Effect of Honokiol on Cytochrome P450 and UDP-Glucuronosyltransferase Enzyme Activities in Human Liver Microsomes




Abstract
:1. Introduction
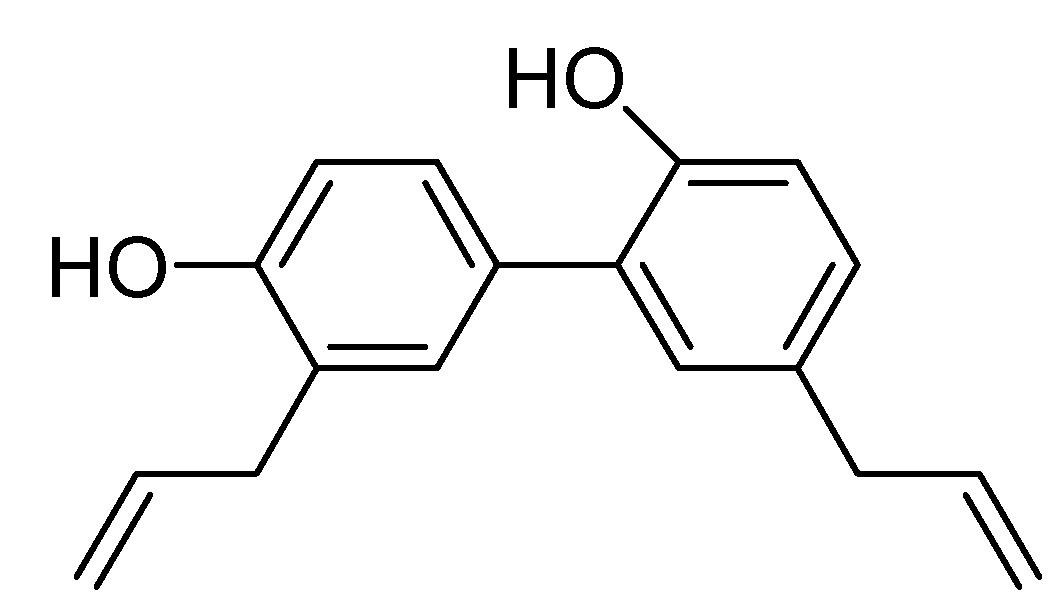
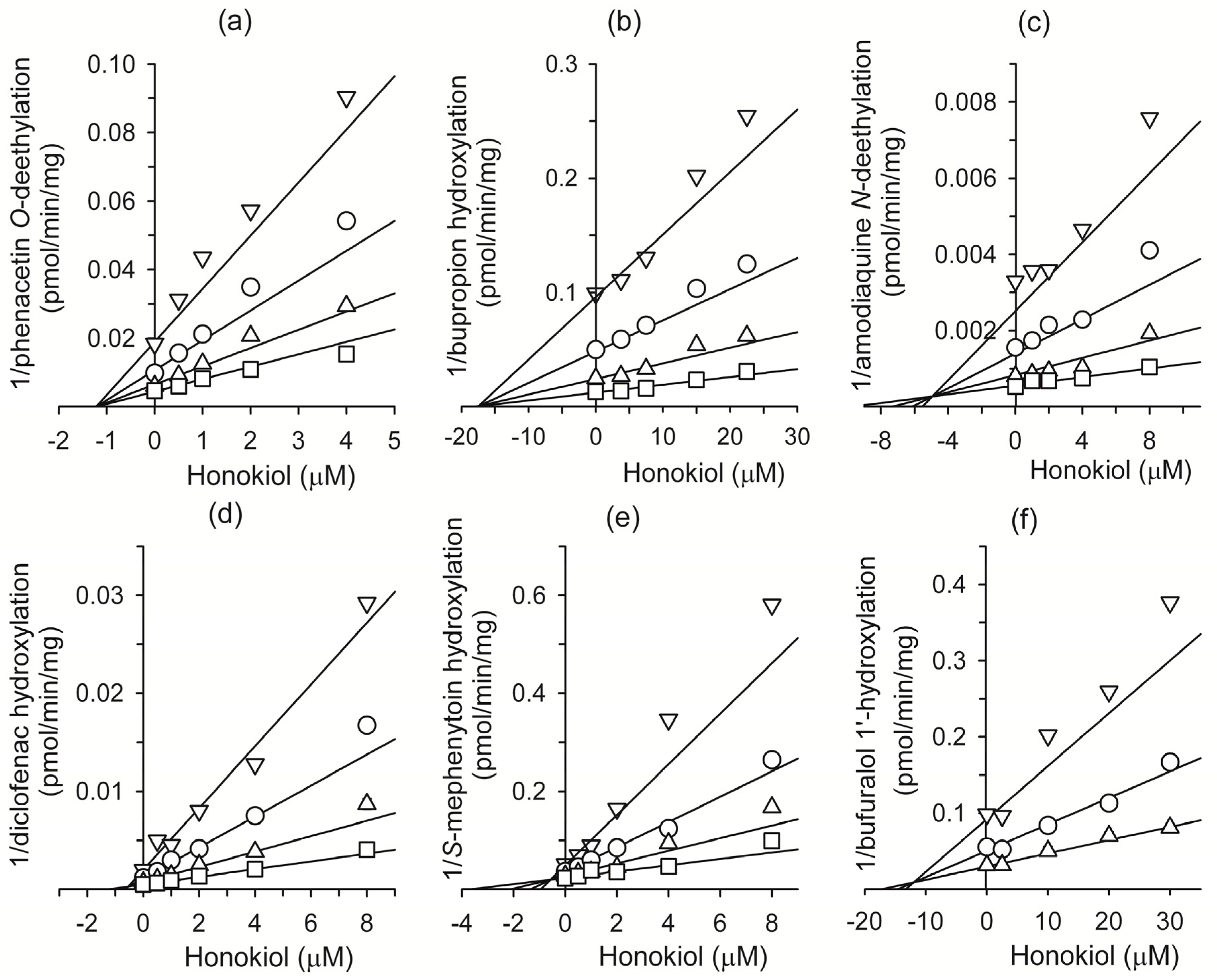
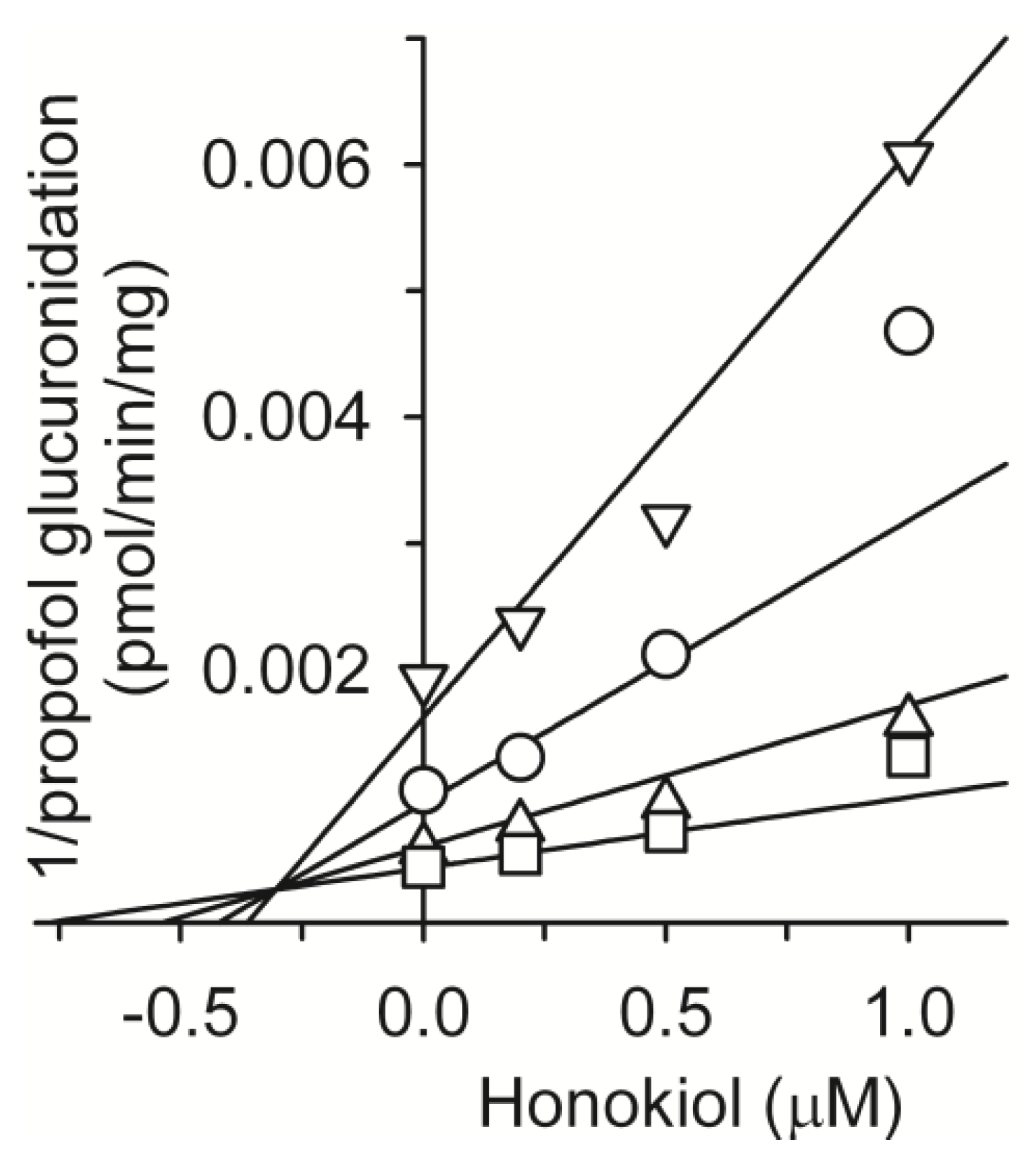
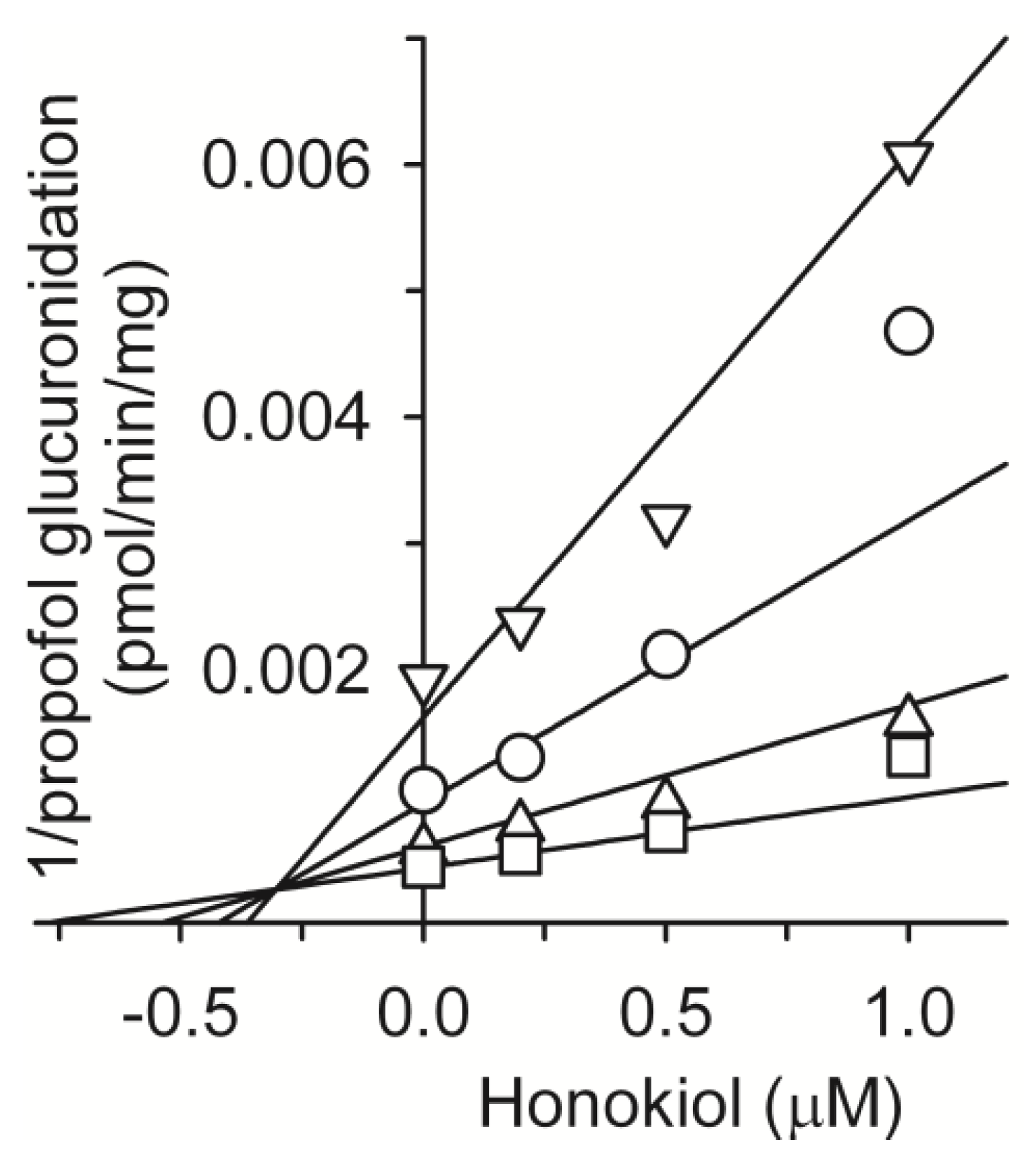
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
| CYP activity | CYP | IC50 (μM) of honokiol | |
|---|---|---|---|
| no preincubation | with preincubation * | ||
| Phenacetin O-deethylation | 1A2 | 2.1 | 4.7 |
| Coumarin 7-hydroxylation | 2A6 | No inhibition | No inhibition |
| Bupropion hydroxylation | 2B6 | 13.8 | 20.8 |
| Amodiaquine N-deethylation | 2C8 | 8.9 | 15.5 |
| Diclofenac 4-hydroxylation | 2C9 | 4.1 | 3.9 |
| S-Mephenytoin 4'-hydroxylation | 2C19 | 2.2 | 2.9 |
| Bufuralol 1'-hydroxylation | 2D6 | 14.0 | 38.1 |
| Midazolam 1'-hydroxylation | 3A4 | 97.3 | 45.8 |
| Enzymes | Marker reactions | Ki (μM) | Inhibition mode |
|---|---|---|---|
| CYP1A2 | Phenacetin O-deethylation | 1.2 | noncompetitive |
| CYP2B6 | Bupropion hydroxylation | 17.5 | competitive |
| CYP2C8 | Amodiaquine N-deethylation | 4.9 | competitive |
| CYP2C9 | Diclofenac 4-hydroxylation | 0.54 | competitive |
| CYP2C19 | S-Mephenytoin 4'-hydroxylation | 0.57 | competitive |
| CYP2D6 | Bufuralol 1'-hydroxylation | 12.0 | competitive |
| UGT1A9 | Propofol glucuronidation | 0.3 | competitive |

| UGT | Marker enzyme | IC50 (μM) |
|---|---|---|
| UGT1A1 | 17β-estradiol 3-glucuronidation | 50.5 |
| UGT1A4 | trifluoperazine N-glucuronidation | 158.1 |
| UGT1A9 | propofol glucuronidation | 0.96 |
| UGT2B7 | azidothymidine glucuronidation | 36.4 |
3. Experimental
3.1. Materials
3.2. Inhibitory Effect of Honokiol on Eight Major CYP Activities in Human Liver Microsomes
3.3. Inhibitory Effect of Honokiol on Four UGT Activities in Human Liver Microsomes
3.4. Kinetic Analysis
3.5. Data Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Cui, H.S.; Huang, L.S.; Sok, D.E.; Shin, J.; Kwon, B.M.; Youn, U.J.; Bae, K. Protective action of honokiol, administered orally, against oxidative stress in brain of mice challenged with NMDA. Phytomedicine 2007, 14, 696–700. [Google Scholar] [CrossRef]
- Dikalov, S.; Losik, T.; Arbiser, J.L. Honokiol is a potent scavenger of superoxide and peroxyl radicals. Biochem. Pharmacol. 2008, 76, 589–596. [Google Scholar] [CrossRef]
- Shen, J.L.; Man, K.M.; Huang, P.H.; Chen, W.C.; Chen, D.C.; Cheng, Y.W.; Liu, P.L.; Chou, M.C.; Chen, Y.H. Honokiol and magnolol as multifunctional antioxidative molecules for dermatologic disorders. Molecules 2010, 15, 6452–6465. [Google Scholar] [CrossRef]
- Kim, B.H.; Cho, J.Y. Anti-inflammatory effect of honokiol is mediated by PI3K/Akt pathway suppression. Acta Pharmacol. Sin. 2008, 29, 113–122. [Google Scholar]
- Lee, J.; Jung, E.; Park, J.; Jung, K.; Lee, S.; Hong, S.; Park, E.; Kim, J.; Park, S.; Park, D. Anti-inflammatory effects of magnolol and honokiol are mediated through inhibition of the downstream pathway of MEKK-1 in NF-kappaB activation signaling. Planta Med. 2005, 71, 338–343. [Google Scholar]
- Munroe, M.E.; Arbiser, J.L.; Bishop, G.A. Honokiol, a natural plant product, inhibits inflammatory signals and alleviates inflammatory arthritis. J. Immunol. 2007, 179, 753–763. [Google Scholar]
- Chao, L.K.; Liao, P.C.; Ho, C.L.; Wang, E.I.; Chuang, C.C.; Chiu, H.W.; Hung, L.B.; Hua, K.F. Anti-inflammatory bioactivities of honokiol through inhibition of protein kinase C, mitogen-activated protein kinase, and the NF-kappaB pathway to reduce LPS-induced TNFalpha and NO expression. J. Agric. Food Chem. 2010, 58, 3472–3478. [Google Scholar]
- Munroe, M.E.; Businga, T.R.; Kline, J.N.; Bishop, G.A. Anti-inflammatory effects of the neurotransmitter agonist Honokiol in a mouse model of allergic asthma. J. Immunol. 2010, 185, 5586–5597. [Google Scholar]
- Hu, H.; Zhang, X.X.; Wang, Y.Y.; Chen, S.Z. Honokiol inhibits arterial thrombosis through endothelial cell protection and stimulation of prostacyclin. Acta Pharmacol. Sin. 2005, 26, 1063–1068. [Google Scholar] [CrossRef]
- Hoi, C.P.; Ho, Y.P.; Baum, L.; Chow, A.H. Neuroprotective effect of honokiol and magnolol, compounds from Magnolia officinalis, on beta-amyloid-induced toxicity in PC12 cells. Phytother. Res. 2010, 24, 1538–1542. [Google Scholar] [CrossRef]
- Lin, Y.R.; Chen, H.H.; Ko, C.H.; Chan, M.H. Neuroprotective activity of honokiol and magnolol in cerebellar granule cell damage. Eur. J. Pharmacol. 2006, 537, 64–69. [Google Scholar]
- Matsui, N.; Takahashi, K.; Takeichi, M.; Kuroshita, T.; Noguchi, K.; Yamazaki, K.; Tagashira, H.; Tsutsui, K.; Okada, H.; Kido, Y.; et al. Magnolol and honokiol prevent learning and memory impairment and cholinergic deficit in SAMP8 mice. Brain Res. 2009, 1305, 108–117. [Google Scholar] [CrossRef]
- Lin, Y.R.; Chen, H.H.; Lin, Y.C.; Ko, C.H.; Chan, M.H. Antinociceptive actions of honokiol and magnolol on glutamatergic and inflammatory pain. J. Biomed. Sci. 2009, 16, 94. [Google Scholar] [CrossRef]
- Xu, Q.; Yi, L.T.; Pan, Y.; Wang, X.; Li, Y.C.; Li, J.M.; Wang, C.P.; Kong, L.D. Antidepressant-like effects of the mixture of honokiol and magnolol from the barks of Magnolia officinalis in stressed rodents. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 715–725. [Google Scholar]
- Hahm, E.R.; Arlotti, J.A.; Marynowski, S.W.; Singh, S.V. Honokiol, a constituent of oriental medicinal herb magnolia officinalis, inhibits growth of PC-3 xenografts in vivo in association with apoptosis induction. Clin. Cancer Res. 2008, 14, 1248–1257. [Google Scholar] [CrossRef]
- Liu, H.; Zang, C.; Emde, A.; Planas-Silva, M.D.; Rosche, M.; Kuhnl, A.; Schulz, C.O.; Elstner, E.; Possinger, K.; Eucker, J. Anti-tumor effect of honokiol alone and in combination with other anti-cancer agents in breast cancer. Eur. J. Pharmacol. 2008, 591, 43–51. [Google Scholar] [CrossRef]
- Fried, L.E.; Arbiser, J.L. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid. Redox Signal 2009, 11, 1139–1148. [Google Scholar] [CrossRef]
- Steinmann, P.; Walters, D.K.; Arlt, M.J.; Banke, I.J.; Ziegler, U.; Langsam, B.; Arbiser, J.; Muff, R.; Born, W.; Fuchs, B. Antimetastatic activity of honokiol in osteosarcoma. Cancer 2012, 118, 2117–2127. [Google Scholar] [CrossRef]
- Tian, W.; Xu, D.; Deng, Y.C. Honokiol, a multifunctional tumor cell death inducer. Pharmazie 2012, 67, 811–816. [Google Scholar]
- Kapoor, S. Attenuation of tumor growth by honokiol: An evolving role in oncology. Drug Discov. Ther. 2012, 6, 327–328. [Google Scholar]
- Kaushik, G.; Ramalingam, S.; Subramaniam, D.; Rangarajan, P.; Protti, P.; Rammamoorthy, P.; Anant, S.; Mammen, J.M. Honokiol induces cytotoxic and cytostatic effects in malignant melanoma cancer cells. Am. J. Surg. 2012, 204, 868–873. [Google Scholar] [CrossRef]
- Chang, J. Medicinal herbs: Drugs or dietary supplements? Biochem. Pharmacol. 2000, 59, 211–219. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Tan, Q.R.; Tong, Y.; Wang, X.Y.; Wang, H.H.; Ho, L.M.; Wong, H.K.; Feng, Y.B.; Wang, D.; Ng, R.; et al. An epidemiological study of concomitant use of Chinese medicine and antipsychotics in schizophrenic patients: Implication for herb-drug interaction. PLoS One 2011, 6, e17239. [Google Scholar] [CrossRef] [Green Version]
- Girard, L.; Vohra, S. Herbal Medicine: Biomolecular and Clinical Aspects; Benzie, I.F.F., Wachtel-Galor, S., Eds.; CRC Press: Boca Raton, FL, USA, 2011; Chapter 21; p. 320. [Google Scholar]
- Zhou, S.F.; Zhou, Z.W.; Li, C.G.; Chen, X.; Yu, X.; Xue, C.C.; Herington, A. Identification of drugs that interact with herbs in drug development. Drug Discov. Today 2007, 12, 664–673. [Google Scholar] [CrossRef] [Green Version]
- He, S.M.; Chan, E.; Zhou, S.F. ADME properties of herbal medicines in humans: Evidence, challenges and strategies. Curr. Pharm. Des. 2011, 17, 357–407. [Google Scholar] [CrossRef]
- Mohamed, M.E.; Frye, R.F. Effects of herbal supplements on drug glucuronidation. Review of clinical, animal, and in vitro studies. Planta Med. 2011, 77, 311–321. [Google Scholar]
- Na, D.H.; Ji, H.Y.; Park, E.J.; Kim, M.S.; Liu, K.H.; Lee, H.S. Evaluation of metabolism-mediated herb-drug interactions. Arch. Pharm. Res. 2011, 34, 1829–1842. [Google Scholar] [CrossRef]
- Pao, L.H.; Hu, O.Y.; Fan, H.Y.; Lin, C.C.; Liu, L.C.; Huang, P.W. Herb-drug interaction of 50 Chinese herbal medicines on CYP3A4 activity in vitro and in vivo. Am. J. Chin. Med. 2012, 40, 57–73. [Google Scholar] [CrossRef]
- Borrelli, F.; Izzo, A.A. Herb-drug interactions with St John’s wort (Hypericumperforatum): An update on clinical observations. AAPS J. 2009, 11, 710–727. [Google Scholar] [CrossRef]
- Wu, J.W.; Lin, L.C.; Tsai, T.H. Drug-drug interactions of silymarin on the perspective of pharmacokinetics. J. Ethnopharmacol. 2009, 121, 185–193. [Google Scholar] [CrossRef]
- He, S.M.; Yang, A.K.; Li, X.T.; Du, Y.M.; Zhou, S.F. Effects of herbal products on the metabolism and transport of anticancer agents. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1195–1213. [Google Scholar] [CrossRef]
- Chen, X.W.; Serag, E.S.; Sneed, K.B.; Liang, J.; Chew, H.; Pan, S.Y.; Zhou, S.F. Clinical herbal interactions with conventional drugs: From molecules to maladies. Curr. Med. Chem. 2011, 18, 4836–4850. [Google Scholar] [CrossRef]
- Le Goff-Klein, N.; Koffel, J.C.; Jung, L.; Ubeaud, G. In vitro inhibition of simvastatin metabolism, a HMG-CoAreductase inhibitor in human and rat liver by bergamottin, a component of grapefruit juice. Eur. J. Pharm. Sci. 2003, 18, 31–35. [Google Scholar] [CrossRef]
- Bailey, D.G.; Dresser, G.K.; Bend, J.R. Bergamottin, lime juice, and red wine as inhibitors of cytochrome P450 3A4 activity: Comparison with grapefruit juice. Clin. Pharmacol. Ther. 2003, 73, 529–537. [Google Scholar] [CrossRef]
- Johnston, P.E.; Milstone, A. Probable interaction of bergamottin and cyclosporine in a lung transplant recipient. Transplantation 2005, 79, 746. [Google Scholar]
- Joo, J.; Lee, D.; Wu, Z.; Shin, J.H.; Lee, H.S.; Kwon, B.M.; Huh, T.L.; Kim, Y.W.; Lee, S.J.; Kim, T.W.; et al. In vitro metabolism of obovatol and its effect on cytochrome P450 enzyme activities in human liver microsomes. Biopharm. Drug Dispos. 2013, 34, 195–202. [Google Scholar]
- Miners, J.O.; Bowalgaha, K.; Elliot, D.J.; Baranczewski, P.; Knights, K.M. Characterization of niflumic acid as a selective inhibitor of human liver microsomal UDP-glucuronosyltransferase 1A9: Application to the reaction phenotyping of acetaminophen glucuronidation. Drug Metab. Dispos. 2011, 39, 644–652. [Google Scholar]
- Tougou, K.; Gotou, H.; Ohno, Y.; Nakamura, A. Stereoselectiveglucuronidation and hydroxylation of etodolac by UGT1A9 and CYP2C9 in man. Xenobiotica 2004, 34, 449–461. [Google Scholar] [CrossRef]
- Lautala, P.; Ethell, B.T.; Taskinen, J.; Burchell, B. The specificity of glucuronidation of entacapone and tolcapone by recombinant human UDP-glucuronosyltransferases. Drug Metab. Dispos. 2000, 28, 1385–1389. [Google Scholar]
- Chu, X.Y.; Liang, Y.; Cai, X.; Cuevas-Licea, K.; Rippley, R.K.; Kassahun, K.; Shou, M.; Braun, M.P.; Doss, G.A.; Anari, M.R.; et al. Metabolism and renal elimination of gaboxadol in humans: Role of UDP-glucuronosyltransferases and transporters. Pharm. Res. 2009, 26, 459–468. [Google Scholar] [CrossRef]
- Borlak, J.; Gasparic, A.; Locher, M.; Schupke, H.; Hermann, R. N-Glucuronidation of the antiepileptic drug retigabine: Results from studies with human volunteers, heterologously expressed human UGTs, human liver, kidney, and liver microsomal membranes of Crigler-Najjar type II. Metabolism 2006, 55, 711–721. [Google Scholar] [CrossRef]
- Luukkanen, L.; Taskinen, J.; Kurkela, M.; Kostiainen, R.; Hirvonen, J.; Finel, M. Kinetic characterization of the 1A subfamily of recombinant human UDP-glucuronosyltransferases. Drug Metab. Dispos. 2005, 33, 1017–1026. [Google Scholar] [CrossRef]
- Newton, D.J.; Wang, R.W.; Lu, A.Y. Cytochrome P450 inhibitors. Evaluation of specificities in the in vitro metabolism of therapeutic agents by human liver microsomes. Drug Metab. Dispos. 1995, 23, 154–158. [Google Scholar]
- Zhou, S.F.; Zhou, Z.W.; Yang, L.P.; Cai, J.P. Substrates, inducers, inhibitors and structure-activity relationships of human Cytochrome P450 2C9 and implications in drug development. Curr. Med. Chem. 2009, 16, 3480–3675. [Google Scholar] [CrossRef]
- Cytochrome P450 Drug Interaction Table. Available online: http://medicine.iupui.edu/clinpharm/ddis/ (accessed on 12 January 2009).
- Sand, P.G.; Dreiseitel, A.; Stang, M.; Schreier, P.; Oehme, A.; Locher, S.; Hajak, G. Cytochrome P450 2C19 inhibitory activity of common berry constituents. Phytother. Res. 2010, 24, 304–307. [Google Scholar]
- Mei, L.; Zhang, L.; Dai, R. An inhibition study of beauvericin on human and rat cytochrome P450 enzymes and its pharmacokinetics in rats. J. Enzyme Inhib. Med. Chem. 2009, 24, 753–762. [Google Scholar]
- Ji, H.Y.; Liu, K.H.; Lee, H.; Im, S.R.; Shim, H.J.; Son, M.; Lee, H.S. Corydaline inhibits multiple cytochrome P450 and UDP-glucuronosyltransferase enzyme activities in human liver microsomes. Molecules 2011, 16, 6591–6602. [Google Scholar]
- Ji, H.Y.; Kim, S.Y.; Kim, D.K.; Jeong, J.H.; Lee, H.S. Effects of eupatilin and jaceosidin on cytochrome p450 enzyme activities in human liver microsomes. Molecules 2010, 15, 6466–6475. [Google Scholar] [CrossRef]
- Kim, K.A.; Lee, J.S.; Park, H.J.; Kim, J.W.; Kim, C.J.; Shim, I.S.; Kim, N.J.; Han, S.M.; Lim, S. Inhibition of cytochrome P450 activities by oleanolic acid and ursolic acid in human liver microsomes. Life Sci. 2004, 74, 2769–2779. [Google Scholar]
- Jahng, Y.; Kwon, O.K.; Lee, S. In vitro inhibitory effect of luotoninA on human CYP1A. Arch. Pharm. Res. 2012, 35, 2199–2203. [Google Scholar] [CrossRef]
- Kim, H.; Choi, H.K.; Jeong, T.C.; Jahng, Y.; Kim, D.H.; Lee, S.H.; Lee, S. Selective inhibitory effects of mollugin on CYP1A2 in human liver microsomes. Food Chem. Toxicol. 2013, 51, 33–37. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Zhang, Y.J.; Guo, Y.L.; Li, W.J.; Yu, C. Astragaloside IV inhibited the activity of CYP1A2 in liver microsomes and influenced theophylline pharmacokinetics in rats. J. Pharm. Pharmacol. 2013, 65, 149–155. [Google Scholar] [CrossRef]
- Lai, X.S.; Yang, L.P.; Li, X.T.; Liu, J.P.; Zhou, Z.W.; Zhou, S.F. Human CYP2C8: Structure, substrate specificity, inhibitor selectivity, inducers and polymorphisms. Curr. Drug. Metab. 2009, 10, 1009–1047. [Google Scholar] [CrossRef]
- Pang, C.Y.; Mak, J.W.; Ismail, R.; Ong, C.E. In vitro modulatory effects of flavonoids on human cytochrome P450 2C8 (CYP2C8). Naunyn Schmiedebergs Arch. Pharmacol. 2012, 385, 495–502. [Google Scholar]
- Walsky, R.L.; Obach, R.S.; Gaman, E.A.; Gleeson, J.P.; Proctor, W.R. Selective inhibition of human cytochrome P4502C8 by montelukast. Drug Metab. Dispos. 2005, 33, 413–418. [Google Scholar]
- Ogilvie, B.W.; Zhang, D.; Li, W.; Rodrigues, A.D.; Gipson, A.E.; Holsapple, J.; Toren, P.; Parkinson, A. Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of CYP2C8: Implications for drug-drug interactions. Drug Metab. Dispos. 2006, 34, 191–197. [Google Scholar]
- Sample Availability: Not Available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jeong, H.-U.; Kong, T.Y.; Kwon, S.S.; Hong, S.-W.; Yeon, S.H.; Choi, J.-H.; Lee, J.Y.; Cho, Y.Y.; Lee, H.S. Effect of Honokiol on Cytochrome P450 and UDP-Glucuronosyltransferase Enzyme Activities in Human Liver Microsomes. Molecules 2013, 18, 10681-10693. https://doi.org/10.3390/molecules180910681
Jeong H-U, Kong TY, Kwon SS, Hong S-W, Yeon SH, Choi J-H, Lee JY, Cho YY, Lee HS. Effect of Honokiol on Cytochrome P450 and UDP-Glucuronosyltransferase Enzyme Activities in Human Liver Microsomes. Molecules. 2013; 18(9):10681-10693. https://doi.org/10.3390/molecules180910681
Chicago/Turabian StyleJeong, Hyeon-Uk, Tae Yeon Kong, Soon Sang Kwon, Sung-Woon Hong, Sung Hum Yeon, Jun-Ho Choi, Jae Young Lee, Yong Yeon Cho, and Hye Suk Lee. 2013. "Effect of Honokiol on Cytochrome P450 and UDP-Glucuronosyltransferase Enzyme Activities in Human Liver Microsomes" Molecules 18, no. 9: 10681-10693. https://doi.org/10.3390/molecules180910681