Coupling Bioorthogonal Chemistries with Artificial Metabolism: Intracellular Biosynthesis of Azidohomoalanine and Its Incorporation into Recombinant Proteins

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
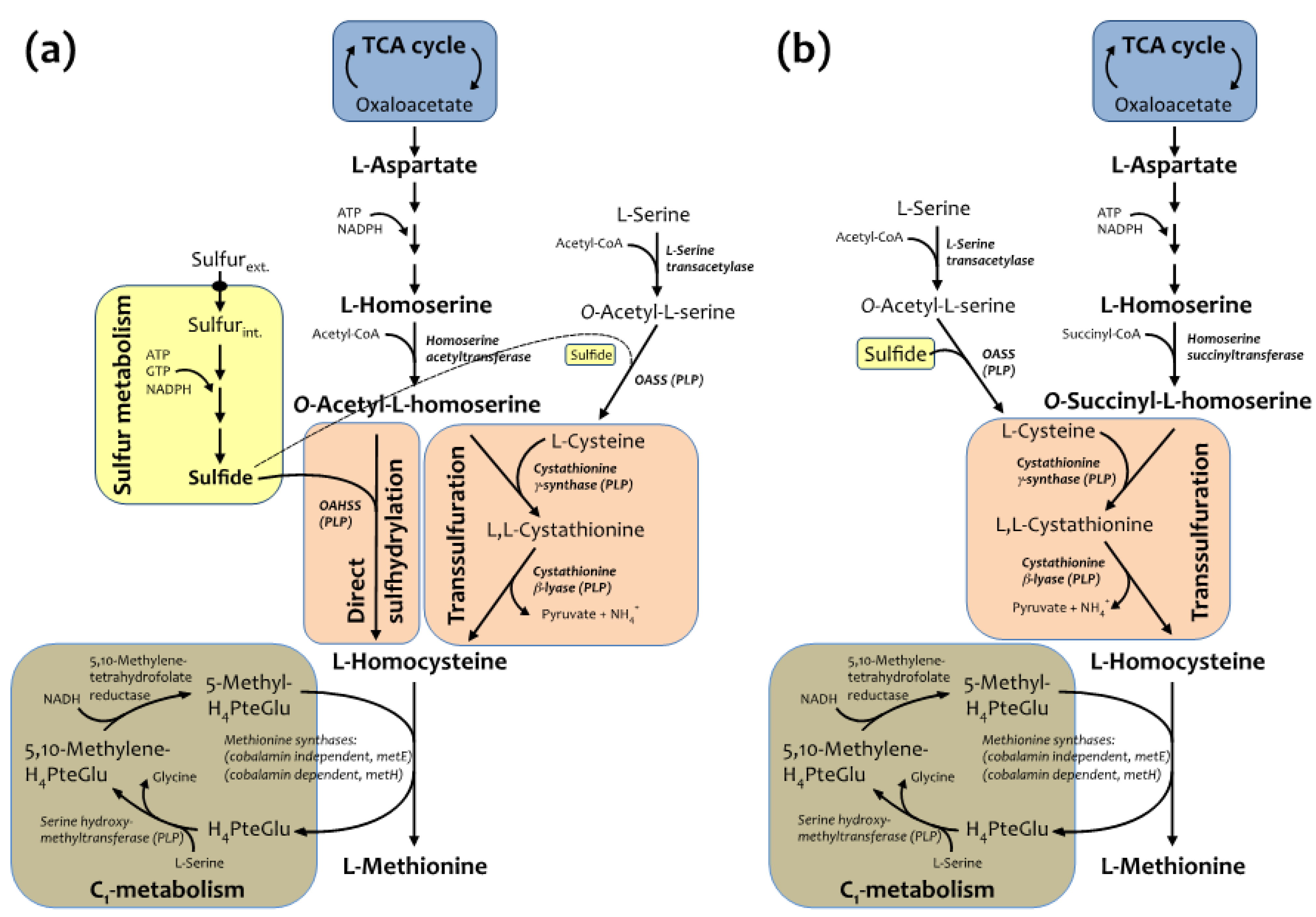
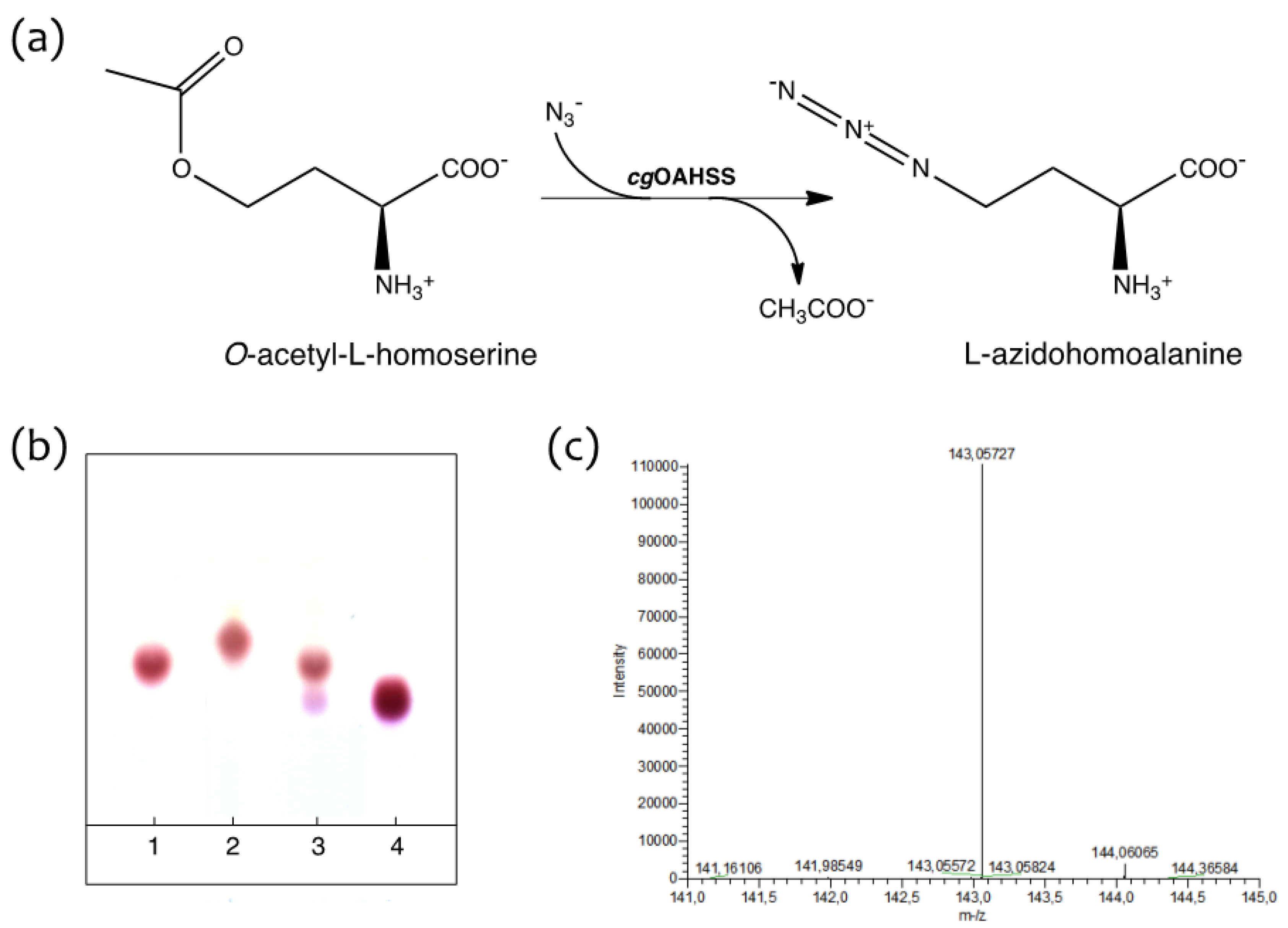
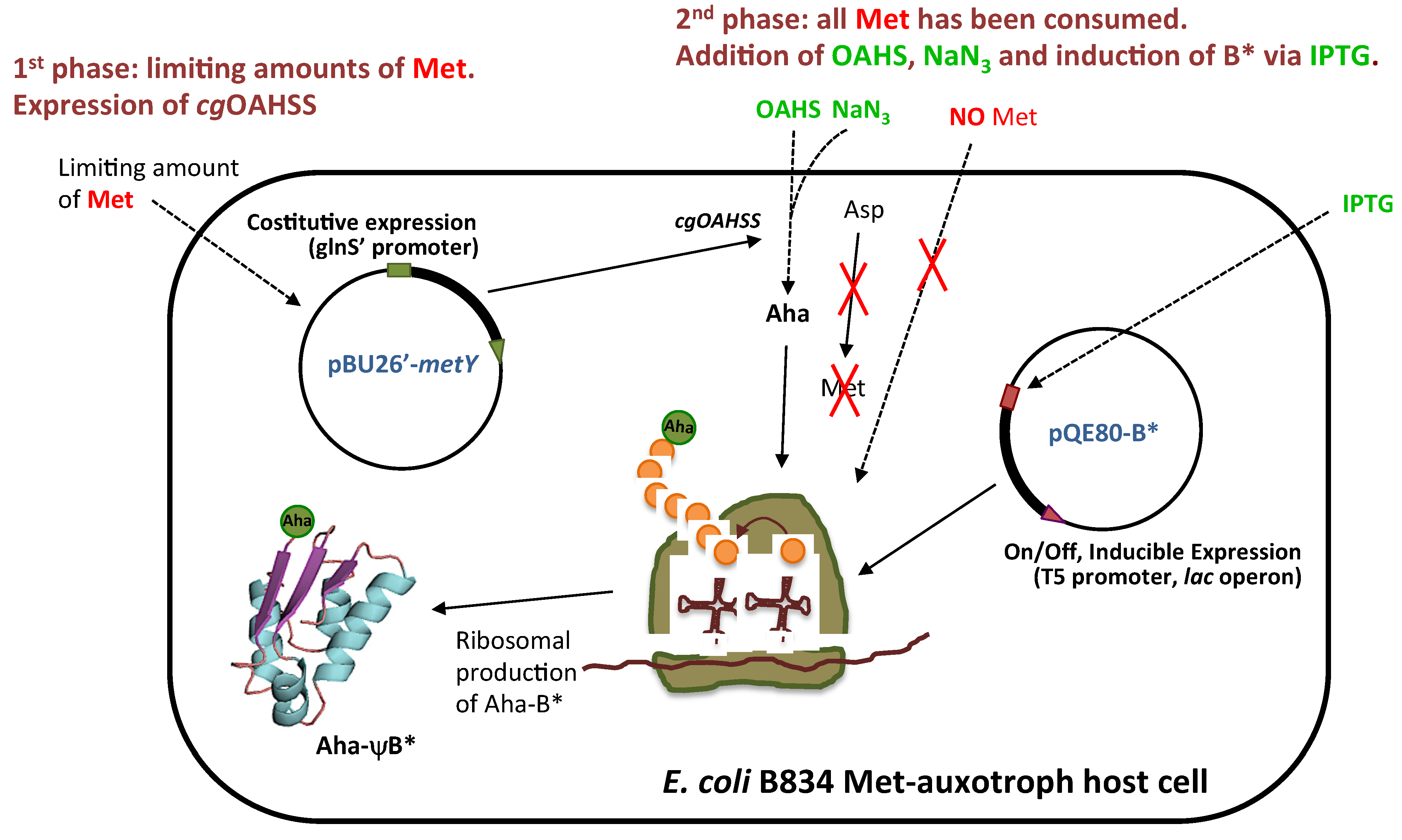
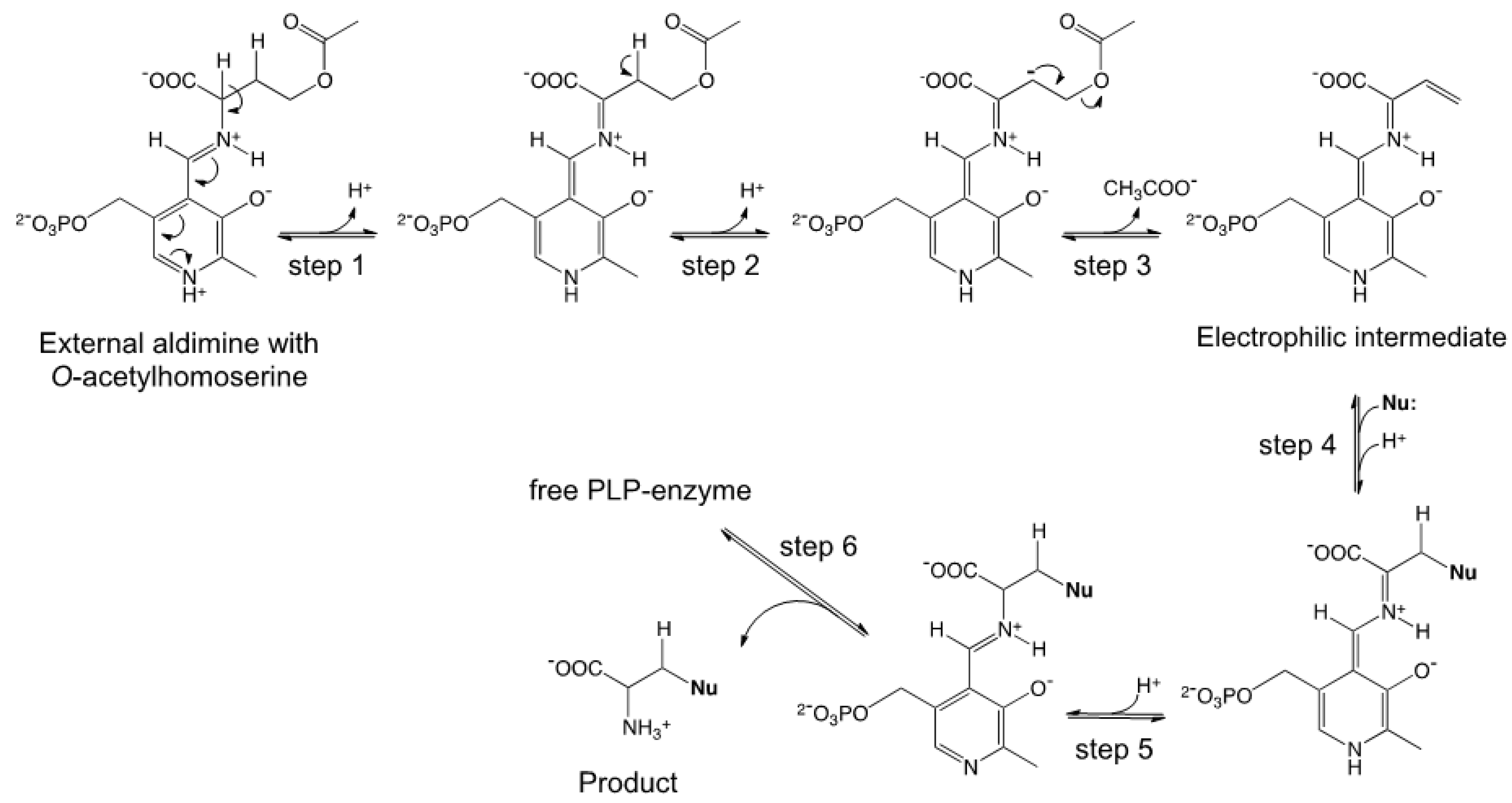
2.1. Semisynthetic Production of l-Azidohomoalanine



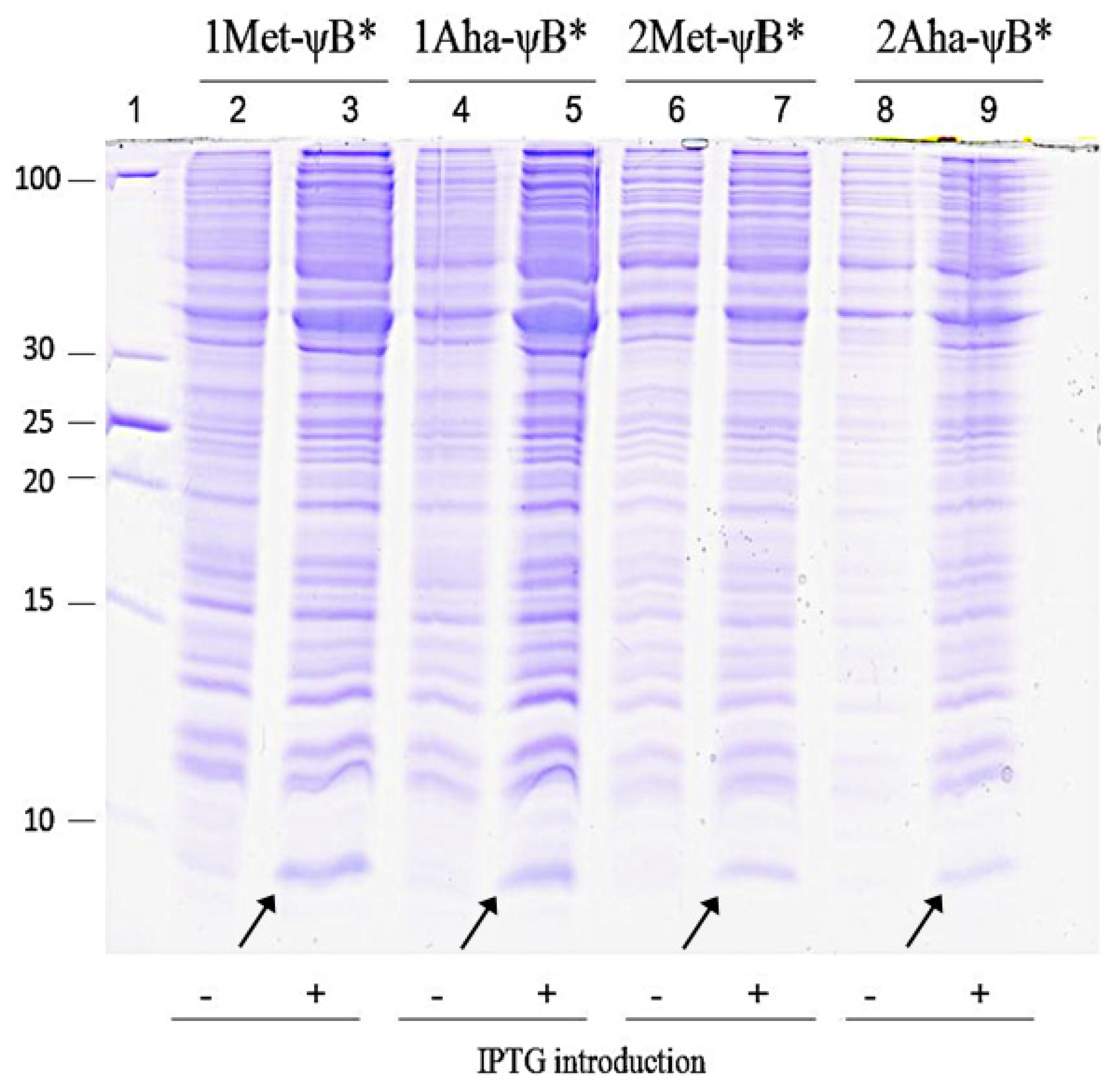
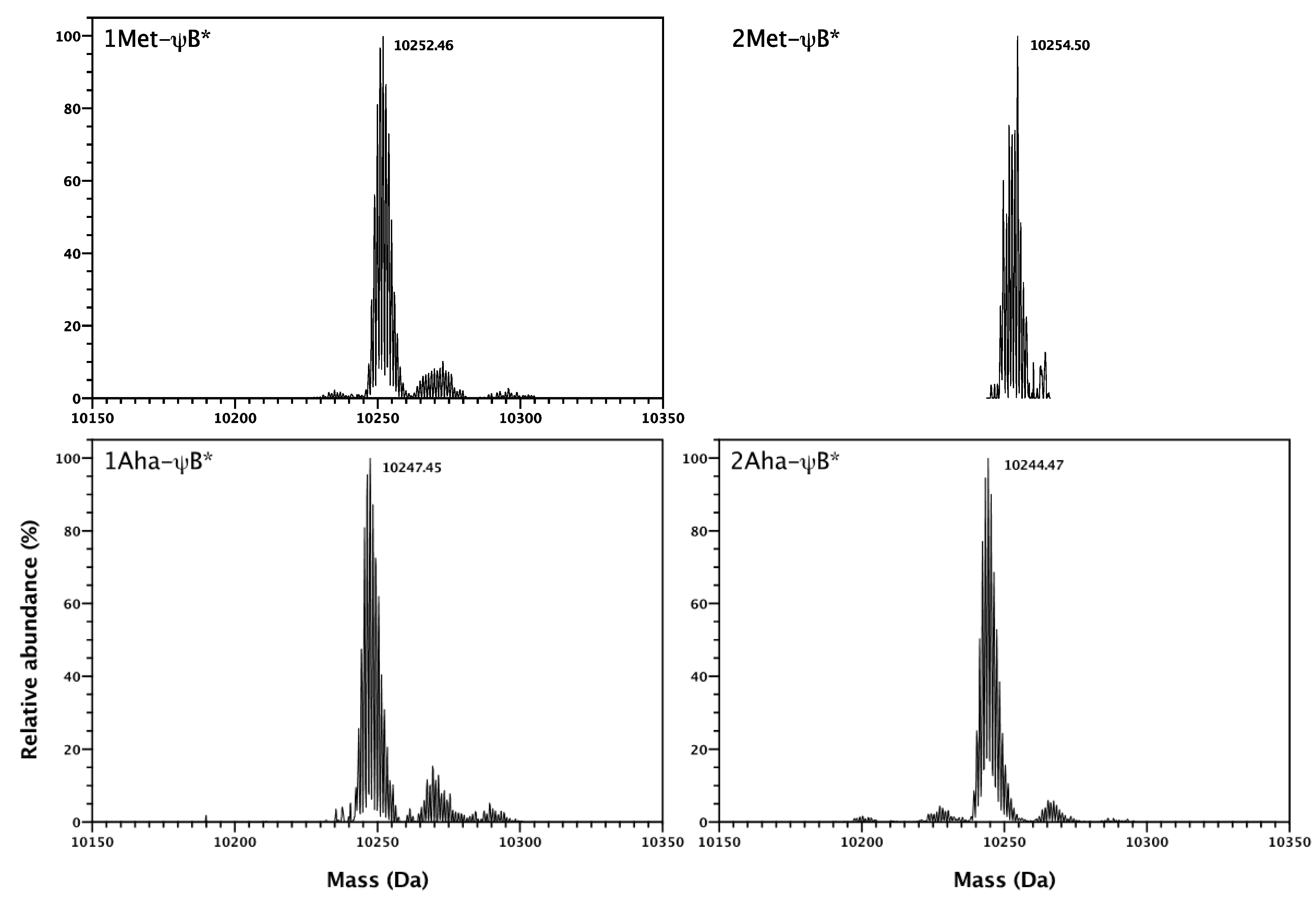
2.2. In Vivo Incorporation of l-Azidohomoalanine into Barstar




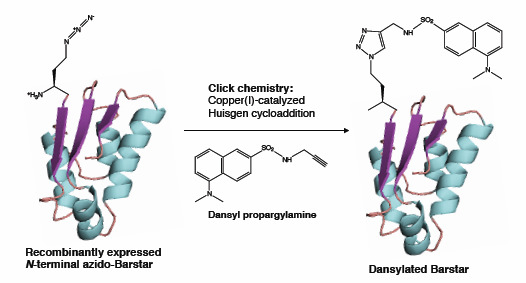
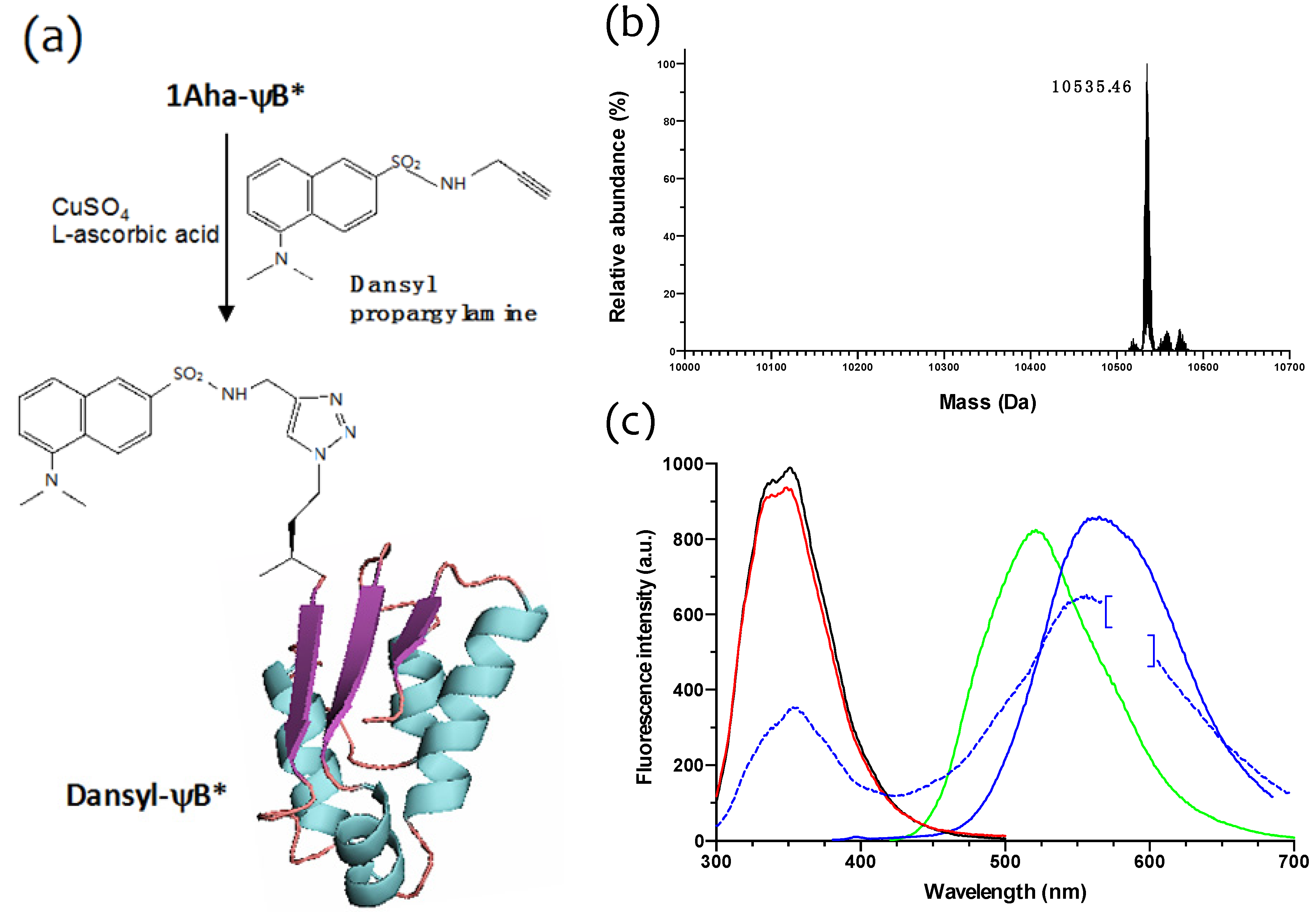
2.3. Click Chemistry Derivatization of 1Aha-ψB*
3. Experimental
3.1. Chemical Synthesis and Analysis
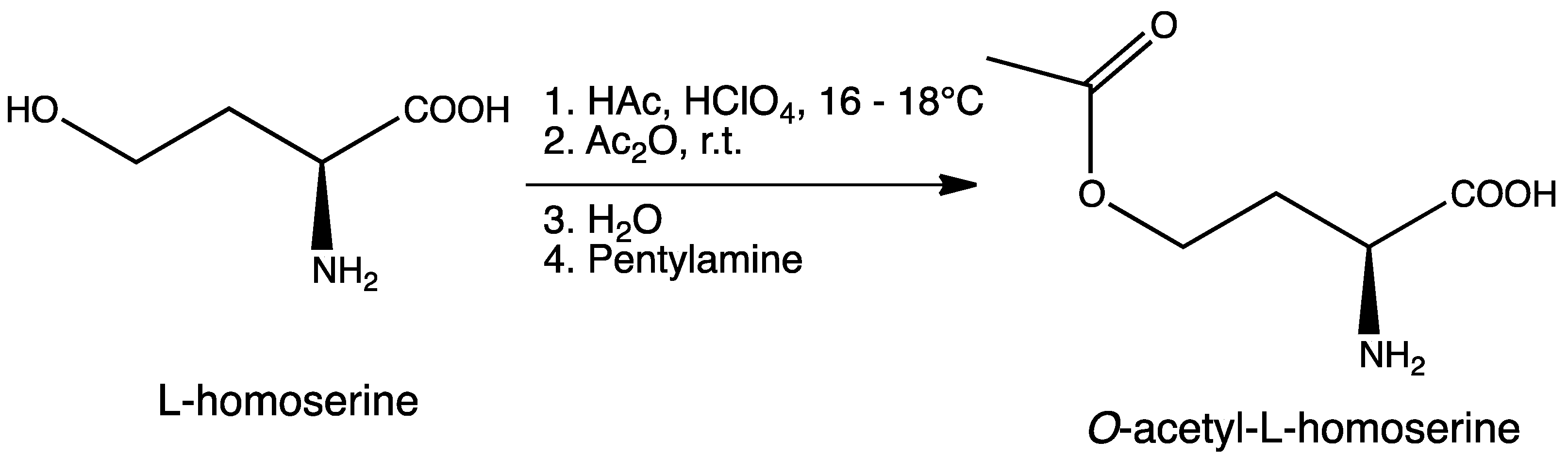
3.2. Synthesis of O-Acetyl-l-homoserine

3.3. Synthesis of l-Azidohomoalanine
3.4. Molecular Biology and Plasmid Construction
3.5. Purification of C. glutamicum O-Acetylhomoserine Sulfhydrylase
3.6. Expression and Purification of the Met- and Aha- Variants of Barstar
3.7. Analysis of Barstar Variants
3.8. Copper(I)-catalyzed Huisgen [3+2] Cycloaddition
4. Conclusions
Abbreviations
| ncAA | non-canonical amino acids |
| Aha | l-azidohomoalanine |
| Met | l-methionine |
| Oahs | O-acetyl-l-homoserine |
| PLP | pyridoxal 5'-phosphate |
| OAHSS | O-acetyl-l-homoserine sulfhydrylase |
| cg | Corynebacterium glutamicum |
| ψB* | ψ–bastar, “pseudo wild-type” form of barstar from Bacillus amyloliquefaciens |
| PMTs | posttranslational modifications |
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Graves, D.J.; Martin, B.L.; Wang, J.H.C. Co- and Post-Translational Modification of Proteins: Chemical Principles and Biological Effects; Oxford University Press: New York, NY, USA, 1994. [Google Scholar]
- Prabakaran, S.; Lippens, G.; Steen, H.; Gunawardena, J. Post-translational modification: nature’s escape from genetic imprisonment and the basis for dynamic information encoding. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012, 4, 565–583. [Google Scholar] [CrossRef]
- Budisa, N. Engineering the Genetic Code: Expanding the Amino Acid Repertoire for the Design of Novel Proteins; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Budisa, N. Expanded genetic code for the engineering of ribosomally synthetized and post-translationally modified peptide natural products (ripps). Curr. Opin. Biotechnol. 2013, 24, 591–598. [Google Scholar] [CrossRef]
- Budisa, N. Prolegomena to future experimental efforts on genetic code engineering by expanding its amino acid repertoire. Angew. Chem. Int. Ed. Engl. 2004, 43, 6426–6463. [Google Scholar] [CrossRef]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. Engl. 2009, 48, 6974–6998. [Google Scholar] [CrossRef]
- Moroder, L.; Budisa, N. Synthetic biology of protein folding. ChemPhysChem 2010, 11, 1181–1187. [Google Scholar]
- Lepthien, S.; Merkel, L.; Budisa, N. In vivo double and triple labeling of proteins using synthetic amino acids. Angew. Chem. Int. Ed. Engl. 2010, 49, 5446–5450. [Google Scholar] [CrossRef]
- Liu, W.R.; Wang, Y.S.; Wan, W. Synthesis of proteins with defined posttranslational modifications using the genetic noncanonical amino acid incorporation approach. Mol. Biosyst. 2011, 7, 38–47. [Google Scholar] [CrossRef]
- Huisgen, R. 1,3-Dipolar cycloadditions. Past and future. Angew. Chem. Int. Ed. Engl. 1963, 2, 565–598. [Google Scholar] [CrossRef]
- Tornoe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Dong, S.; Moroder, L.; Budisa, N. Protein iodination by click chemistry. ChemBioChem 2009, 10, 1149–1151. [Google Scholar] [CrossRef]
- Hwang, B.J.; Park, S.D.; Kim, Y.; Kim, P.; Lee, H.S. Biochemical analysis on the parallel pathways of methionine biosynthesis in corynebacterium glutamicum. J. Microbiol. Biotechnol. 2007, 17, 1010–1017. [Google Scholar]
- Bolten, C.J.; Schroder, H.; Dickschat, J.; Wittmann, C. Towards methionine overproduction in corynebacterium glutamicum-methanethiol and dimethyldisulfide as reduced sulfur sources. J. Microbiol. Biotechnol. 2010, 20, 1196–1203. [Google Scholar] [CrossRef]
- Brzywczy, J.; Paszewski, A. Cloning and characterization of the kluyveromyces lactis homocysteine synthase gene. Yeast 1999, 15, 1403–1409. [Google Scholar]
- Yamagata, S. Roles of O-acetyl-l-homoserine sulfhydrylases in micro-organisms. Biochimie 1989, 71, 1125–1143. [Google Scholar] [CrossRef]
- Chocat, P.; Esaki, N.; Tanaka, H.; Kenji Soda, K. Synthesis of selenocystine and selenohomocystine with O-acetylhomoserine sulfhydrylase. Agric. Biol. Chem. 1985, 49, 1143–1150. [Google Scholar] [CrossRef]
- Maier, T.H. Semisynthetic production of unnatural l-alpha-amino acids by metabolic engineering of the cysteine-biosynthetic Pathway. Nat. Biotechnol. 2003, 21, 422–427. [Google Scholar] [CrossRef]
- Krishnamoorthy, K.; Begley, T.P. Protein thiocarboxylate-dependent methionine biosynthesis in wolinella succinogenes. J. Am. Chem. Soc. 2011, 133, 379–386. [Google Scholar] [CrossRef]
- Bickler, S.W.; Ring, J.; de Maio, A. Sulfur amino acid metabolism limits the growth of children living in environments of poor sanitation. Med. Hypotheses 2011, 77, 380–382. [Google Scholar] [CrossRef]
- Amadasi, A.; Bertoldi, M.; Contestabile, R.; Bettati, S.; Cellini, B.; di Salvo, M.L.; Borri-Voltattorni, C.; Bossa, F.; Mozzarelli, A. Pyridoxal 5'-phosphate enzymes as targets for therapeutic agents. Curr. Med. Chem. 2007, 14, 1291–1324. [Google Scholar] [CrossRef]
- Lepthien, S.; Hoesl, M.G.; Merkel, L.; Budisa, N. Azatryptophans endow proteins with intrinsic blue fluorescence. Proc. Natl. Acad. Sci. USA 2008, 105, 16095–16100. [Google Scholar] [CrossRef]
- Tran, T.H.; Krishnamoorthy, K.; Begley, T.P.; Ealick, S.E. A novel mechanism of sulfur transfer catalyzed by O-acetylhomoserine sulfhydrylase in the methionine-biosynthetic pathway of wolinella succinogenes. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 831–838. [Google Scholar] [CrossRef]
- Di Salvo, M.L.; Scarsdale, J.N.; Kazanina, G.; Contestabile, R.; Schirch, V.; Wright, H.T. Structure-based mechanism for early PLP-mediated steps of rabbit cytosolic serine hydroxymethyltransferase reaction. Biomed. Res. Int. 2013, 2013, 458571. [Google Scholar]
- Di Salvo, M.L.; Florio, R.; Paiardini, A.; Vivoli, M.; D’Aguanno, S.; Contestabile, R. Alanine racemase from Tolypocladium inflatum: A key PLP-dependent enzyme in cyclosporin biosynthesis and a model of catalytic promiscuity. Arch. Biochem. Biophys. 2013, 529, 55–65. [Google Scholar] [CrossRef]
- Florio, R.; di Salvo, M.L.; Vivoli, M.; Contestabile, R. Serine hydroxymethyltransferase: A model enzyme for mechanistic, structural, and evolutionary studies. Biochim. Biophys. Acta 2011, 1814, 1489–1496. [Google Scholar]
- Ryu, Y.; Schultz, P.G. Efficient Incorporation of unnatural amino acids into proteins in Escherichia coli. Nat. Methods 2006, 3, 263–265. [Google Scholar] [CrossRef]
- Link, A.J.; Vink, M.K.; Tirrell, D.A. Preparation of the functionalizable methionine surrogate azidohomoalanine via copper-catalyzed diazo transfer. Nat. Protoc. 2007, 2, 1879–1883. [Google Scholar] [CrossRef]
- Zhang, M.M.; Tsou, L.K.; Charron, G.; Raghavan, A.S.; Hang, H.C. Tandem fluorescence imaging of dynamic S-acylation and protein turnover. Proc. Natl. Acad. Sci. USA 2010, 107, 8627–8632. [Google Scholar]
- Merkel, L.; Cheburkin, Y.; Wiltschi, B.; Budisa, N. In vivo chemoenzymatic control of n-terminal processing in recombinant human epidermal growth factor. ChemBioChem 2007, 8, 2227–2232. [Google Scholar] [CrossRef]
- Van Kasteren, S.I.; Kramer, H.B.; Jensen, H.H.; Campbell, S.J.; Kirkpatrick, J.; Oldham, N.J.; Anthony, D.C.; Davis, B.G. Expanding the diversity of chemical protein modification allows post-translational mimicry. Nature 2007, 446, 1105–1109. [Google Scholar] [CrossRef]
- Merkel, L.; Beckmann, H.S.; Wittmann, V.; Budisa, N. Efficient N-terminal glycoconjugation of proteins by the N-end rule. ChemBioChem 2008, 9, 1220–1224. [Google Scholar] [CrossRef]
- Nolting, B.; Golbik, R.; Fersht, A.R. Submillisecond events in protein folding. Proc. Natl. Acad. Sci. USA 1995, 92, 10668–10672. [Google Scholar] [CrossRef]
- Nagai, S.; Flavin, M. Acetylhomoserine. An intermediate in the fungal biosynthesis of methionine. J. Biol. Chem. 1967, 242, 3884–3895. [Google Scholar]
- Silva-Rocha, R.; Martinez-Garcia, E.; Calles, B.; Chavarria, M.; Arce-Rodriguez, A.; de Las Heras, A.; Paez-Espino, A.D.; Durante-Rodriguez, G.; Kim, J.; Nikel, P.I.; et al. The Standard European Vector Architecture (SEVA): A coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res. 2013, 41, D666–D675. [Google Scholar] [CrossRef]
- Vivoli, M.; Angelucci, F.; Ilari, A.; Morea, V.; Angelaccio, S.; di Salvo, M.L.; Contestabile, R. Role of a conserved active site cation-pi interaction in Escherichia coli serine hydroxymethyltransferase. Biochemistry 2009, 48, 12034–12046. [Google Scholar] [CrossRef]
- Bae, J.H.; Alefelder, S.; Kaiser, J.T.; Friedrich, R.; Moroder, L.; Huber, R.; Budisa, N. Incorporation of beta-selenolo[3,2-b]Pyrrolyl-Alanine into proteins for phase determination in protein X-Ray crystallography. J. Mol. Biol. 2001, 309, 925–936. [Google Scholar] [CrossRef]
- Christen, E.H.; Gubeli, R.J.; Kaufmann, B.; Merkel, L.; Schoenmakers, R.; Budisa, N.; Fussenegger, M.; Weber, W.; Wiltschi, B. Evaluation of bicinchoninic acid as a ligand for copper(I)-catalyzed azide-alkyne bioconjugations. Org. Biomol. Chem. 2012, 10, 6629–6632. [Google Scholar] [CrossRef]
- Mehl, R.A.; Anderson, J.C.; Santoro, S.W.; Wang, L.; Martin, A.B.; King, D.S.; Horn, D.M.; Schultz, P.G. Generation of a bacterium with a 21 amino acid genetic code. J. Am. Chem Soc. 2003, 125, 935–939. [Google Scholar]
- Giese, C.; Lepthien, S.; Metzner, L.; Brandsch, M.; Budisa, N.; Lilie, H. Intracellular uptake and inhibitory activity of aromatic fluorinated amino acids in human breast cancer cells. ChemMedChem 2008, 3, 1449–1456. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds and plasmid constructs are available from the authors through Material Transfer Agreement.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ma, Y.; Biava, H.; Contestabile, R.; Budisa, N.; Di Salvo, M.L. Coupling Bioorthogonal Chemistries with Artificial Metabolism: Intracellular Biosynthesis of Azidohomoalanine and Its Incorporation into Recombinant Proteins. Molecules 2014, 19, 1004-1022. https://doi.org/10.3390/molecules19011004
Ma Y, Biava H, Contestabile R, Budisa N, Di Salvo ML. Coupling Bioorthogonal Chemistries with Artificial Metabolism: Intracellular Biosynthesis of Azidohomoalanine and Its Incorporation into Recombinant Proteins. Molecules. 2014; 19(1):1004-1022. https://doi.org/10.3390/molecules19011004
Chicago/Turabian StyleMa, Ying, Hernán Biava, Roberto Contestabile, Nediljko Budisa, and Martino Luigi Di Salvo. 2014. "Coupling Bioorthogonal Chemistries with Artificial Metabolism: Intracellular Biosynthesis of Azidohomoalanine and Its Incorporation into Recombinant Proteins" Molecules 19, no. 1: 1004-1022. https://doi.org/10.3390/molecules19011004