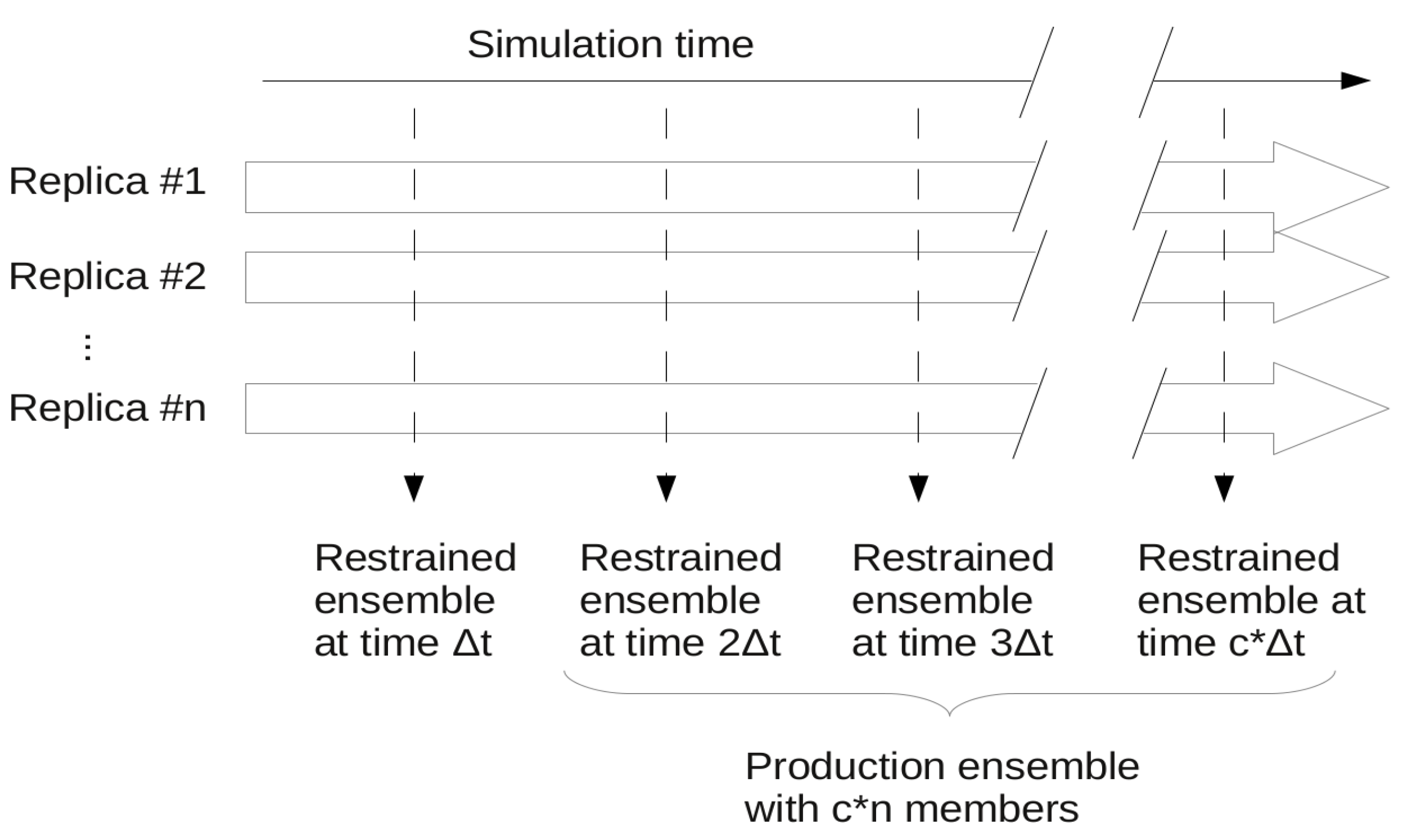
3.1.6. Protocols for Restraining

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protocol (ensemble restraining) | Parameters used and number of replicas (under parentheses) | Reference | |||
|---|---|---|---|---|---|
| NOEs | S2 order parameters | J-couplings | RDCs | ||
| DER (Dynamic Ensemble Refinement) | √ (8) | √ (8) | [34] | ||
| DER modified | √ (8) | √ (16) | [35] | ||
| MUMO (minimal under-restraining minimal over-restraining) | √ (2) | √ (8) | [21] | ||
| EROS | √ (8) | [38] | |||
| ERNST | √ (2) | √ (64) | [39] | ||
3.2. Methods for Conformer Generation and Selection
3.2.1. Conformer Generation
3.2.2. Conformer Selection
4. Evaluation and Properties of Dynamic Structural Ensembles
4.1. Validation of Dynamic Ensembles
4.2. Analysis of Ensemble-Based Representations
4.3. Do Dynamic Ensembles Reflect “Reality”?
5. Alternative Approaches
5.1. Use of Time-Averaged Restraints
5.2. Methods without Restraining
6. Example Applications
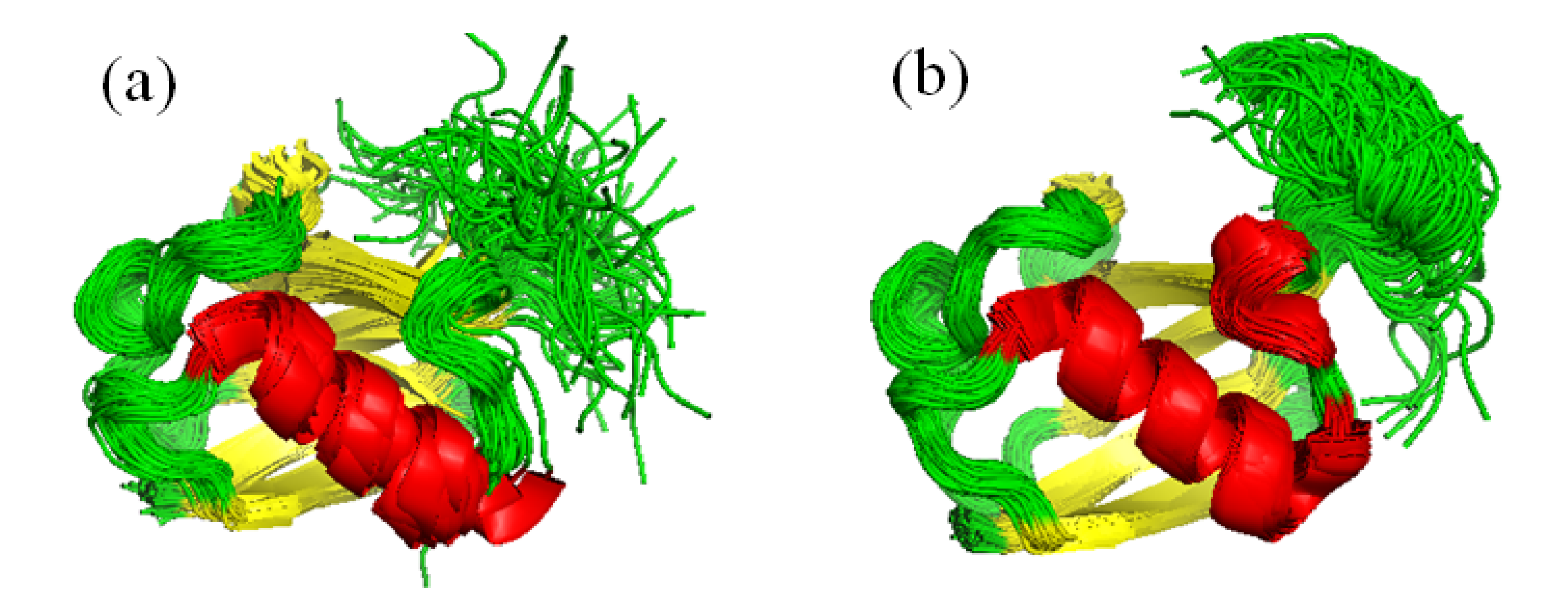
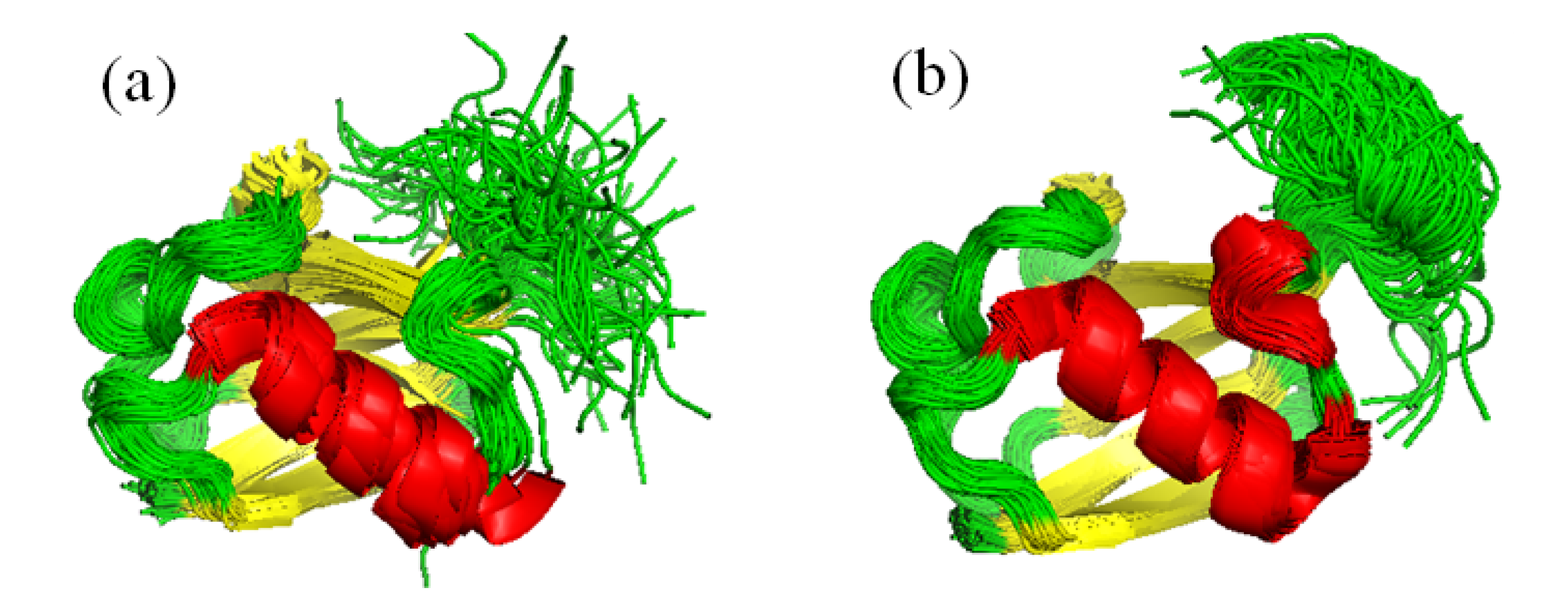
6.1. Partner Binding and Correlated Motions in Ubiquitin
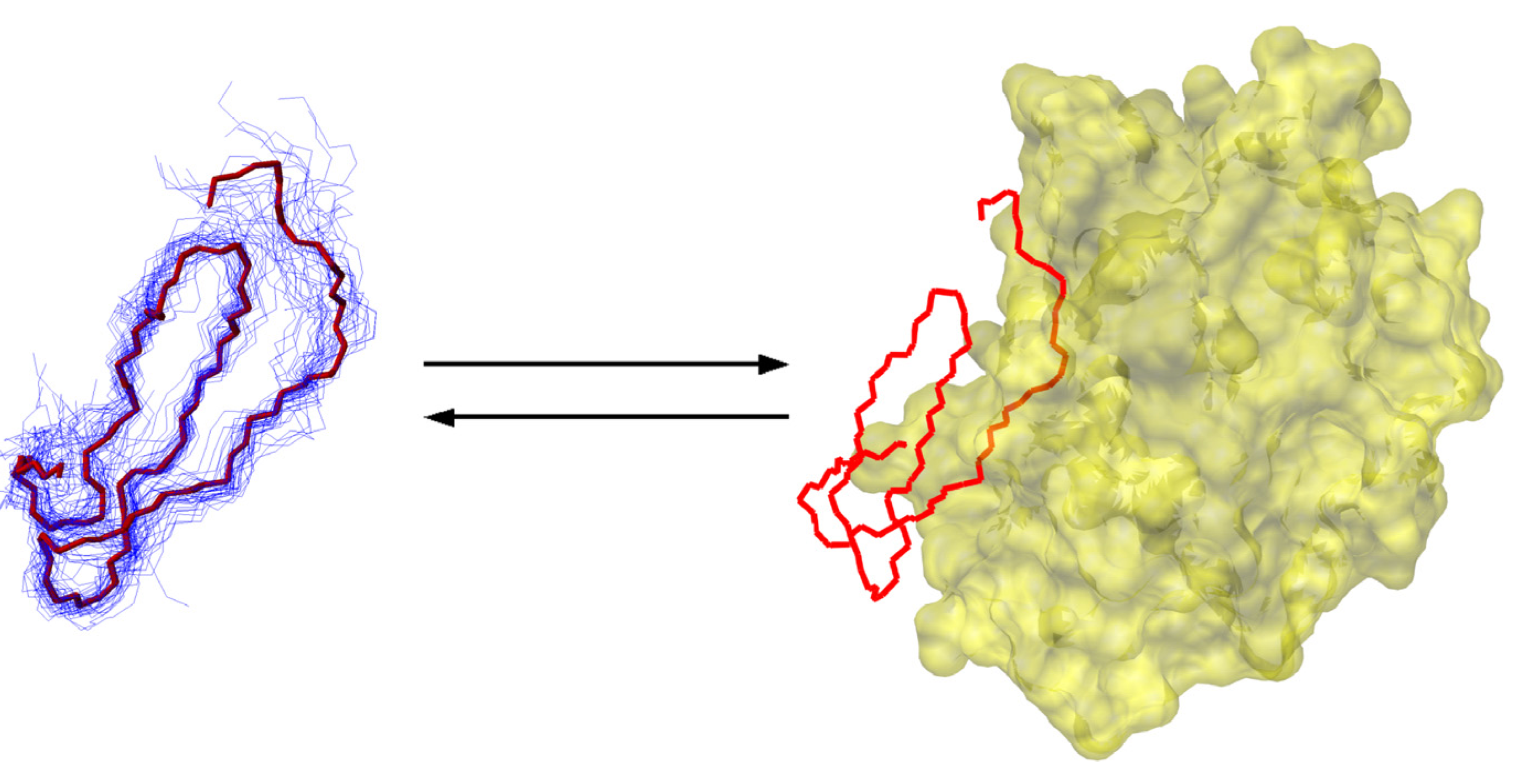
6.2. Small, Canonical Serine Protease Inhibitors
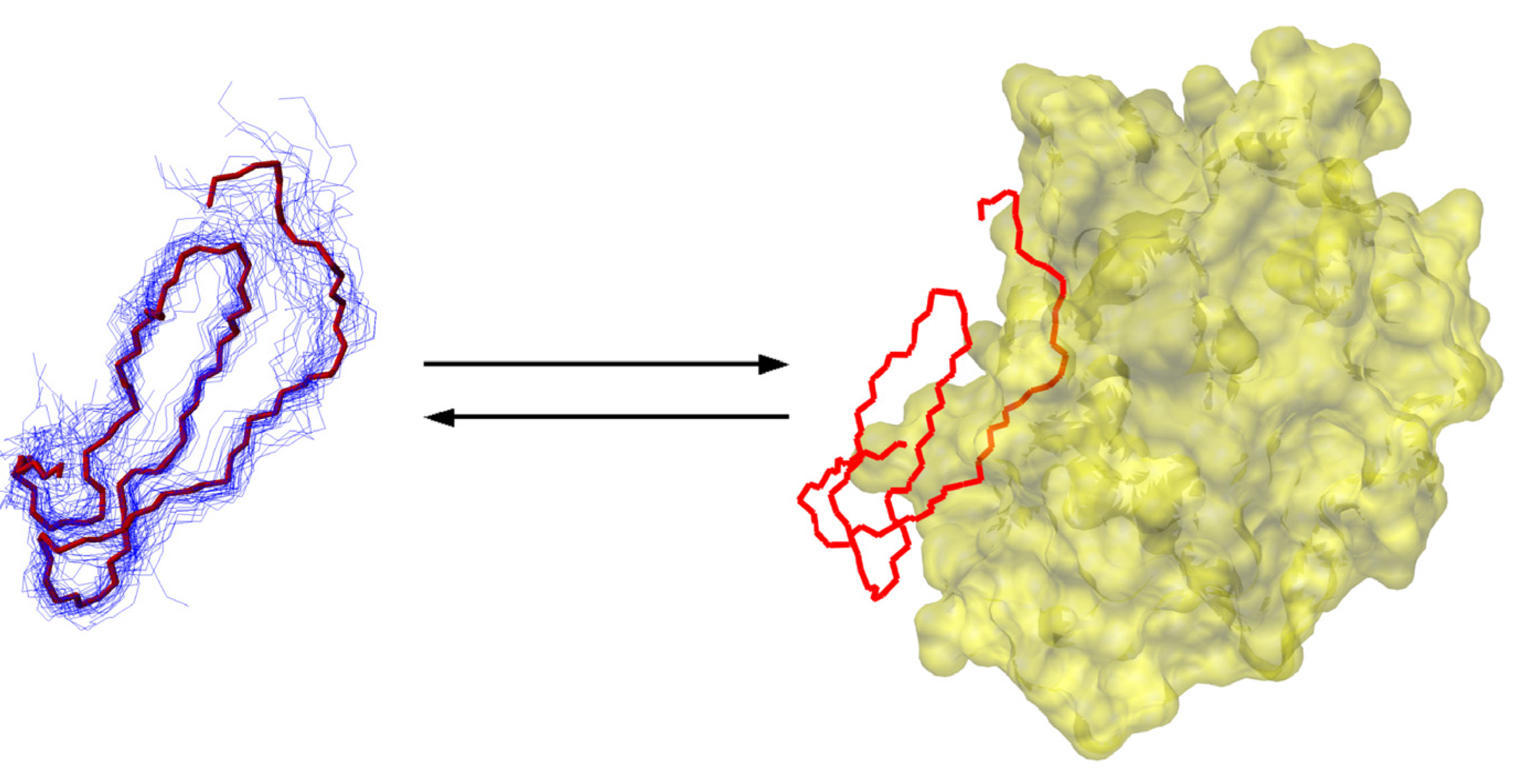
6.3. The Intrinsically Disordered Protein ACTR
6.4. The Transmembrane Helix of Vpu
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Markwick, P.R.; Malliavin, T.; Nilges, M. Structural biology by NMR: structure, dynamics, and interactions. PLoS Comp. Biol. 2008, 4, e1000168. [Google Scholar] [CrossRef]
- Boehr, D.D.; Nussinov, R.; Wright, P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009, 5, 789–796. [Google Scholar] [CrossRef]
- Fenwick, R.B.; Esteban-Martín, S.; Salvatella, X. Understanding biomolecular motion, recognition, and allostery by use of conformational ensembles. Eur. Biophys. J. 2011, 40, 1339–1355. [Google Scholar] [CrossRef]
- Gáspári, Z.; Perczel, A. Protein dynamics as reported by NMR. Annu. Rep. NMR Spect. 2010, 71, 35–75. [Google Scholar] [CrossRef]
- Gáspári, Z.; Várnai, P.; Szappanos, B.; Perczel, A. Reconciling the lock-and-key and dynamic views of canonical serine protease inhibitor action. FEBS Lett. 2010, 584, 203–206. [Google Scholar] [CrossRef]
- Dyson, J.H.; Wright, P.E. [12] Nuclear magnetic resonance methods for elucidation of structure and dynamics in disordered states. Meth. Enzymol. 2001, 339, 258–270. [Google Scholar] [CrossRef]
- Ferreon, A.C.M.; Ferreon, J.C.; Wright, P.E.; Deniz, A.A. Modulation of allostery by protein intrinsic disorder. Nature 2013, 498, 390–394. [Google Scholar] [CrossRef]
- Laskowski, R.A. Structural Quality Assurance. In Structural Bioinformatics, 2nd Ed. ed; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Wütrich, K. NMR of Proteins and Nucleic Acids; Wiley: Hoboken, NJ, USA, 1986. [Google Scholar]
- Bürgi, R.; Pitera, J.; van Gunsteren, W.F. Assessing the effect of conformational averaging on the measured values of observables. J. Biomol. NMR 2001, 19, 305–320. [Google Scholar] [CrossRef]
- Allison, J.R. Assessing and refining molecular dynamics simulations of proteins with nuclear magnetic resonance data. Biophys. Rev. 2012, 4, 189–203. [Google Scholar] [CrossRef]
- Best, R.B.; Vendruscolo, M. Structural interpretation of hydrogen exchange protection factors in proteins: Characterization of the native state fluctuations of CI2. Structure 2006, 14, 97–106. [Google Scholar] [CrossRef]
- Paci, E.; Karplus, M. Forced unfolding of fibronectin type 3 modules: an analysis by biased molecular dynamics simulations. J. Mol. Biol. 1999, 288, 441–459. [Google Scholar] [CrossRef]
- Jarymowycz, V.A.; Stone, M.J. Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem. Rev. 2006, 106, 1624–1671. [Google Scholar] [CrossRef]
- Schwieters, C.D.; Kuszewski, J.J.; Clore, M.G. Using Xplor–NIH for NMR molecular structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 2006, 48, 47–62. [Google Scholar] [CrossRef]
- Best, R.B.; Vendruscolo, M. Determination of protein structures consistent with NMR order parameters. J. Am. Chem. Soc. 2004, 126, 8090–8091. [Google Scholar] [CrossRef]
- Batta, G.; Barna, T.; Gáspári, Z.; Sándor, S.; Kövér, K.E.; Binder, U.; Sarg, B.; Kaiserer, L.; Chhillar, A.K.; Eigentler, A.; et al. Functional aspects of the solution structure and dynamics of PAF—A highly‐stable antifungal protein from Penicillium chrysogenum. FEBS J. 2009, 276, 2875–2890. [Google Scholar] [CrossRef]
- Bonvin, A.M.; Brünger, A.T. Conformational variability of solution nucelar magnetic resonance structures. J. Mol. Biol. 1995, 250, 80–93. [Google Scholar] [CrossRef]
- Kemmink, J.; van Mierlo, C.P.; Scheek, R.M.; Creighton, T.E. Local structure due to an aromatic-amide interaction observed by 1H-nuclear magnetic resonance spectroscopy in peptides related to the N terminus of bovine pancreatic trypsin inhibitor. J. Mol. Biol. 1993, 230, 312–322. [Google Scholar] [CrossRef]
- Bonvin, A.M.; Boelens, R.; Kaptein, R. Time-and ensemble-averaged direct NOE restraints. J. Biomol. NMR 1994, 4, 143–149. [Google Scholar]
- Richter, B.; Gsponer, J.; Várnai, P.; Salvatella, X.; Vendruscolo, M. The MUMO (minimal under-restraining minimal over-restraining) method for the determination of native state ensembles of proteins. J. Biomol. NMR 2007, 37, 117–135. [Google Scholar] [CrossRef]
- Lakomek, N.A.; Lange, O.; Walter, K.F.; Fares, C.; Egger, D.; Lunkenheimer, P.; Meiler, J.; Grubmüller, H.; Becker, S.; de Groot, B.L.; et al. Residual dipolar couplings as a tool to study molecular recognition of ubiquitin. Biochem. Soc. Trans. 2008, 36, 1433–1437. [Google Scholar] [CrossRef]
- Higman, V.A.; Boyd, J.; Smith, L.J.; Redfield, C. Residual dipolar couplings: are multiple independent alignments always possible. J. Biomol. NMR 2011, 49, 53–60. [Google Scholar] [CrossRef]
- Hess, B.; Scheek, R.M. Orientation restraints in molecular dynamics simulations using time and ensemble averaging. J. Magn. Reson. 2003, 164, 19–27. [Google Scholar] [CrossRef]
- Louhivuori, M.; Otten, R.; Lindorff-Larsen, K.; Annila, A. Conformational fluctuations affect protein alignment in dilute liquid crystal media. J. Am. Chem. Soc. 2006, 128, 4371–4376. [Google Scholar] [CrossRef]
- Montalvao, R.W.; de Simone, A.; Vendruscolo, M. Determination of structural fluctuations of proteins from structure-based calculations of residual dipolar couplings. J. Biomol. NMR 2012, 53, 281–292. [Google Scholar] [CrossRef]
- De Simone, A.R.W.; Montalvao, R.W.; Vendruscolo, M. Determination of conformational equilibria in proteins using residual dipolar couplings. J. Chem. Theory Comput. 2011, 7, 4189–4195. [Google Scholar] [CrossRef]
- Shen, Y.; Lange, O.; Delaglio, F.; Rossi, P.; Aramini, J.M.; Liu, G.; Eletsky, A.; Wu, Y.; Singarapu, K.K.; Lemak, A.; et al. Consistent blind protein structure generation from NMR chemical shift data. Proc. Natl. Acad. Sci. USA 2008, 105, 4685–4690. [Google Scholar] [CrossRef]
- Neal, S.; Nip, A.M.; Zhang, H.; Wishart, D.S. Rapid and accurate calculation of protein 1H, 13C and 15N chemical shifts. J. Biomol. NMR 2003, 26, 215–240. [Google Scholar] [CrossRef]
- Robustelli, P.; Kohlhoff, K.; Cavalli, A.; Vendruscolo, M. Using NMR chemical shifts as structural restraints in molecular dynamics simulations of proteins. Structure 2010, 18, 923–933. [Google Scholar] [CrossRef]
- Camilloni, C.; Cavalli, A.; Vendruscolo, M. Assessment of the use of NMR chemical shifts as replica-averaged structural restraints in molecular dynamics simulations to characterise the dynamics of proteins. J. Phys. Chem. B 2013, 117, 1838–1843. [Google Scholar] [CrossRef]
- Camilloni, C.; Robustelli, P.; Simone, A.D.; Cavalli, A.; Vendruscolo, M. Characterization of the conformational equilibrium between the two major substates of RNase a using NMR chemical shifts. J. Am. Chem. Soc. 2012, 134, 3968–3971. [Google Scholar] [CrossRef]
- Dedmon, M.M.; Lindorff-Larsen, K.; Christodoulou, J.; Vendruscolo, M.; Dobson, C.M. Mapping long-range interactions in α-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J. Am. Chem. Soc. 2005, 127, 476–477. [Google Scholar]
- Lindorff-Larsen, K.; Best, R.B.; DePristo, M.A.; Dobson, C.M.; Vendruscolo, M. Simultaneous determination of protein structure and dynamics. Nature 2005, 433, 128–132. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Best, R.B.; Vendruscolo, M. Interpreting dynamically-averaged scalar couplings in proteins. J. Biomol. NMR 2005, 32, 273–280. [Google Scholar] [CrossRef]
- Allison, J.R.; Várnai, P.; Dobson, C.M.; Vendruscolo, M. Determination of the free energy landscape of α-synuclein using spin label nuclear magnetic resonance measurements. J. Am. Chem. Soc. 2009, 131, 18314–18326. [Google Scholar] [CrossRef]
- Im, W.; Jo, S.; Kim, T. An ensemble dynamics approach to decipher solid-state NMR observables of membrane proteins. Biochim. Biophys. Acta 2012, 1818, 252–262. [Google Scholar] [CrossRef]
- Lange, O.F.; Lakomek, N.A.; Farès, C.; Schröder, G.F.; Walter, K.F.; Becker, S.; de Groot, B.L. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science 2008, 320, 1471–1475. [Google Scholar] [CrossRef]
- Fenwick, R.B.; Esteban-Martín, S.; Richter, B.; Lee, D.; Walter, K.F.; Milovanovic, D.; Becker, S.; Lakomek, N.A.; Griesinger, C.; Salvatella, X. Weak long-range correlated motions in a surface patch of ubiquitin involved in molecular recognition. J. Am. Chem. Soc. 2011, 133, 10336–10339. [Google Scholar] [CrossRef]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef]
- Fisher, C.K.; Stultz, C.M. Constructing ensembles for intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2011, 21, 426–431. [Google Scholar] [CrossRef]
- Ozenne, V.; Bauer, F.; Salmon, L.; Huang, J.R.; Jensen, M.R.; Segard, S.; Bernadó, P.; Charavay, C.; Blackledge, M. Flexible-meccano: A tool for the generation of explicit ensemble descriptions of intrinsically disordered proteins and their associated experimental observables. Bioinformatics 2012, 28, 1463–1470. [Google Scholar] [CrossRef]
- Feldman, H.J.; Hogue, C.W. Probabilistic sampling of protein conformations: New hope for brute force? Proteins 2002, 46, 8–23. [Google Scholar] [CrossRef]
- Petoukhov, M.V.; Svergun, D.I. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys J. 2005, 89, 1237–1250. [Google Scholar] [CrossRef]
- Bernadó, P.; Mylonas, E.; Petoukhov, M.V.; Blackledge, M.; Svergun, D.I. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007, 129, 5656–5664. [Google Scholar] [CrossRef]
- Bertini, I.; Ferella, L.; Luchinat, C.; Parigi, G.; Petoukhov, M.V.; Ravera, E.; Rosato, A.; Svergun, D.I. MaxOcc: A web portal for maximum occurrence analysis. J. Biomol. NMR 2012, 53, 271–280. [Google Scholar] [CrossRef]
- Louhivuori, M.; Pääkkönen, K.; Fredriksson, K.; Permi, P.; Lounila, J.; Annila, A. On the origin of residual dipolar couplings from denatured proteins. J. Am. Chem. Soc. 2003, 125, 15647–15650. [Google Scholar] [CrossRef]
- Louhivuori, M.; Fredriksson, K.; Pääkkönen, K.; Permi, P.; Annila, A. Alignment of chain-like molecules. J. Biomol. NMR 2004, 29, 517–524. [Google Scholar] [CrossRef]
- Jha, A.K.; Colubri, A.; Freed, K.F.; Sosnick, T.R. Statistical coil model of the unfolded state: Resolving the reconciliation problem. Proc. Natl. Acad. Sci. USA 2005, 102, 13099–13104. [Google Scholar] [CrossRef]
- Bernadó, P.; Blanchard, L.; Timmins, P.; Marion, D.; Ruigrok, R.W.; Blackledge, M. A structural model for unfolded proteins from residual dipolar couplings and small-angle x-ray scattering. Proc. Natl. Acad. Sci. USA 2005, 102, 17002–17007. [Google Scholar] [CrossRef]
- Meier, S.; Grzesiek, S.; Blackledge, M. Mapping the conformational landscape of urea-denatured ubiquitin using residual dipolar couplings. J. Am. Chem. Soc. 2007, 129, 9799–9807. [Google Scholar] [CrossRef]
- Mukrasch, M.D.; Markwick, P.; Biernat, J.; von Bergen, M.; Bernadó, P.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M.; Blackledge, M. Highly populated turn conformations in natively unfolded tau protein identified from residual dipolar couplings and molecular simulation. J. Am. Chem. Soc. 2007, 129, 5235–5243. [Google Scholar] [CrossRef]
- Choy, W.Y.; Forman-Kay, J.D. Calculation of ensembles of structures representing the unfolded state of an SH3 domain. J. Mol. Biol. 2001, 308, 1011–1032. [Google Scholar] [CrossRef]
- Krzeminski, M.; Marsh, J.A.; Neale, C.; Choy, W.Y.; Forman-Kay, J.D. Characterization of disordered proteins with ENSEMBLE. Bioinformatics 2013, 29, 398–399. [Google Scholar] [CrossRef]
- Nodet, G.; Salmon, L.; Ozenne, V.; Meier, S.; Jensen, M.R.; Blackledge, M. Quantitative description of backbone conformational sampling of unfolded proteins at amino acid resolution from NMR residual dipolar couplings. J. Am. Chem. Soc. 2009, 131, 17908–17918. [Google Scholar] [CrossRef]
- Salmon, L.; Nodet, G.; Ozenne, V.; Yin, G.; Jensen, M.R.; Zweckstetter, M.; Blackledge, M. NMR characterization of long-range order in intrinsically disordered proteins. J. Am. Chem. Soc. 2010, 132, 8407–8418. [Google Scholar]
- Jensen, M.R.; Communie, G.; Ribeiro, E.A.; Martinez, N.; Desfosses, A.; Salmon, L.; Mollica, L.; Gabel, F.; Jamin, M.; Longhi, S.; et al. Intrinsic disorder in measles virus nucleocapsids. Proc. Natl. Acad. Sci. USA 2011, 108, 9839–9844. [Google Scholar] [CrossRef]
- Guerry, P.; Salmon, L.; Mollica, L.; Ortega Roldan, J.L.; Markwick, P.; van Nuland, N.A.; Blackledge, M. Mapping the population of protein conformational energy sub‐states from NMR dipolar couplings. Angew. Chem. Int. Ed. 2013, 52, 3181–3185. [Google Scholar] [CrossRef]
- Ángyán, A.F.; Szappanos, B.; Perczel, A.; Gáspári, Z. CoNSEnsX: An ensemble view of protein structures and NMR-derived experimental data. BMC Struct. Biol. 2010, 10, 39. [Google Scholar] [CrossRef]
- Gáspári, Z.; Ángyán, A.F; Dhir, S.; Franklin, D.; Perczel, A.; Pintar, A.; Pongor, S. Probing dynamic protein ensembles with atomic proximity measures. Curr. Prot. Pept. Sci. 2010, 11, 515–522. [Google Scholar] [CrossRef]
- Dhulesia, A.; Gsponer, J.; Vendruscolo, M. Mapping of two networks of residues that exhibit structural and dynamical changes upon binding in a PDZ domain protein. J. Am. Chem. Soc. 2008, 130, 8931–8939. [Google Scholar] [CrossRef]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Ferkinghoff-Borg, J. Similarity measures for protein ensembles. PLoS One 2009, 4, e4203. [Google Scholar] [CrossRef]
- Rieping, W.; Habeck, M.; Nilges, M. Inferential structure determination. Science 2005, 309, 303–306. [Google Scholar] [CrossRef]
- Fisher, C.K.; Huang, A.; Stultz, C.M. Modeling intrinsically disordered proteins with bayesian statistics. J. Am. Chem. Soc. 2010, 132, 14919–14927. [Google Scholar] [CrossRef]
- Pitera, J.W.; Chodera, J.D. On the use of experimental observations to bias simulated ensembles. J. Chem. Theory Comput. 2012, 8, 3445–3451. [Google Scholar] [CrossRef]
- Cavalli, A.; Camilloni, C.; Vendruscolo, M. Molecular dynamics simulations with replica-averaged structural restraints generate structural ensembles according to the maximum entropy principle. J. Chem. Phys. 2013, 138, 094112. [Google Scholar] [CrossRef]
- Roux, B.; Weare, J. On the statistical equivalence of restrained-ensemble simulations with the maximum entropy method. J. Chem. Phys. 2013, 138, 084107. [Google Scholar] [CrossRef]
- Torda, A.E.; Scheek, R. M.; van Gunsteren, W.F. Time-dependent distance restraints in molecular dynamics simulations. Chem. Phys. Lett. 1989, 157, 289–294. [Google Scholar] [CrossRef]
- Torda, A.E.; Brunne, R.M.; Huber, T.; Kessler, H.; van Gunsteren, W.F. Structure refinement using time-averaged J-coupling constant restraints. J. Biomol. NMR 1993, 3, 55–66. [Google Scholar]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef]
- Li, D.W.; Brüschweiler, R. NMR‐based protein potentials. Angew. Chem. Int. Ed. 2010, 49, 6778–6780. [Google Scholar] [CrossRef]
- Markwick, P.R.; Bouvignies, G.; Salmon, L.; McCammon, J.A.; Nilges, M.; Blackledge, M. Toward a unified representation of protein structural dynamics in solution. J. Am. Chem. Soc. 2009, 131, 16968–16975. [Google Scholar]
- Markwick, P.R.; Cervantes, C.F.; Abel, B.L.; Komives, E.A.; Blackledge, M.; McCammon, J.A. Enhanced conformational space sampling improves the prediction of chemical shifts in proteins. J. Am. Chem. Soc. 2010, 132, 1220–1221. [Google Scholar]
- Salmon, L.; Bascom, G.; Andricioaei, I.; Al-Hashimi, H.M. A general method for constructing atomic-resolution RNA ensembles using NMR residual dipolar couplings: the basis for interhelical motions revealed. J. Am. Chem. Soc. 2013, 135, 5457–5466. [Google Scholar]
- Li, D.W.; Brüschweiler, R. Iterative optimization of molecular mechanics force fields from NMR data of full-length proteins. J. Chem. Theory Comput. 2011, 7, 1773–1782. [Google Scholar] [CrossRef]
- Iesmantavicius, V.; Jensen, M.R.; Ozenne, V.; Blackledge, M.; Poulsen, F.M.; Kjaergaard, M. Modulation of the intrinsic helix propensity of an intrinsically disordered protein reveals long-range helix-helix interactions. J. Am. Chem. Soc. 2013, 135, 10155–10163. [Google Scholar]
- Jo, S.; Im, W. Transmembrane helix orientation and dynamics: insights from ensemble dynamics with solid-state NMR observables. Biophys. J. 2011, 100, 2913–2921. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
F. Ángyán, A.; Gáspári, Z. Ensemble-Based Interpretations of NMR Structural Data to Describe Protein Internal Dynamics. Molecules 2013, 18, 10548-10567. https://doi.org/10.3390/molecules180910548
F. Ángyán A, Gáspári Z. Ensemble-Based Interpretations of NMR Structural Data to Describe Protein Internal Dynamics. Molecules. 2013; 18(9):10548-10567. https://doi.org/10.3390/molecules180910548
Chicago/Turabian StyleF. Ángyán, Annamária, and Zoltán Gáspári. 2013. "Ensemble-Based Interpretations of NMR Structural Data to Describe Protein Internal Dynamics" Molecules 18, no. 9: 10548-10567. https://doi.org/10.3390/molecules180910548