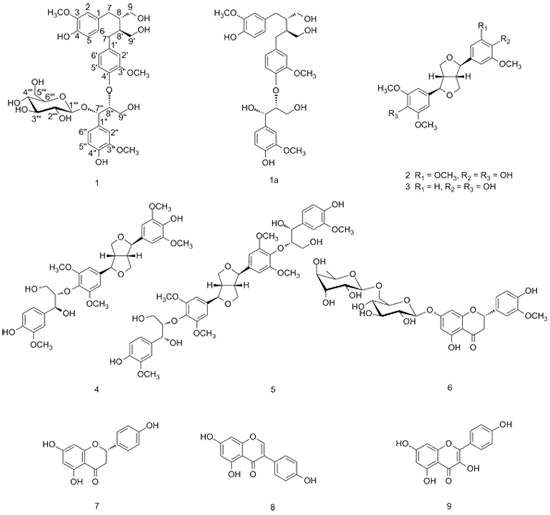
2. Results and Discussion
The EtOH extract of the aerial parts of
U. sinensis was suspended in water and successively partitioned with petroleum ether, CH
2Cl
2, and
n-butanol. The
n-butanol-soluble extract was subjected to column chromatography to afford a new sesquilignan glucoside, urarisinoside A (
1), along with a known compound, 5,4'-dihydroxy-3'-methoxyflavanone-7-(6''
O-
β-
l-rhamnopyranosyl)-
β-D-glucopyranoside (
6) [
9]. The CH
2Cl
2-soluble extract was subjected to repeated column chromatography to yield seven known compounds: (–)-syringaresinol (
2) [
10], (–)-medioresinol (
3) [
11], (–)-(7
R,7'
R,7''
S,8
S,8'
S,8''
S)-4',4''-dihydroxy-3,3',3'',5,5'-pentamethoxy-7,9':7',9-diepoxy-4,8''-oxy-8,8'-sesquineolignan-7'',9''-diol (
4) [
12], (+)-(7
R,7'
R,7''
R,7'''
R,8
S,8'
S,8''
S,8'''
S)-4'',4'''-dihydroxy-3,3',3'',3''',5,5'-hexamethoxy-7,9':7',9-diepoxy-4,8'':4',8'''-bisoxy-8,8'-dineolignan-7'',7''',9'',9''-tetraol (
5) [
12], naringenin (
7) [
13], quercetin (
8) [
14], and kaempferol (
9) [
14]. The known compounds were identified on the basis of NMR spectroscopic analyses and comparison with the data reported in the literature.
Compound
1 was obtained as pale yellow oil with the molecular formula C
36H
48O
15, as evidenced by a pseudo-molecular ion peak [M+Na]
+ at
m/z 743.2863 in the HRESIMS spectrum. The UV spectrum suggested the existence of conjugated groups on the basis of maximum absorption bands at 228 nm and 280 nm. The IR spectrum exhibited the presence of hydroxyl groups (3364 cm
−1) and benzene rings (1603 cm
−1 and 1513 cm
−1). Its
1H-NMR spectrum (
Table 1) showed three ABX spin systems assignable to three sets of 1,3,4-trisubstituted aromatic rings at
δH 6.59 (1H, d,
J = 1.1 Hz), 6.63 (1H, d,
J = 8.0 Hz), and 6.50 (1H, dd,
J = 8.0, 1.1 Hz); 6.61 (1H, d,
J = 1.0 Hz), 6.70 (1H, d,
J = 8.1 Hz), and 6.53 (1H, dd,
J = 8.1, 1.0 Hz); 7.12 (1H, d,
J = 1.1 Hz), 6.74 (1H, d,
J = 8.1 Hz), and 6.86 (1H, dd,
J = 8.1, 1.1 Hz). Its
13C-NMR and DEPT spectral data (
Table 1) revealed the presence of 27 carbons except for the carbon signals due to a glucopyranose moiety (
δc 101.1, 75.3, 77.8, 71.9, 77.8, and 62.8) and three methoxy groups (
δC 56.5, 56.4, and 56.4). Subsequently, 27 carbons were assigned to a sesquilignan moiety, including three groups of aromatic carbons: nine quaternary carbons (
δC 151.4, 149.1, 148.9, 147.6 147.5, 145.6, 136.9, 133.9, and 130.5), and nine unsubstituted aromatic carbons (
δC 122.8, 122.8, 122.0, 118.4, 116.0, 115.9, 114.0, 113.6, and 112.8). The remaining 9 carbons were ascribed to five methylene carbons (
δC 62.2, 62.1, 61.9, 36.1, and 36.0), and four methine carbons (
δC 86.1, 77.9, 44.4, and 44.0). These before mentioned
1H- and
13C-NMR data implied that compound
1 should be a sesquilignan glucoside.
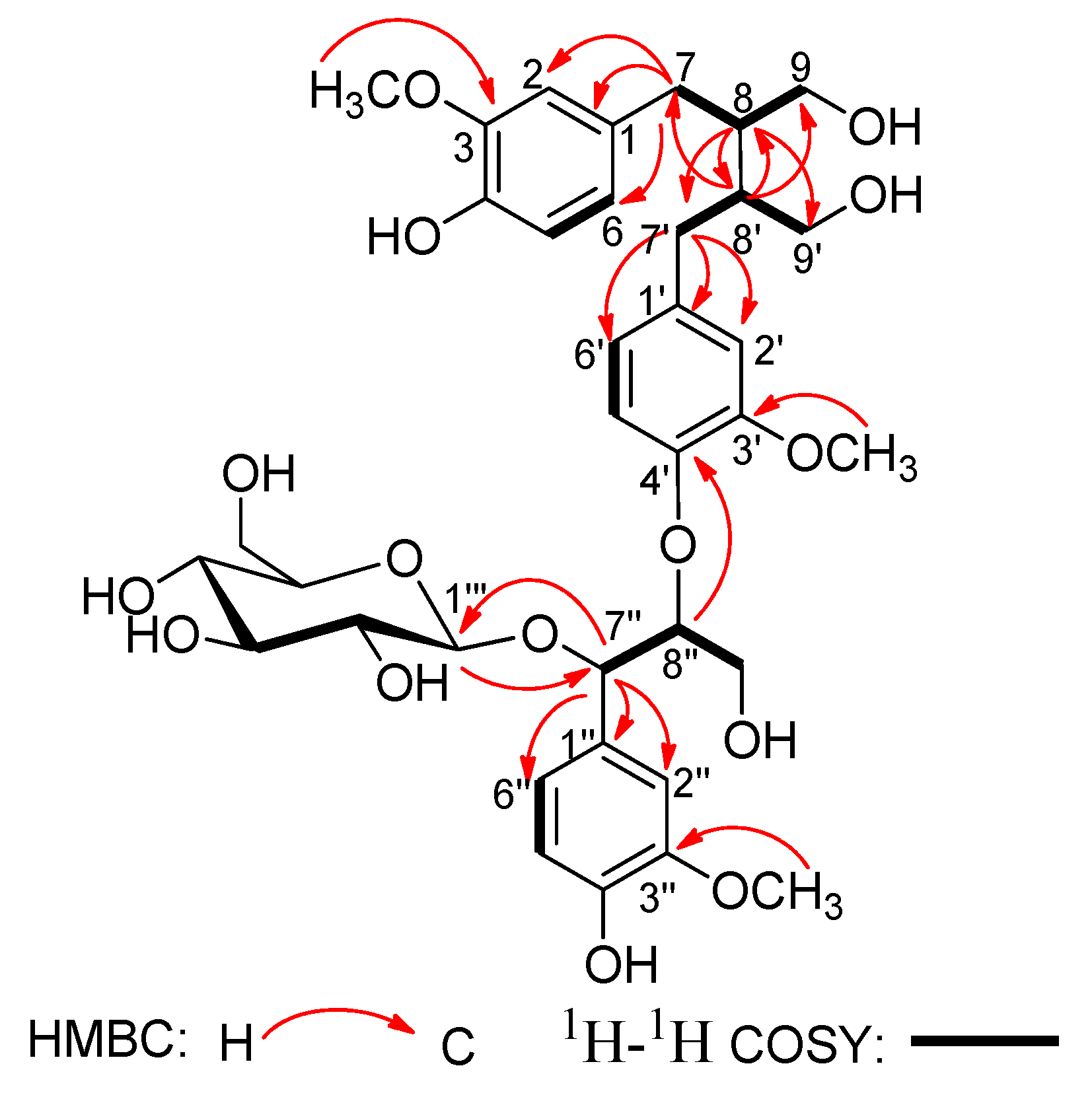
In the
1H-
1H COSY spectrum (
Figure 2), the consecutive cross-peaks starting from the methylene protons at
δH 2.52 (1H, m, H-7a) and 2.62 (1H, m, H-7b) to methine proton at
δH 1.87 (1H, m, H-8), and from H-8 to the methylene protons at
δH 3.56 (1H, dd,
J = 11.2, 4.3Hz, H-9a) and 3.82 (1H, dd,
J = 11.2, 5.1 Hz, H-9b) suggested the linkage of the C
3 fragment of
7CH
2-
8CH-
9CH
2OH. The
1H–
1H COSY correlation of CH
2-7' with CH-8' and the correlation of CH-8' with CH
2-9' revealed the connectivity of the C
3 unit
7'CH
2-
8'CH-
9'CH
2OH. Additionally, the
1H-
1H COSY correlation of the oxygenated methine proton at
δH 5.17 (1H, d,
J = 4.0 Hz, H-7'') with the oxygenated methine proton at
δH 4.30 (1H, m, H-8''), and the correlation of H-8'' with the oxygenated methylene protons at
δH 3.51 (1H, dd,
J = 11.1, 5.1 Hz, H-9''a) and 3.84 (1H, dd,
J = 11.1, 4.2 Hz, H-9''b) disclosed the fragment of the third the C
3 unit
7''CH(O)-
8''CH(O)-
9''CH
2OH. These three C
3 units were connected to the above three sets of 1,3,4-trisubstituted phenyl groups respectively, according to the HMBC correlations (
Figure 2) of H-7 with C-1, C-2, and C-6; of H-7' with C-1', C-2', and C-6'; and of H-7'' with C-1'', C-2'', and C-6''.
Table 1.
1H-NMR (400 MHz) and 13C-NMR (100 MHz) data of compound 1 (in CD3OD, δ in ppm, J in Hz).
Table 1.
1H-NMR (400 MHz) and 13C-NMR (100 MHz) data of compound 1 (in CD3OD, δ in ppm, J in Hz).
| NO. | δH | δC | NO. | δH | δC |
|---|
| 1 | - | 133.9 | 9'b | 3.56 dd (11.2, 4.3) | - |
| 2 | 6.59 d (1.1) | 113.6 | 1'' | - | 130.5 |
| 3 | - | 149.1 | 2'' | 7.12 d (1.1) | 112.8 |
| 4 | - | 145.6 | 3'' | - | 148.9 |
| 5 | 6.63 d (8.0) | 116.0 | 4'' | - | 147.6 |
| 6 | 6.50 dd (8.0, 1.1) | 122.8 | 5'' | 6.74 d (8.1) | 115.9 |
| 7a | 2.52 m | 36.0 | 6'' | 6.86 dd (8.1, 1.1) | 122.0 |
| 7b | 2.62 m | - | 7'' | 5.17 d (4.0) | 77.9 |
| 8 | 1.87 m | 44.0 | 8'' | 4.30 m | 86.1 |
| 9a | 3.56 dd (11.2, 5.1) | 62.1 | 9''a | 3.51 dd (11.1, 5.1) | 61.9 |
| 9b | 3.82 dd (11.2, 4.3) | - | 9''b | 3.84 dd (11.1, 4.2) | - |
| 1' | - | 136.9 | Glc-1''' | 4.15 d (7.4) | 101.1 |
| 2' | 6.61 d (1.0) | 114.0 | 2''' | 3.32 dd (8.9, 7.4) | 75.3 |
| 3' | - | 151.4 | 3''' | 3.27 t (8.9) | 77.8 |
| 4' | - | 147.5 | 4''' | 3.28 m | 71.9 |
| 5' | 6.70 d (8.1) | 118.4 | 5''' | 3.11 m | 77.8 |
| 6' | 6.53 dd (8.1, 1.0) | 122.8 | 6'''a | 3.66 dd (11.8, 4.5) | 62.8 |
| 7'a | 2.52 m | 36.1 | 6'''b | 3.83 dd (11.8, 2.3) | - |
| 7'b | 2.62 m | - | 3-OCH3 | 3.69 s | 56.4 |
| 8' | 1.86 m | 44.4 | 3'-OCH3 | 3.69 s | 56.4 |
| 9'a | 3.52 dd (11.2, 5.1) | 62.2 | 3''-OCH3 | 3.76 s | 56.5 |
Figure 2.
Key HMBC and 1H-1H COSY correlations of compound 1.
Figure 2.
Key HMBC and 1H-1H COSY correlations of compound 1.
Moreover, the HMBC correlation (
Figure 2) between H-8'' and C-4' verified ether linkage between C-8'' and C-4'. Meanwhile, the C-8/C-8' linkage was confirmed by HMBC correlations of H-8 with C-7', C-8' and C-9', and of H-8' with C-7, C-8 and C-9 in the HMBC spectrum. The signals due to methoxy protons were assigned as 3-OCH
3, 3'-OCH
3, and 3''-OCH
3, on the basis of the cross-peaks from H-3-OCH
3 to C-3, H-3'-OCH
3 to C-3', and H-3''-OCH
3 to C-3'' according to the HMBC spectrum. The position of 3-OCH
3, 3'-OCH
3, and 3''-OCH
3 was also corroborated by the correlations between H-3-OCH
3 and H-2, H-3'-OCH
3 and H-2', and H-3''-OCH
3 and H-2'' in the NOESY spectrum (
Figure 3). Thus, all the information mentioned above suggested that compound
1 was a secoisolariciresinol-sesquilignan derivative related to sesquimarocanol B [
15,
17,
18].
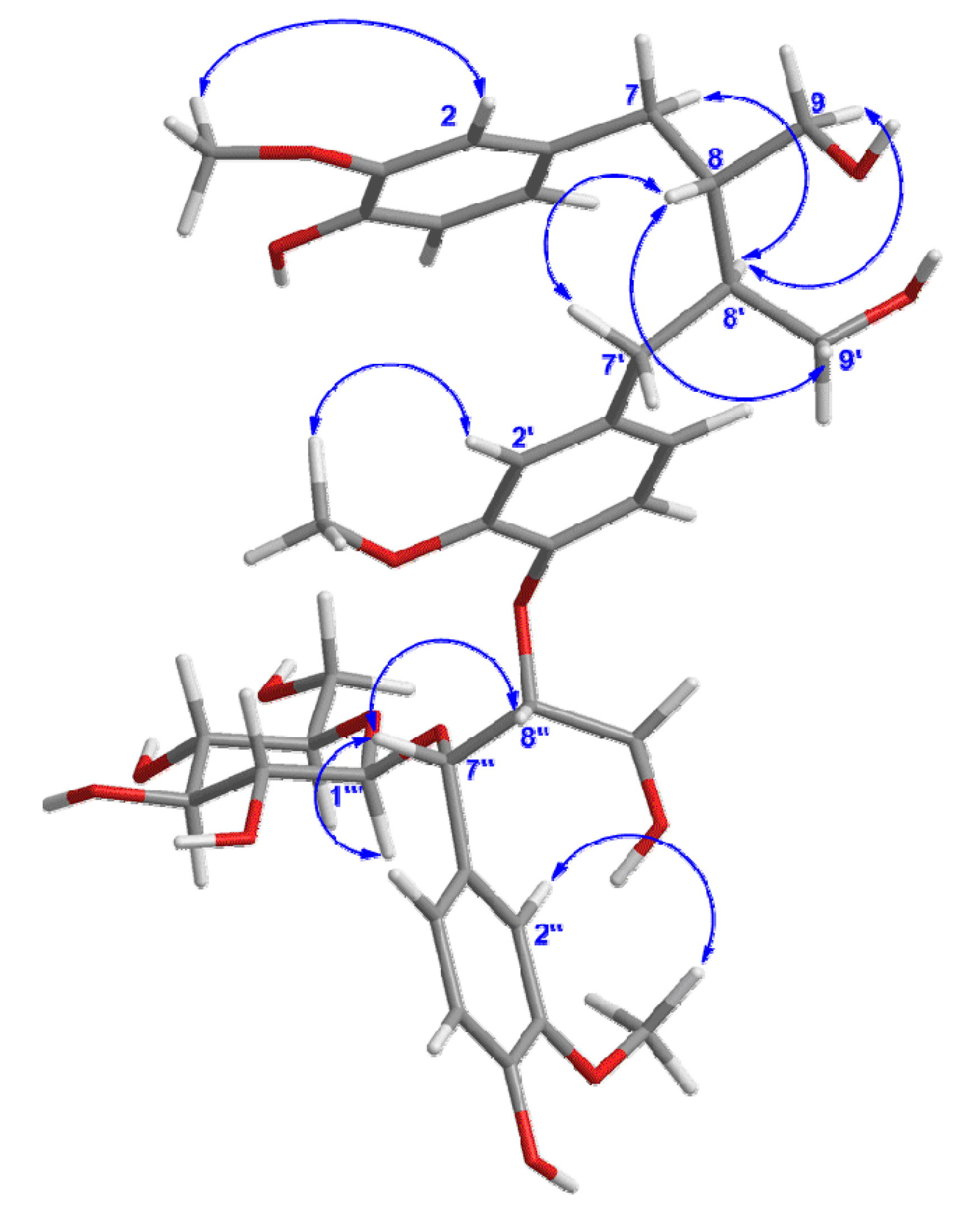
Figure 3.
Key NOESY correlations of compound 1.
Figure 3.
Key NOESY correlations of compound 1.
The remaining proton signals from
δH 3.83 to 3.32 in the
1H-NMR spectrum disclosed a set of
β-glucopyranose moiety with the anomeric proton resonated at
δH 4.15 (1H, d,
J = 7.4 Hz, H-1'''), corresponding to six carbon signals at
δc 101.1, 75.3, 77.8, 71.9, 77.8, 62.8 in
13C-NMR spectrum. The
β-glucose was located at C-7'' as elucidated by the HMBC correlation between Glc-H-1''' and the oxygenated methine carbon C-7'', which was further confirmed by the NOESY correlation between H-1''' and H-7''. Herein,
1 was identified as a secoisolariciresinol-sesquilignan glucoside. Furthermore, the presence of
β-D-glucose was determined by GC analysis of its trimethylsilyl thiazolidine derivative after acid hydrolysis [
16].
The relative configuration of
1 was elucidated by the NOESY (
Figure 3) experiment, together with comparison with data of sesquimarocanol B. Likewise, the NOESY correlations of H-8' with H-7 and H-9, of H-8-with H-7' and H-9', and the absence of the NOESY cross-peak between H-8 and H-8', pointed to the
trans orientations for H-8/H-8' as that of sesquimarocanol B. Similarly, the relative stereochemistry of H-7''/H-8'' was supposed to
cis configuration according to the correlation between H-7'' and H-8''.
Enzymatic hydrolysis of
1 by the cellulase liberated an aglycon
1a, which had the molecular C
30H
38O
10, as deduced from HRESIMS at
m/z 581.2344 [M+Na]
+. The NMR data of
1a was very similar with that of sesquimarocanol B [
15]. However, the coupling constants of H-7''/H-8'' for
1a and sesquimarocanol B were obviously different. In sesquimarocanol B, a large coupling constant (
J = 6.3 Hz) suggested the
threo configuration of H-7''/H-8'' [
17]. Whereas in
1a, a small coupling constant (
J = 4.0 Hz) of H-7''/H-8'', and the proton signal of 7'' at
δH 4.88 further confirmed an
erythro configuration between H-7''/H-8'' in
1, as a stereoisomer of sesquimarocanol B [
19,
20,
21]. In addition, the absolute configuration of C-8'' was determined to be
R, as the CD spectrum showed a negative Cotton effect at 239 nm [
22,
23,
24]. Correspondingly, the absolute configuration of 7'' was elucidated as
S. Finally, the structure of
1 was determined to be (7''
S,8''
R)-(–)-4,4'',9,9',9''-pentahydroxy-3,3',3''-trimethoxy-4',8''-oxy-8,8'-sesquineolignan-7''-
O-
β-D-glucopyranoside, to which the trivial name urariasinoside A was assigned.
In the other respects, previous reports of cytotoxicity activity of secoisolariciresinol-sesquilignan related structures such as sesquimarocanol B hexaacetate [
16], suggest compound
1 might have potential activity as an anticancer agent. Therefore, compound
1 was evaluated for its cytotoxic activities against five human cancer cell lines: HL-60 (human myeloid leukemia), SMMC-7721 (hepatocellular carcinoma), A549 (lung), MCF-7 (breast), and SW480 (colon) by the MTT method [
25]. DDP (cisplatin) and taxol were used as positive controls. However, the bioassay results revealed that compound
1 has no
in vitro cytotoxicity (IC
50 > 40 μM) against any of the five tested cancer cell lines.
3. Experimental
3.1. General Procedures
Optical rotations were measured on a Perkin Elmer PE-341LC polarimeter. UV spectra were measured on a Perkin Elmer Lambda 35 spectrometer. CD spectra were recorded on a JASCO J-810 CD spectrometer. IR spectra were recorded on a Bruker Vertex 70 FT-IR microscope instrument (FT-IRmicroscope transmission). NMR spectra were obtained at 400 MHz for 1H and 100 for 13C, on Bruker AM-400 MHz spectrometers with solvent peaks being used as references. Two-dimensional HSQC, HMBC, COSY, and NOESY experiments were performed using the pulse sequences provided by Bruker. HRESIMS data were measured using an API QSTAR Pulsar spectrometer. Column chromatography was performed using polyamide (10–30 mesh, Taizhou Luqiao Sijia Biochemical Plastics Factory, Taizhou, China), silica gel (100–200, 200–300 mesh, H, Qingdao Marine Chemical Inc., China), ODS (50 μm, YMC, Kyoto, Japan) and Sephadex LH-20 (Pharmacia Biotech AB, Uppsala Sweden). HPLC separation was performed on an instrument consisting of an Ultimate 3000 controller, an Ultimate 3000 pump, and an Ultimate 3000 UV detector with an YMC (250 × 10 mm, 5 μm) preparative column. GC analysis was performed on an Agilent Technologies 7820A GC instrument, OV-17 column (30 m × 0.32 mm × 0.5 μm, Lanzhou Zhongke Antai Analysis Technology Co. Ltd., Lanzhou, China), hydrogen-flame ionization detector. Enzymatic hydrolysis was treated with cellulase (Shanghai Yuanye Biology & Technology Co. Ltd., Shanghai, China). TLC was carried out on precoated silica gel GF254 plates. Spots were visualized under UV light (254 or 356 nm) or by spraying with 5% H2SO4 in 95% EtOH followed by heating.
3.2. Plant Material
The aerial parts of U. sinensis were collected from Shiyan, Hubei Province, China in September 2010 and identified by Dr. Jianping Wang of School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology. The voucher specimen (1009) was deposited in the herbarium of Hubei Key Laboratory of Natural Medicinal Chemistry and Resource Evaluation, School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology.
3.3. Extraction and Isolation
The air-dried aerial parts of U. sinensis (45 kg) were extracted three times with 95% EtOH (50 L) at room temperature for 24 hThe dried EtOH extract (4.0 kg) was suspended in H2O and then partitioned successively with petroleum ether (10.0 L × 3), CH2Cl2 (10.0 L × 3) and n-BuOH (10.0 L × 3). The n-butanol portion (375 g) was fractionated by column chromatography over polyamide (6 Kg) using H2O–EtOH of increasing polarity (100:0 (50 L × 3), 10:90 (50 L × 3), 30:70 (50 L × 3), 0:100 (50 L × 3), v/v). The eluates were combined together on the basis of TLC analysis. Then, the fractions eluted with pure water (the extract 172 g) were further subjected to silica gel column chromatography (CHCl3–MeOH–H2O 15:1:0 to 3:1:0.1, v/v/v) to afford seven fractions (Fr.1–7). Fr.6 (11.7 g) was subjected to silica gel column chromatography (CHCl3–MeOH–H2O 5:1:0.1 to 3:1:0.1, v/v/v) once again to give four subfractions (Fr.6.1–6.4). Fr.6.3 was subjected to a Sephadex LH-20 column eluted with MeOH to give six subfractions (Fr.6.3.1–6.3.6). Fr.6.3.4 was purified by silica gel H column chromatography (CHCl3–MeOH–H2O 3:1:0.1, v/v/v) to obtain compound 6 (10.5 mg). Fr.6.3.3 was subjected to ODS column chromatography eluted with 50% MeOH in H2O, and then purified by semi-preparative HPLC (37% MeOH in H2O, flow rate 1.8 mL/min, wavelength 212 nm) to yield compound 1 (5.0 mg, retention time 37 min).
The CH2Cl2 extract (150 g) was applied to silica gel column chromatography eluted with petroleum ether–acetone (10:1 to 1:1, v/v) to afford ten fractions (Fr.A–J). Compound 10 (20.0 mg) was recrystallized from Fr.G using acetone. Fr.G (5.1 g) was subjected to silica gel column chromatography (CHCl3–MeOH 20:1 to 8:1, v/v) to afford seven subfractions (Fr.G1–G7). Fr.G3 and Fr.G5 were subjected to a Sephadex LH-20 column eluted with CH2Cl2–MeOH (1:1, v/v) to give compound 8 (16.4 mg) by recrystallization. Compound 9 (14.5 mg) was obtained by recrystallization in acetone from Fr.G3.
Fr.H (20.0 g) was subjected to silica gel column chromatography (CHCl3–MeOH 30:1: to 8:1, v/v) to afford eight subfractions (Fr.H1–H8). Fr.H2 was applied to MCI gel column eluted with MeOH/H2O (8:1, v/v), and then was subjected to a Sephadex LH-20 column eluted with MeOH to give seven subfractions (Fr.H2.1–2.7). Fr.H2.2 was further purified by semi-preparative HPLC (MeOH–MeCN–H2O 35:15:50, flow rate 2.5 mL/min, wavelength 254 nm) to yield compound 2 (17.5 mg, retention time 20 min) and compound 3 (8.0 mg, retention time 23 min).
Fr.I (6.0 g) was subjected to silica gel column chromatography (CHCl3–MeOH 15:1: to 7:1, v/v) to afford nine subfractions (Fr.I1–I9). Fr.I1 was subjected to a Sephadex LH-20 column eluted with CH2Cl2–MeOH (1:1, v/v) to give four subfractions (Fr.I1.1–1.4). Fr.I1.2 was subjected to ODS column chromatography eluted with 50% MeOH in H2O, and then purified by semi-preparative HPLC (MeOH–MeCN–H2O 20:18:62, flow rate 2.5 mL/min, wavelength 212 nm) to yield compound 4 (7.2 mg, retention time 58 min).
Fr.J (25.0 g) was subjected to silica gel column chromatography (CHCl3–MeOH 12:1 to 8:1, v/v) to afford four subfractions (Fr.J1–J8). Fr.J2 was subjected to a Sephadex LH-20 column eluted with CH2Cl2–MeOH (1:1, v/v) to remove chlorophyll, and then further subjected to ODS column chromatography eluted with 50% MeOH in H2O to give two subfractions (Fr.J2.1–2.2). Fr.J2.1 was subjected to silica gel column chromatography (CHCl3–Me2CO–MeOH 30:15:0.1, v/v/v) and then purified by semi-preparative HPLC (MeOH–MeCN–H2O 29:21:50, flow rate 2 mL/min, wavelength 254 nm) to yield compound 5 (2.3 mg, retention time 30 min).
Urariasinoside A [(7''S,8''R)-(–)-4,4'',9,9',9''-pentahydroxy-3,3',3''-trimethoxy-4',8''-oxy-8,8'-sesqui-neolignan-7''-O-β-D-glucopyranoside] (
1). Pale yellow oil. [α]
20D: –29 (c = 0.79, MeOH); UV (MeOH) λ
max (logε) nm: 280 (3.32), 228 (3.68), 207 (4.03); CD (MeOH, nm) λ
max (Δε) 211 (+ 11.48), 229 (+ 1.30), 239 (− 3.74), 281 (+ 2.71), 297 (+ 1.37), 322 (+ 1.83); IR (KBr) ν
max 3364, 2935, 1603, 1513, 1453, 1423, 1369, 1268, 1224, 1155, 1127, 1075, 1029 cm
−1;
1H- and
13C-NMR: see
Table 1; HRESIMS
m/z: 743.2863 ([M+Na]
+, C
36H
48O
15Na
+, calc. 743.2891).
3.4. Determination of the Absolute Configuration of Sugar Unit in 1
A solution of 1 (1.0 mg), in 2 M aqueous CF3COOH (2.0 mL) was heated at 100 °C for 3 h in a water bath. The reaction mixture was diluted in H2O (4.0 mL) and extracted with EtOAc (4.0 mL × 3), then the aqueous layer was concentrated to remove CF3COOH. The residue was dissolved in pyridine (1.0 mL), to which L-cysteine methyl ester hydrochloride (2.0 mg) in pyridine (1.0 mL) was added. Then, the mixture was kept at 60 °C for 2 h. After the reaction mixture was dried in vacuo, the residue was trimethylsilylated with 1-trimethylsilylimidazole (0.2 mL) at 60 °C for 2 h in a water bath. Finally, the mixture was partitioned between hexane and H2O (0.3 mL each) and the hexane extract was analyzed by gas chromatography (GC) under the following conditions: column temperature, 250 °C; injection temperature, 250 °C; carrier N2 gas; flow rate 1.0 mL/min. In the acid hydrolysate of 1, D-Glucose was confirmed by comparison of the retention times of their derivatives with those of D-glucose and L-glucose derivatives prepared in a similar way, which showed retention times of 13.66 and 14.34 min, respectively.
3.5. Enzymatic Hydrolysis of 1
A solution of 1 (2.67 mg) in 0.1 M acetate buffer (pH 4.0, 2.0 mL) was treated with cellulase (3.0 mg) and then the reaction mixture was stirred at 40 °C for 12 h. The reaction mixture was diluted in H2O (4.0 mL) and extracted with EtOAc (4.0 mL × 3). After that, the EtOAc extract was further purified by semi-preparative HPLC (50% MeOH in H2O, flow rate 1.8 mL/min, wavelength 212 nm) to obtain compounds 1a (1.2 mg, retention time 18.3 min).
Erythro-(–)-secoisolariciresinol-sesquilignan (1a). Pale yellow oil. [α]20D: –7.1 (c = 0.028, MeOH); 1H-NMR (CD3OD, 400 MHz) δH: 7.03 (1H, d, J = 1.8 Hz, 2''-H), 6.93 (1H, d, J = 8.2 Hz, 5''-H), 6.86 (1H, dd, J = 8.2, 1.8 Hz, 6''-H), 6.76 (1H, d, J = 8.1 Hz, 5-H), 6.68 (1H, d, J = 1.8 Hz, 2-H), 6.66 (1H, d, J = 8.0 Hz, 5'-H), 6.64 (1H, d, J = 1.9 Hz, 2'-H), 6.63 (1H, dd, J = 8.1, 1.8 Hz, 6-H), 6.54 (1H, dd, J = 8.0, 1.9 Hz, 6'-H), 4.88 (1H, d, J = 4.0 Hz, 7''-H), 4.20 (1H, m, 8''-H ), 3.86 (1H, d, J = 11.5 Hz, 9''-Ha), 3.82 (3H, s, 3''-OCH3), 3.77 (3H, s, 3-OCH3), 3.75 (3H, s, 3'-OCH3), 3.72 (1H, J = 11.5, 4.1 Hz, 9''-Hb), 3.69 (1H, overlapped, 9-Hb), 3.58 (2H, m, 9-Ha, 9'-Hb), 3.46 (1H, dd, J = 11.8, 5.2 Hz, 9'-Ha), 2.67 (2H, m, 7,7'-Ha), 2.60 (2 H, m, 7, 7'-Hb ), 2.06 (2H, m, 8,8'-H); HRESIMS m/z: 581.2344 ([M+Na] +, C30H38O10Na +, calc. 581.2363).
3.6. MTT Cytotoxicity Assay
Compound
1 was tested against five human cancer cell lines [HL-60 and SMMC-7721, A549 (lung), MCF-7 (breast), and SW480 (colon)] and a normal cell line (BEAS-2B). The antiproliferative assay was performed by the MTT colorimetric method as described previously [
25]. Briefly, adherent cells were seeded into 96-well tissue culture plates with density of 1 × 10
5 cells/mL. After 12 h, cells were treated with the medium containing different concentrations of test compounds for 48 h. Then, attached cells were incubated with MTT (15 μL, 5 mg/mL, 1 h) and subsequently solubilized in DMSO. The optical density of absorbency at 595 nm was then measured using a microplate reader. Experiments were performed in triplicate, and the values are the averages of three (n = 3) independent experiments. DDP (cisplatin, Sigma, San Francisco, CA, USA) and taxol were used as the positive control.
4. Conclusions
Studies carried out on the EtOH extracts of U. sinensis, revealed the presence of a new sesquilignan glucoside, urariasinoside A (1), was isolated from the aerial parts of U. sinensis together with eight known compounds, including two lignans (–)-syringaresinol (2), (–)-medioresinol (3); a sesquilignan (–)-(7R,7'R,7''S,8S,8'S,8''S)-4',4''-dihydroxy-3,3',3'',5,5'-pentamethoxy-7,9':7',9-diepoxy-4,8''-oxy-8,8'-sesquineolignan-7'',9''-diol (4); a dilignan (+)-(7R,7'R,7''R,7'''R,8S,8'S,8''S,8'''S)-4'',4'''-dihydroxy-3,3',3'',3''',5,5'-hexamethoxy-7,9':7',9-diepoxy-4,8'':4',8'''-bisoxy-8,8'-dineolignan-7'',7''',9'',9'''-tetraol (5), and four flavonoids derivatives 5,4'-dihydroxy-3'-methoxyflavanone-7-(6''-O-β-l-rhamnopyranosyl)-β-d-glucopyranoside (6), naringenin (7), quercetin (8), and kaempferol (9). Compounds 2–9 were isolated from U. sinensis for the first time. Cytotoxicity assays revealed that compound 1 was inactive (IC50 > 40 μM) against the HL-60, SMMC-7721, A549, MCF-7, and SW480 cell lines.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}

