Two New Sphingolipids from the Leaves of Piper betle L.

Abstract
:1. Introduction
2. Results and Discussion
2.1. Structure Analysis of Pipercerebroside A

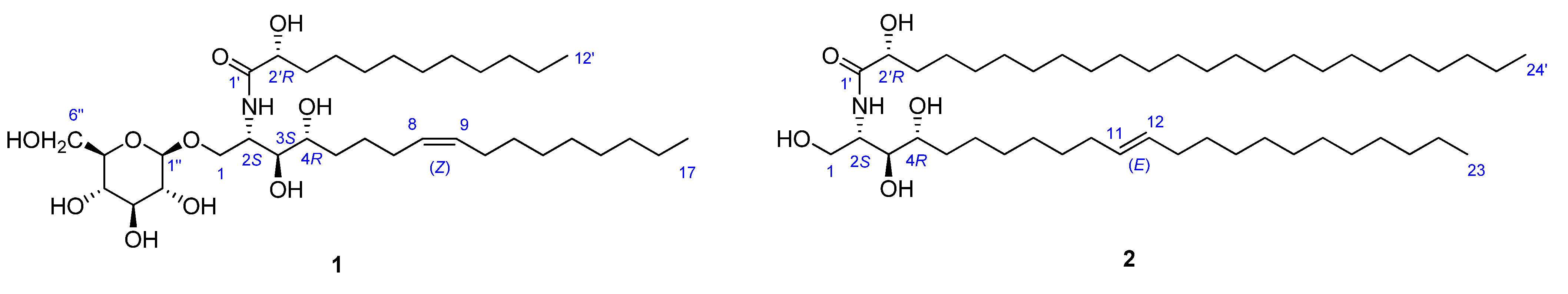{kind=link}
{kind=link}
| No. | 1 | No. | 2 | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | |||
| 1a | 3.66 (dd, 1H, 10.5, 4.0 Hz)3.96 (dd, 1H, 10.5, 4.0 Hz) | 69.5 t | 1a | 4.42 (dd, 1H, 10.0, 4.5 Hz)4.50 (dd, 1H, 10.0, 4.5 Hz) | 62.9 t | |
| 1b | 1b | |||||
| 2 | 3.97 (m, 1H) | 50.4 d | 2 | 5.16 (m, 1H) | 54.0 d | |
| 3 | 3.53 (m, 1H) | 74.3 d | 3 | 4.35 (m, 1H) | 77.1 d | |
| 4 | 3.46 (m, 1H) | 71.1 d | 4 | 4.30 (m, 1H) | 73.9 d | |
| 5a | 1.48 (m, 1H) | 32.1 t | 5a | 2.02 (m, 1H) | 34.2 t | |
| 5b | 1.90 (m, 1H) | 5b | 2.19 (m, 1H) | |||
| 6 | 1.20–1.30 (m, 2H) | 26.1 t | 6 | 1.96 (m, 2H) | 27.5 t | |
| 7 | 1.97 (m, 2H) | 27.4 t | 7–9 | 1.26–1.32 (m, 6H) | 30.5–30.9 t | |
| 8 | 5.35 (dt, 1H, 10.0,5.0 Hz) | 130.3 d | 10a | 2.05 (m, 1H) | 34.2 t | |
| 9 | 5.28 (dt, 1H, 10.0,5.0 Hz) | 129.5 d | 10b | 2.29 (m, 1H) | ||
| 10 | 1.97 (m, 2H) | 27.2 t | 11 | 5.52 (dt, 1H, 15.0, 6.0 Hz) | 131.7 | |
| 11–14 | 1.20–1.30 (m, 8H) | 29.0–29.6t | 12 | 5.52 (dt, 1H, 15.0, 6.0 Hz) | 131.6 | |
| 15 | 1.20–1.30 (m, 2H) | 31.8 t | 13a | 2.03 (m, 1H) | 33.8 t | |
| 16 | 1.20–1.30 (m, 2H) | 22.6 t | 13b | 2.17 (m, 1H) | ||
| 17 | 0.83 (t, 3H, 6.5 Hz) | 14.2 q | 14–20 | 1.26–1.32 (m, 14H) | 30.5–30.9 t | |
| NH | 7.55 (d, 1H, 8.6 Hz) | 21 | 1.26–1.32 (m, 2H) | 33.0 t | ||
| 1′ | 174.3 s | 22 | 1.26–1.32 (m, 2H) | 23.8 t | ||
| 2′ | 3.81 (dd, 1H, 7.5, 4.0 Hz) | 71.5 d | 23 | 0.87 (t, 3H, 6.5 Hz) | 15.1 q | |
| 3′a | 1.48 (m, 1H) | 34.8 t | NH | 8.58 (d, 1H, 9.0 Hz) | ||
| 3′b | 2.00 (m, 1H) | 1′ | 174.9 s | |||
| 4′ | 1.20–1.30 (m, 2H) | 24.9 t | 2′ | 4.56 (dd, 1H, 7.5, 4.0 Hz) | 73.7 d | |
| 5′–9′ | 1.20–1.30 (m, 10H) | 29.0–29.6t | 3′a | 2.05 (m, 1H) | 34.8 t | |
| 10′ | 1.20–1.30 (m, 2H) | 31.8 t | 3′b | 2.21 (m, 1H) | ||
| 11′ | 1.20–1.30 (m, 2H) | 22.6 t | 4′a | 1.65 (m, 1H) | ||
| 12′ | 0.83 (t, 3H, 6.5 Hz) | 14.2 q | 4′b | 1.96 (m, 1H) | 27.4 t | |
| 1′′ | 4.14 (d, 1H, 7.6 Hz), | 103.9 | 5′–21′ | 1.26–1.32 (m, 32H) | 30.5–30.9 t | |
| 2′′ | 2.95 (t, 1H, 8.0 Hz) | 73.9 | 22′ | 1.26–1.32 (m, 2H) | 33.0 t | |
| 3′′ | 3.13 (m, 1H) | 77.3 | 23′ | 1.26–1.32 (m, 2H) | 23.8 t | |
| 4′′ | 3.02 (m, 1H) | 70.4 | 24′ | 0.87 (t, 3H, 6.5 Hz) | 15.1 q | |
| 5′′ | 3.10 (m, 1H) | 76.9 | ||||
| 6′′a | 3.56 (dd, 1H, 12.0,5.0Hz) | 61.5 | ||||
| 6′′b | 3.70 (m, 1H) | |||||
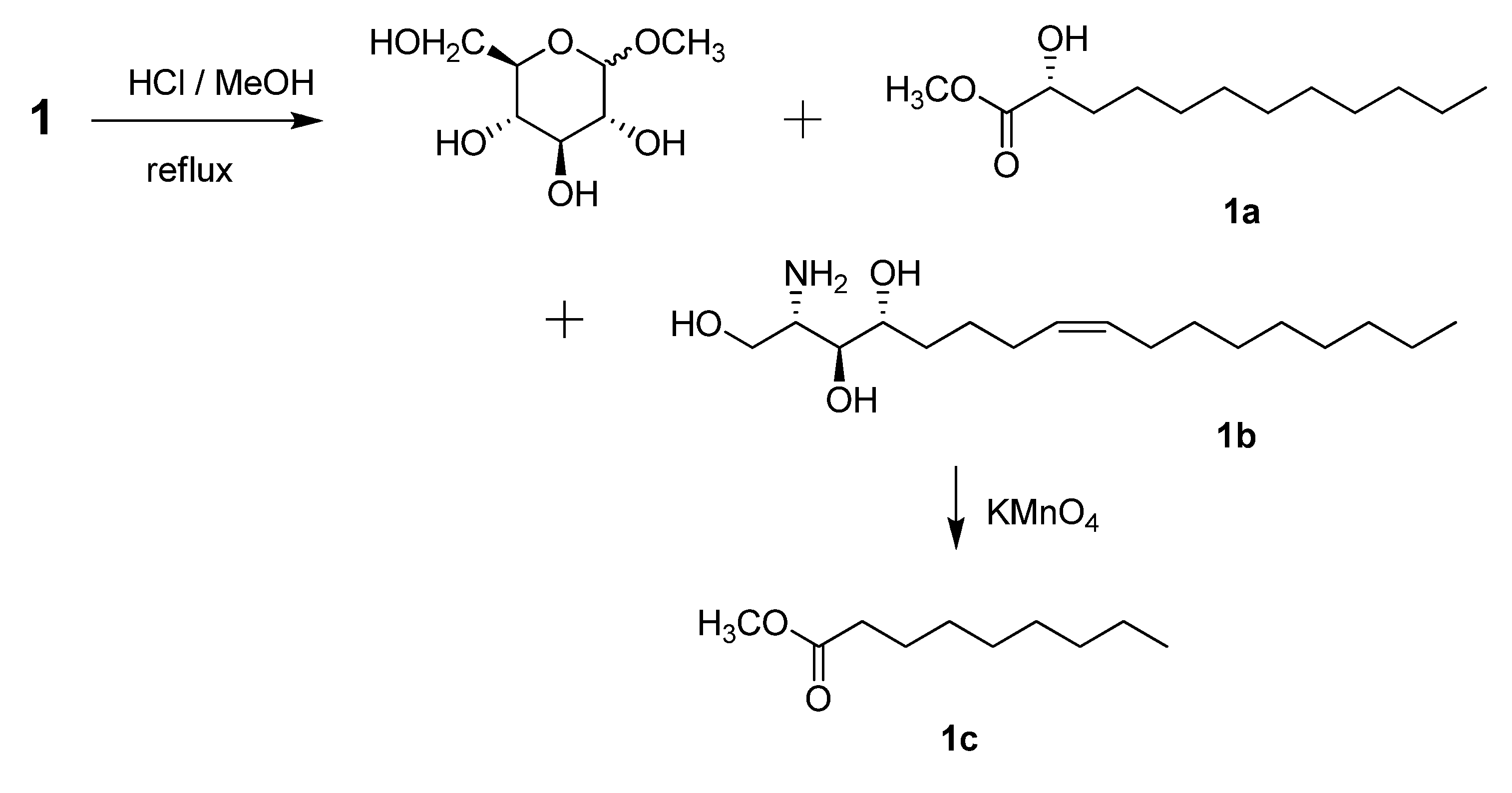
 −1.2 (c 0.07, CHCl3) by means of GC/MS analysis, and the absolute configuration of C-2′ was determined to be R from the specific rotation [11]. The phytosphingosine part is thus a C17 aliphatic amino alcohol unit with three hydroxyls, an amino group, and an olefinic bond. The 2S, 3S, and 4R configurations of the ceramide moieties were assigned by comparing the specific rotation +9.6 (c 0.11, pyridine)], 1H-NMR, and 13C-NMR data of compound 1 with those of the known synthetic ceramide (2S,3S,4R)-2-[(2′R)-2′-hydroxytetracosanoylamino]-1,3,4-hexadecanetriol [12] and the natural ceramide 1-O-β-d-glucopyranosyl-(2S,3S,4R,8E)-2-[(2′R)-2′-hydroxybehenoylamino]-8-octadecene-1,3,4-triol [13]. To determine the position of the olefinic bond in the dihydrosphingosine moiety, KMnO4 oxidation [14] was performed on compound 1b to yield nonanoic acid (1a) (Scheme 1), which was methylated and detected by GC/MS. This indicated the location of the olefinic bond between C-8 and C-9. The ∆ [8,9] olefinic bond was confirmed to have a (Z)-configuration as evidenced by the vicinal coupling constants (J = 10.0 Hz), together with the chemical shifts of C-7 (δ 27.4) and C-10 (δ 27.2). Analysis of the 1H-1H COSY, HMQC, and HMBC spectra led to the assignment of proton and carbon signals for 1. Methanolysis of 1 yielded a fatty acid methyl ester 1a and a long-chain base 1b (Scheme 1). Compound 1a was identified as 2-hydroxydodecanoic acid methyl ester −1.2 (c 0.07, CHCl3) by means of GC/MS analysis, and the absolute configuration of C-2′ was determined to be R from the specific rotation [11]. The phytosphingosine part is thus a C17 aliphatic amino alcohol unit with three hydroxyls, an amino group, and an olefinic bond. The 2S, 3S, and 4R configurations of the ceramide moieties were assigned by comparing the specific rotation +9.6 (c 0.11, pyridine)], 1H-NMR, and 13C-NMR data of compound 1 with those of the known synthetic ceramide (2S,3S,4R)-2-[(2′R)-2′-hydroxytetracosanoylamino]-1,3,4-hexadecanetriol [12] and the natural ceramide 1-O-β-d-glucopyranosyl-(2S,3S,4R,8E)-2-[(2′R)-2′-hydroxybehenoylamino]-8-octadecene-1,3,4-triol [13]. To determine the position of the olefinic bond in the dihydrosphingosine moiety, KMnO4 oxidation [14] was performed on compound 1b to yield nonanoic acid (1a) (Scheme 1), which was methylated and detected by GC/MS. This indicated the location of the olefinic bond between C-8 and C-9. The ∆ [8,9] olefinic bond was confirmed to have a (Z)-configuration as evidenced by the vicinal coupling constants (J = 10.0 Hz), together with the chemical shifts of C-7 (δ 27.4) and C-10 (δ 27.2) .
−1.2 (c 0.07, CHCl3) by means of GC/MS analysis, and the absolute configuration of C-2′ was determined to be R from the specific rotation [11]. The phytosphingosine part is thus a C17 aliphatic amino alcohol unit with three hydroxyls, an amino group, and an olefinic bond. The 2S, 3S, and 4R configurations of the ceramide moieties were assigned by comparing the specific rotation +9.6 (c 0.11, pyridine)], 1H-NMR, and 13C-NMR data of compound 1 with those of the known synthetic ceramide (2S,3S,4R)-2-[(2′R)-2′-hydroxytetracosanoylamino]-1,3,4-hexadecanetriol [12] and the natural ceramide 1-O-β-d-glucopyranosyl-(2S,3S,4R,8E)-2-[(2′R)-2′-hydroxybehenoylamino]-8-octadecene-1,3,4-triol [13]. To determine the position of the olefinic bond in the dihydrosphingosine moiety, KMnO4 oxidation [14] was performed on compound 1b to yield nonanoic acid (1a) (Scheme 1), which was methylated and detected by GC/MS. This indicated the location of the olefinic bond between C-8 and C-9. The ∆ [8,9] olefinic bond was confirmed to have a (Z)-configuration as evidenced by the vicinal coupling constants (J = 10.0 Hz), together with the chemical shifts of C-7 (δ 27.4) and C-10 (δ 27.2). Analysis of the 1H-1H COSY, HMQC, and HMBC spectra led to the assignment of proton and carbon signals for 1. Methanolysis of 1 yielded a fatty acid methyl ester 1a and a long-chain base 1b (Scheme 1). Compound 1a was identified as 2-hydroxydodecanoic acid methyl ester −1.2 (c 0.07, CHCl3) by means of GC/MS analysis, and the absolute configuration of C-2′ was determined to be R from the specific rotation [11]. The phytosphingosine part is thus a C17 aliphatic amino alcohol unit with three hydroxyls, an amino group, and an olefinic bond. The 2S, 3S, and 4R configurations of the ceramide moieties were assigned by comparing the specific rotation +9.6 (c 0.11, pyridine)], 1H-NMR, and 13C-NMR data of compound 1 with those of the known synthetic ceramide (2S,3S,4R)-2-[(2′R)-2′-hydroxytetracosanoylamino]-1,3,4-hexadecanetriol [12] and the natural ceramide 1-O-β-d-glucopyranosyl-(2S,3S,4R,8E)-2-[(2′R)-2′-hydroxybehenoylamino]-8-octadecene-1,3,4-triol [13]. To determine the position of the olefinic bond in the dihydrosphingosine moiety, KMnO4 oxidation [14] was performed on compound 1b to yield nonanoic acid (1a) (Scheme 1), which was methylated and detected by GC/MS. This indicated the location of the olefinic bond between C-8 and C-9. The ∆ [8,9] olefinic bond was confirmed to have a (Z)-configuration as evidenced by the vicinal coupling constants (J = 10.0 Hz), together with the chemical shifts of C-7 (δ 27.4) and C-10 (δ 27.2) .
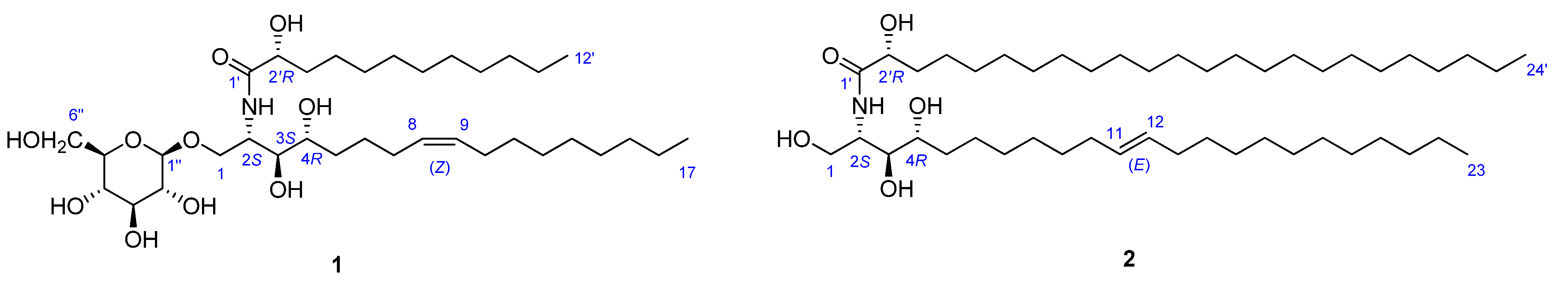
2.2. Structure Analysis of Pipercerebroside B
−1.4 (c 0.09, CHCl3)]} and the absolute configuration of C-2′ was determined to be R as in compound 1. Thus, the LCB part is a C23 aliphatic amino alcohol unit containing three hydroxyls, an amino group and a double bond. The dihydrosphingosine (LCB) moiety was oxidized to yield undecanoic acid, which was methylated and detected by GC/MS. This indicated that the double bond was located at C-11. The 11,12 alkene bond was shown to be trans by the large vicinal coupling constants (J = 15.0). The trans geometry of this double bond was also supported by the chemical shift of C-10 (δ 34.2) and C-13 (δ 33.8).2.3. Cytotoxic Activity of Compounds
3. Experimental
3.1. General
3.2. Plant Resource
3.3. Extraction and Isolation of New Compounds
3.4. Methanolysis, Oxidation and Methylation of Pipercerebroside A
-1.2 (c 0.07, CHCl3); GC-MS: GC, tR 39.41 min, EI-MS m/z: 171 [M − CH3OCO]+ (39), 127 [C9H19]+ (6), 111 (17), 97 (74), 90 [CH3OC(OH)=CH OH]+ (45), 83 (72), 69 (89), 55 (91), 43 (100).3.5. Methanolysis, Oxidation and Methylation of Pipercerebroside B
-1.4 (c 0.09, CHCl3); GC-MS: GC, tR 54.38 min, EI-MS m/z: 398 [M]+ (11), 339 [M − CH3OCO]+ (9), 281 (5), 207 (10), 145 (4), 127 (6), 111 (12), 97 (27), 90 [CH3OC(OH)=CHOH]+ (28), 83 (27), 57 (58), 44 (100). Methyl undecanoate: GC-MS: GC, tR 24.36 min, EI-MS m/z: 200 [M]+ (4), 169 [M − OCH3]+ (5), 157 (4), 143 (7), 101 (6), 87 (47), 74 (100), 69 (8), 55 (16), 43 (22).3.6. Spectral data of New Compounds
+ 9.6 (c 0.11, pyridine); IR (KBr) νmax: 3385, 3178, 2922, 2853, 1653, 1543, 1463, 1402, 1083, 932 cm−1; ESI(+)-MS m/z: 662 [M + H]+, 684 [M + Na]+, 500 [M + H − glu]+; HRESI(+)-MS m/z: 662.4797 [M + H]+ (calcd. for C35H68O10N, 662.4799). + 3.7 (c 0.19, pyridine); IR (CH3OH) νmax: 3480, 2940, 2860, 1640, 1506, and 1297 cm−1; ESI(+)-MS m/z: 738 [M + H]+; HRESI(+)-MS m/z: 738.6966 [M + H]+ (calcd. for C46H92NO5, 738.6975).3.7. Bioassay
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Wu, Z.Y. Piper betle. In Flora of China; Science Press: Beijing, China, 1982; Volume 20, pp. 42–45. [Google Scholar]
- Nagori, K.; Singh, M.K.; Alexander, A.; Kumar, T.; Dewangan, D.; Badwaik, H.; Tripathi, D.K. Piper betle L.:A review on its ethnobotany, phytochemistry, pharmacological profile and profiling by new hyphenated technique DART-MS (Direct Analysis in Real Time Mass Spectrometry). J. Pharm. Res. 2011, 4, 2991–2997. [Google Scholar]
- Rai, M.P.; Thilakchand, K.R.; Palatty, P.L.; Rao, P.; Rao, S.; Bhat, H.P.; Baliga, M.S. Piper betel Linn (betel vine), the maligned Southeast Asian medicinal plant possesses cancer preventive effects: Time to reconsider the wronged opinion. Asian Pac. J. Cancer Prev. 2011, 12, 2149–2156. [Google Scholar]
- Kumar, N.; Misra, P.; Dube, A.; Bhattacharya, S.; Dikshit, M.; Ranade, S. Piper betle Linn. A maligned Pan—Asiatic plant with an array of pharmacological activities and prospects for drug discovery. Curr. Sci. 2010, 99, 922–931. [Google Scholar]
- Huang, X.Z.; Yin, Y.; Huang, W.Q.; Sun, K.Z.; Cheng, C.M.; Bai, L.; Dai, Y. Studies on alkaloids and lignins from stems of Piper betle L. China J. Chin. Mat. Med. 2010, 35, 15–18. [Google Scholar]
- Li, H.Y.; Matsunaga, S.; Fusetani, N. Halicylindrosides, antifungal and cytotoxic cerebrosides from the marine sponge Halichondria cylindrata. Tetrahedron 1995, 51, 2273–2280. [Google Scholar] [CrossRef]
- Natori, T.; Morita, M.; Akimoto, K. Agelasphins, novel antitumor and immunostimulatory cerebrosides from the marine sponge Agelas Mauritianus. Tetrahedron 1994, 50, 2771–2784. [Google Scholar] [CrossRef]
- Dong, J.Y.; Li, R.; He, H.P.; Zhang, K.Q. Nematicidal sphingolipids from the freshwater fungus Paraniesslia sp. YMF1.01400. Eur. J. Lipid Sci. Technol. 2005, 107, 779–785. [Google Scholar] [CrossRef]
- Jia, A.Q.; Yang, X.; Wang, W.X.; Jia, Y.H. Glycocerebroside bearing a novel long-chain base from Sagina japonica (Caryophyllaceae). Fitoterapia 2010, 81, 540–545. [Google Scholar] [CrossRef]
- Cateni, F.; Zilic, J.; Falsone, G.; Scialino, G.; Banfi, E. New cerebrosides from Euphorbia peplis L.: Antimicrobial activity evaluation. Bioorg. Med. Chem. Lett. 2003, 13, 4345–4350. [Google Scholar] [CrossRef]
- Kang, S.S.; Kim, J.S.; Xu, Y.N.; Kim, Y.H. Isolation of a new cerebroside from the root bark of Aralia elata. J. Nat. Prod. 1999, 62, 1059–1060. [Google Scholar] [CrossRef]
- Sugiyama, S.; Honda, M.; Higuchi, R.; Komori, T. Biologically active glycosides from asteroidea. XXVI. Stereochemistry of the four diastereomers of ceramide and ceramide lactoside. Liebigs Ann. Chem. 1991, 4, 349–356. [Google Scholar]
- Ling, T.; Xia, T.; Wan, X.; Li, D.; Wei, X. Cerebrosides from the roots of Serratula chinensis. Molecules 2006, 11, 677–683. [Google Scholar] [CrossRef]
- Lourenco, A.; Lobo, A.M.; Rodriguez, B.; Jimeno, M.-L. Ceramides from the fungus Phellinus pini. Phytochemistry 1996, 43, 617–620. [Google Scholar]
- Liu, H.; Orjala, J.; Rali, T.; Sticher, O. Glycosides from Stenochlaena palustris. Phytochemistry 1998, 49, 2403–2408. [Google Scholar]
- Huang, X.Z.; Yin, Y.; Dai, J.H.; Liang, H.; Dai, Y.; Bai, L. Two new ceramides from the stems of Piper betle L. Chin. Chem. Lett. 2010, 21, 433–436. [Google Scholar]
- Yang, N.-Y.; Ren, D.-C.; Duan, J.-A.; Xu, X.-H.; Xie, N.; Tian, L.-J. Ceramides and cerebrosides from Ligusticum chuanxiong HORT. Helv. Chim. Acta 2009, 92, 291–297. [Google Scholar] [CrossRef]
- Oueslati, M.H.; Mighri, Z.; Jannet, H.B.; Abreu, P.M. New ceramides from Rantherium suaveolens. Lipids 2005, 40, 1075–1079. [Google Scholar] [CrossRef]
- Kang, S.S.; Kim, J.S.; Son, K.H.; Kim, H.P.; Chang, H.W. Cyclooxygenase-2 inhibitory cerebrosides from phytolaccae radix. Chem. Pharm. Bull. 2001, 49, 321–323. [Google Scholar] [CrossRef]
- Huang, X.Z.; Wang, Y.H.; Yu, S.S.; Fu, G.M.; Hu, Y.C.; Liu, Y.; Fan, L.H. Iridoid glycosides and grayanane diterpenoids from the roots of Craibiodendron henryi. J. Nat. Prod. 2005, 68, 1646–1650. [Google Scholar] [CrossRef]
- Fu, G.M.; Wang, Y.H.; Gao, S.; Tang, M.J.; Yu, S.S. Five new cytotoxic triterpenoid saponins from the roots of Symplocos chinensis. Planta Med. 2005, 71, 666–672. [Google Scholar]
- Sample Availability: Samples of the compounds 1 and 2 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, D.-Z.; Xiong, H.-B.; Tian, K.; Guo, J.-M.; Huang, X.-Z.; Jiang, Z.-Y. Two New Sphingolipids from the Leaves of Piper betle L. Molecules 2013, 18, 11241-11249. https://doi.org/10.3390/molecules180911241
Chen D-Z, Xiong H-B, Tian K, Guo J-M, Huang X-Z, Jiang Z-Y. Two New Sphingolipids from the Leaves of Piper betle L. Molecules. 2013; 18(9):11241-11249. https://doi.org/10.3390/molecules180911241
Chicago/Turabian StyleChen, Duo-Zhi, Hua-Bin Xiong, Kai Tian, Jun-Ming Guo, Xiang-Zhong Huang, and Zhi-Yong Jiang. 2013. "Two New Sphingolipids from the Leaves of Piper betle L." Molecules 18, no. 9: 11241-11249. https://doi.org/10.3390/molecules180911241