Recent Syntheses of 1,2,3,4-Tetrahydroquinolines, 2,3-Dihydro-4(1H)-quinolinones and 4(1H)-Quinolinones using Domino Reactions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
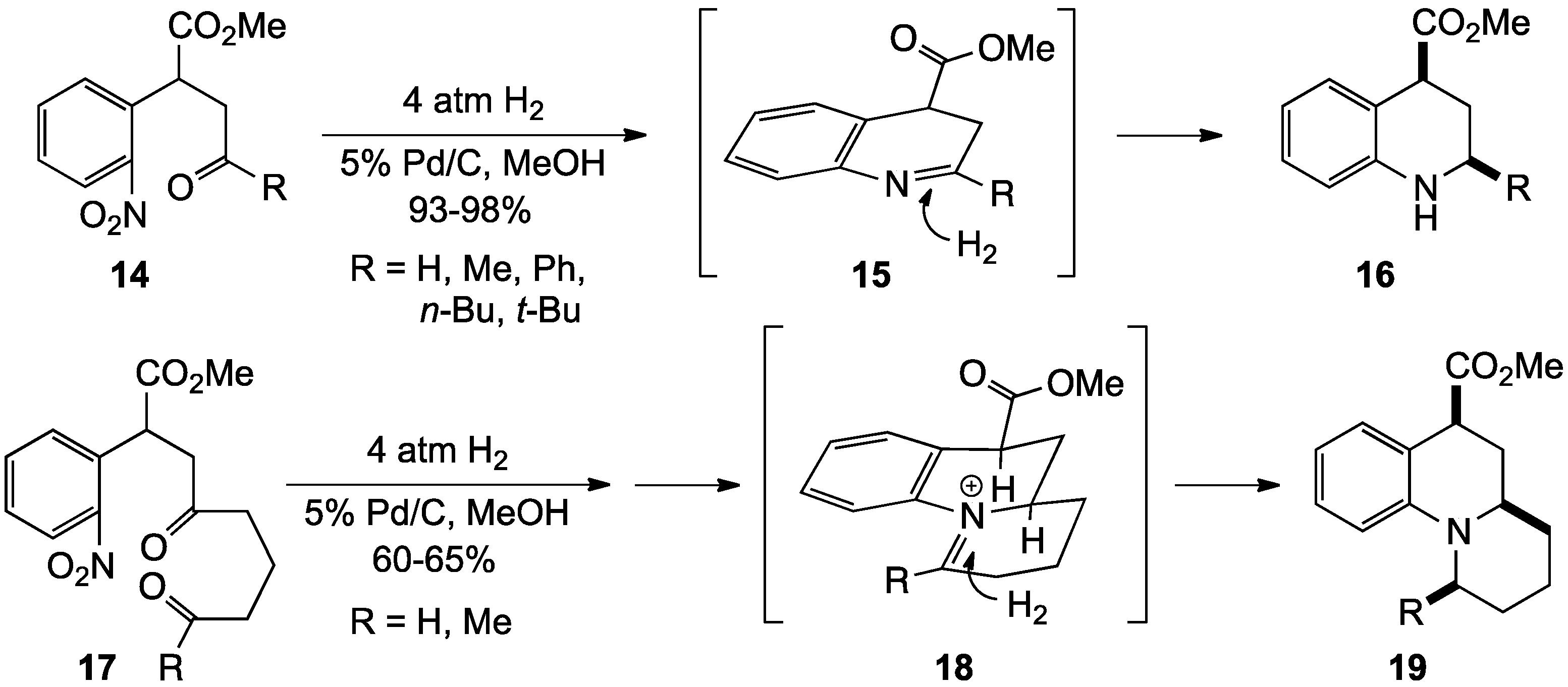{kind=link}
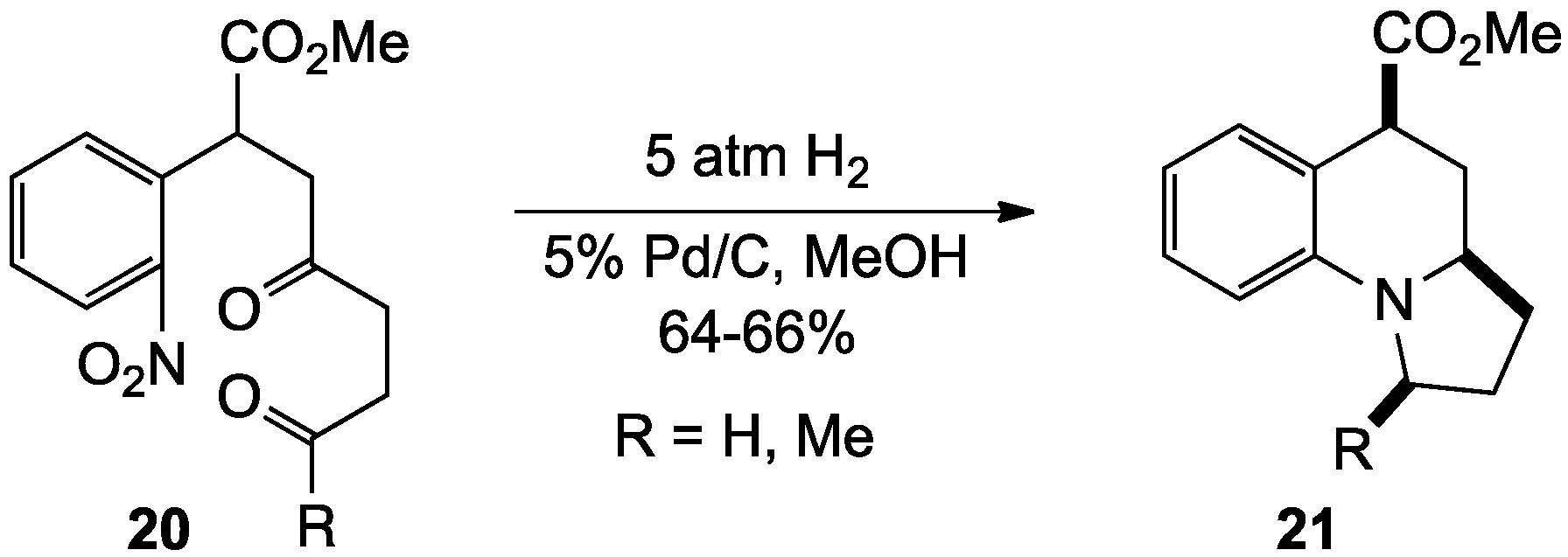{kind=link}
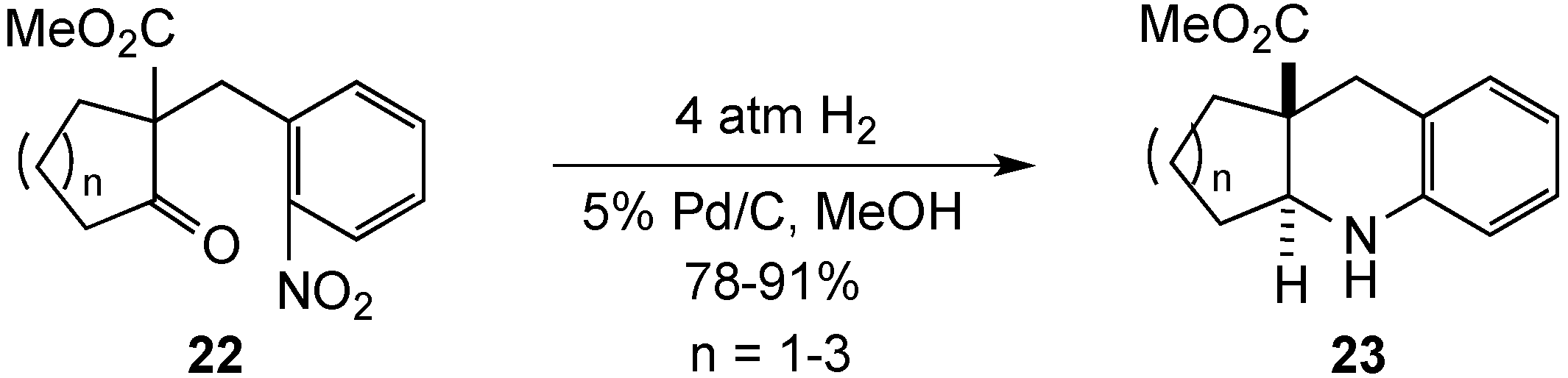{kind=link}
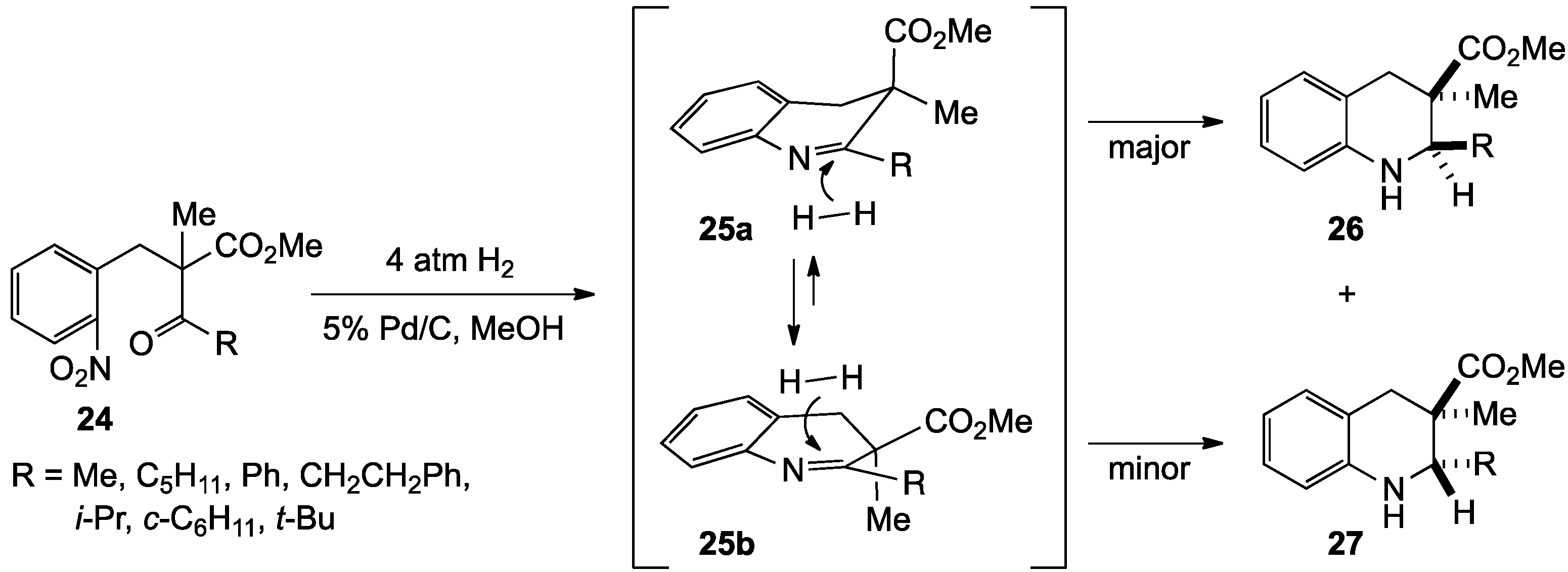{kind=link}
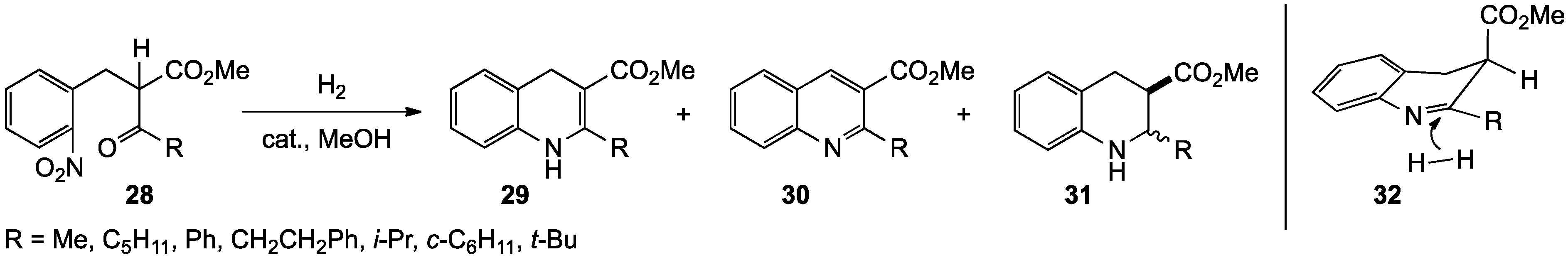{kind=link}
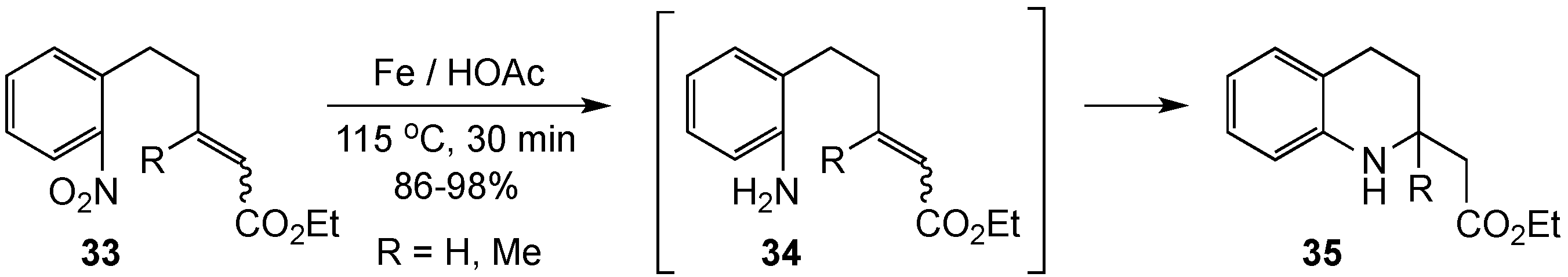{kind=link}
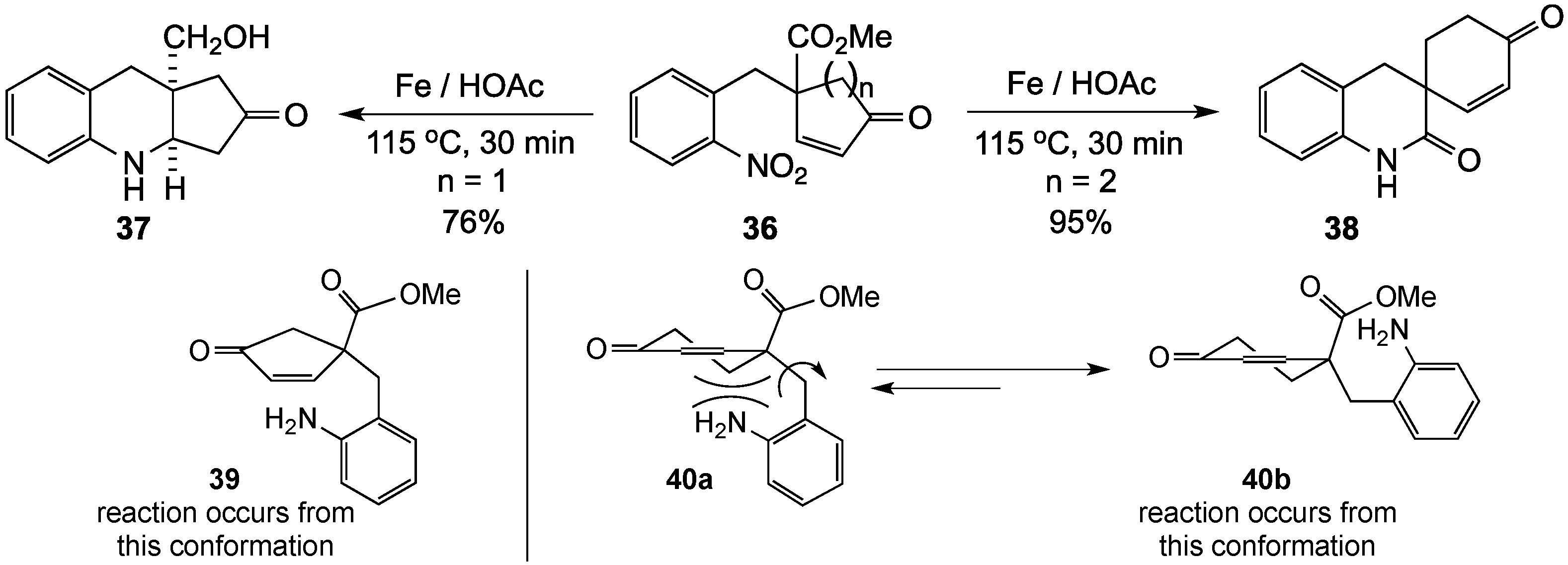{kind=link}
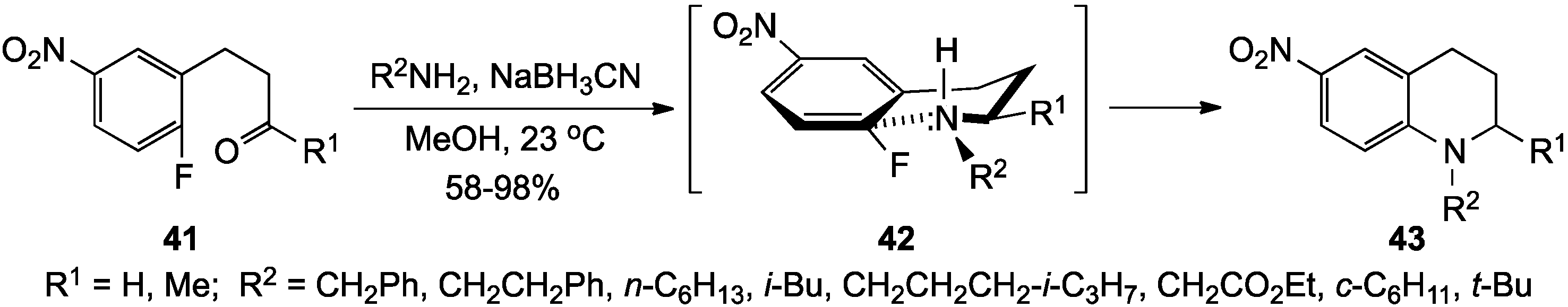{kind=link}
{kind=link}
{kind=link}
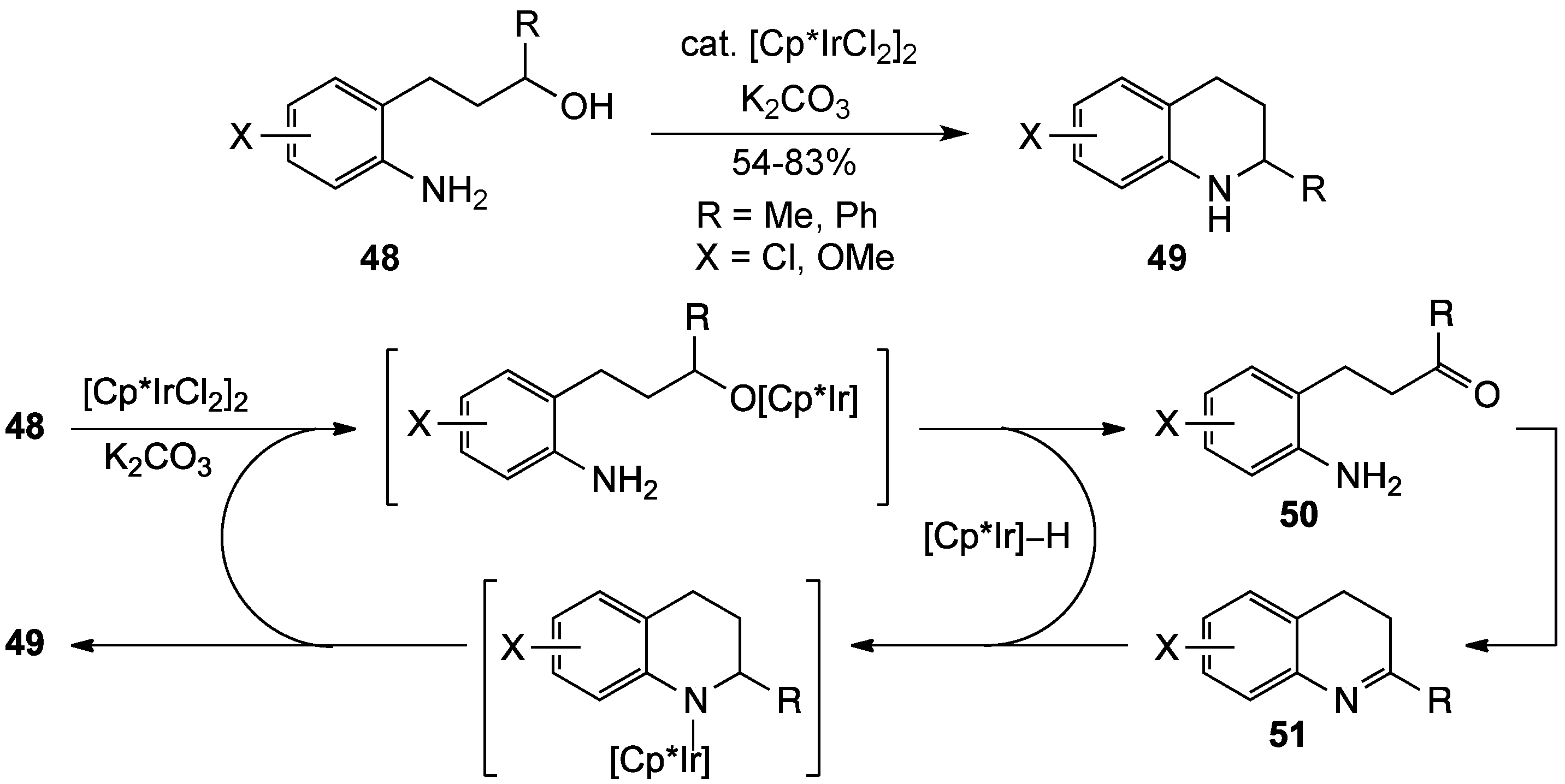{kind=link}
{kind=link}
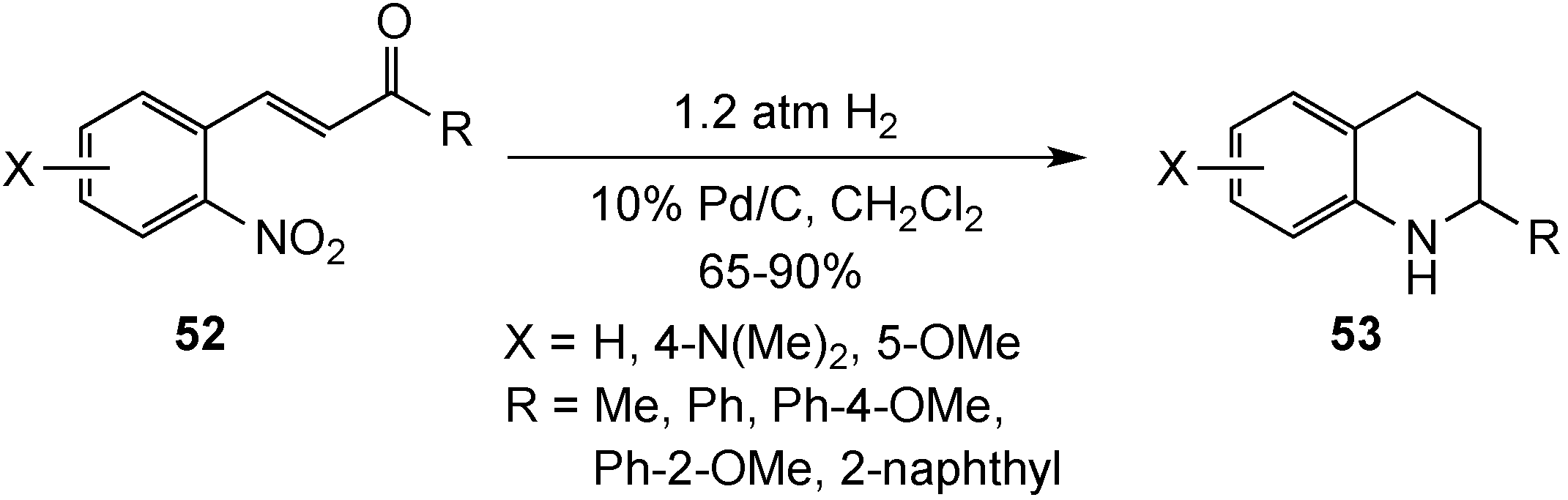
{kind=link}
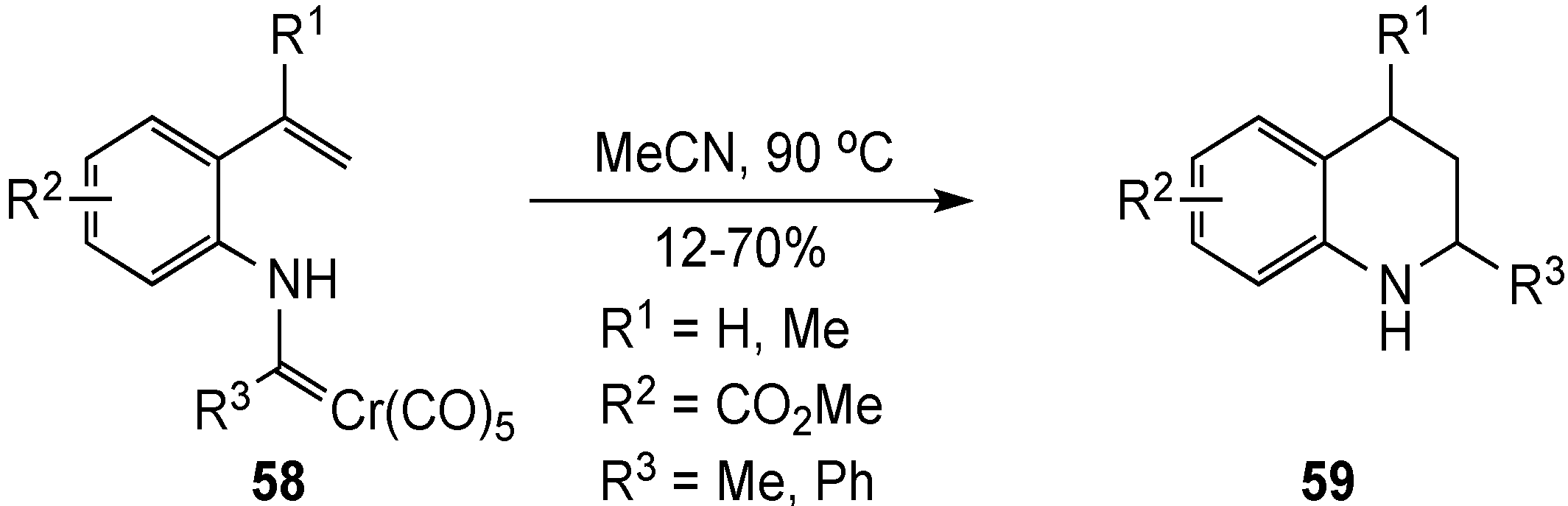{kind=link}
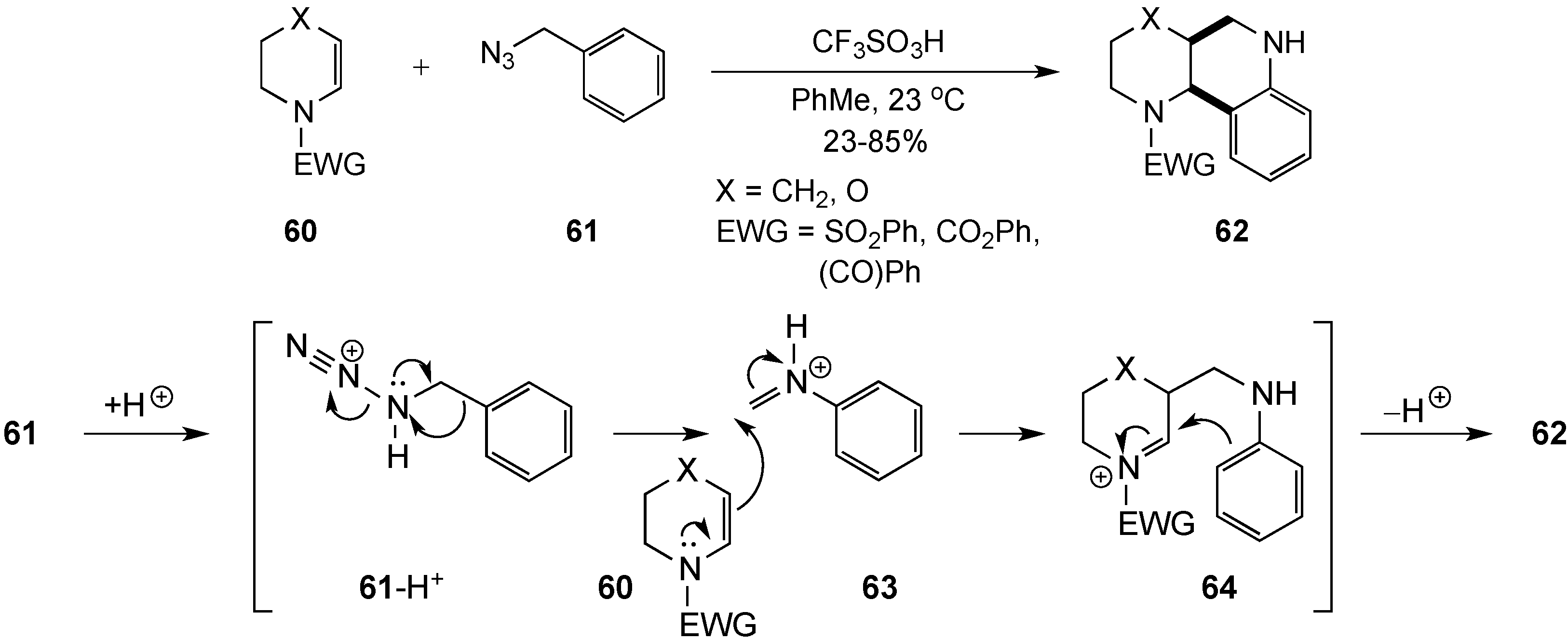{kind=link}
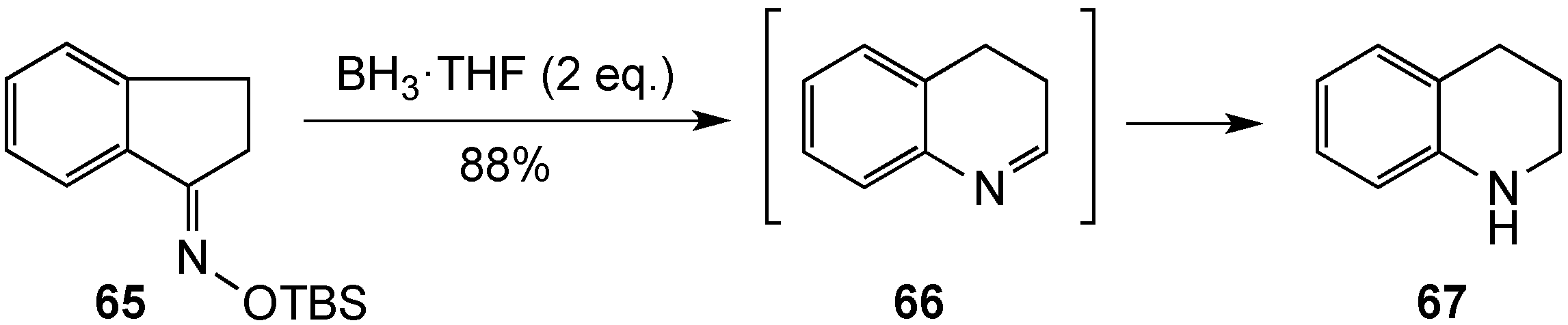{kind=link}
{kind=link}
{kind=link}
{kind=link}
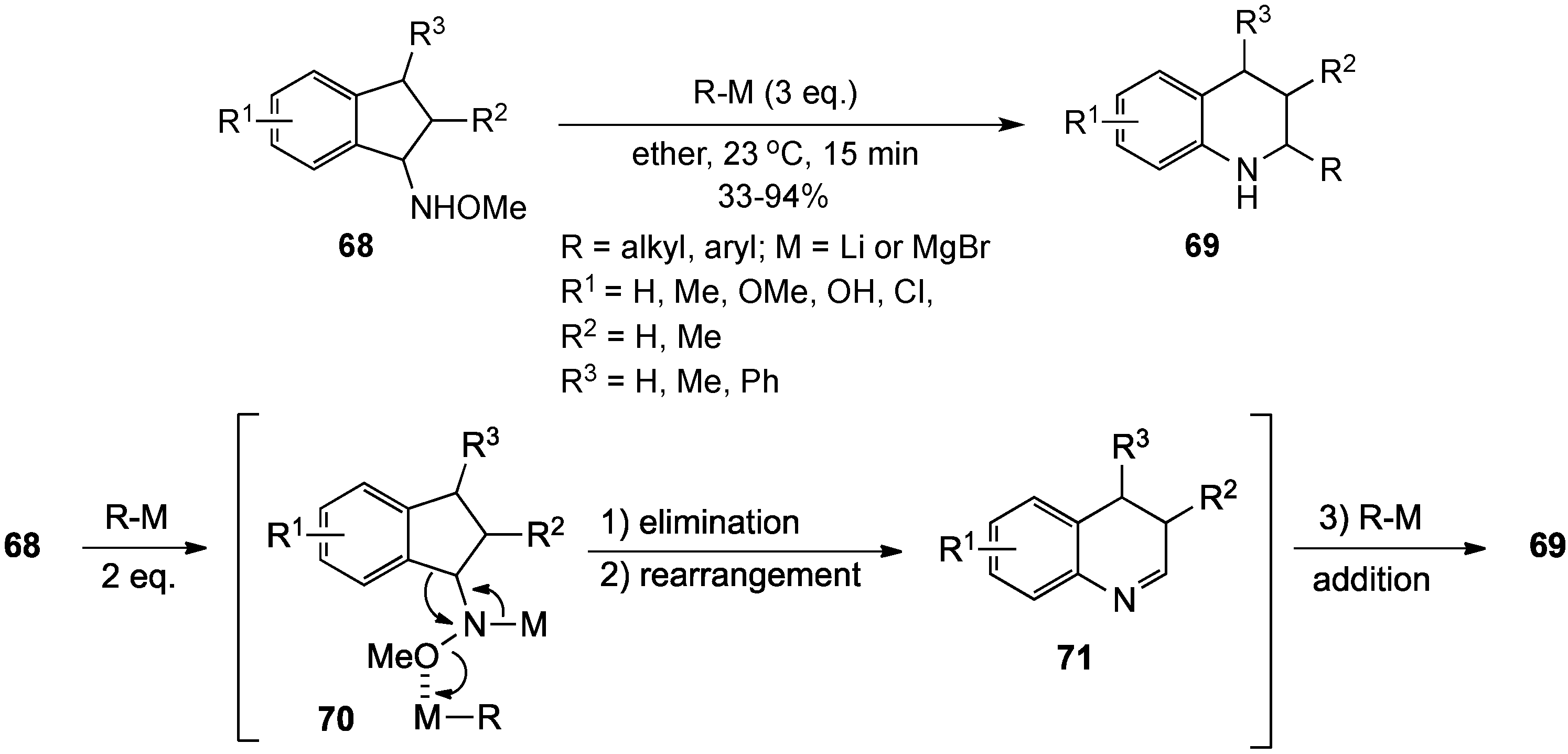
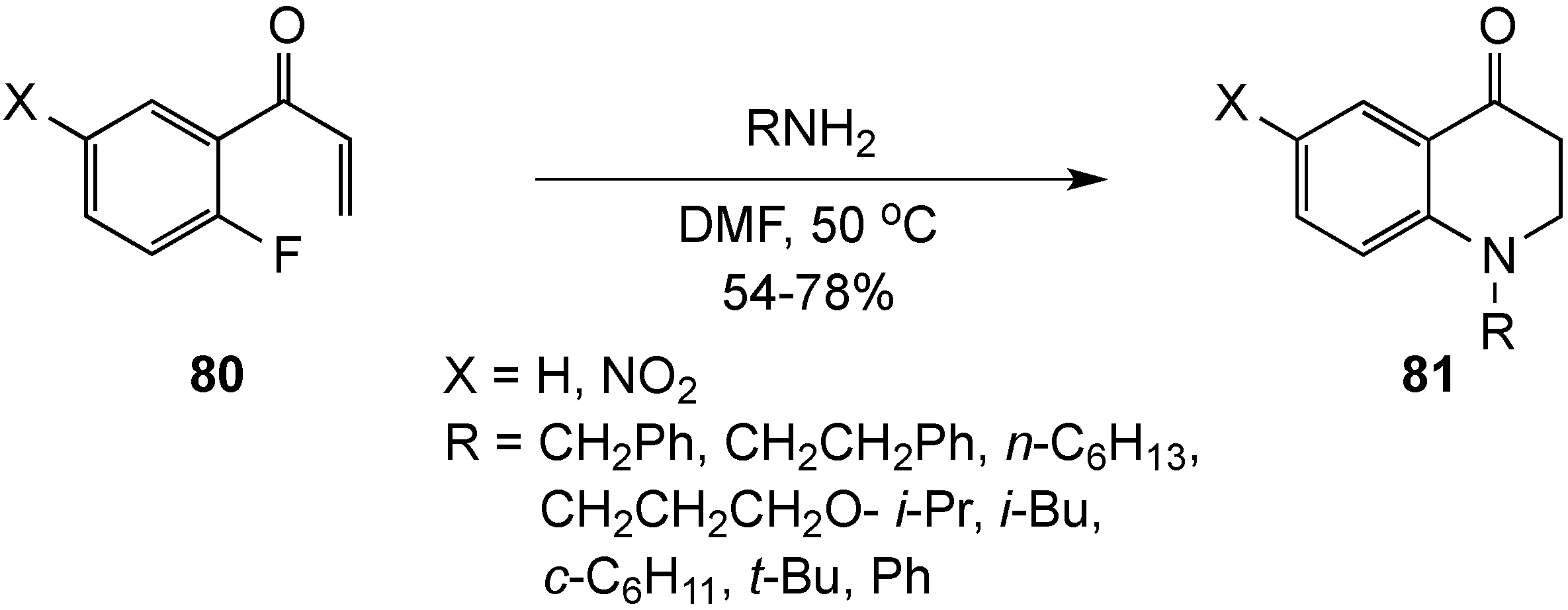{kind=link}
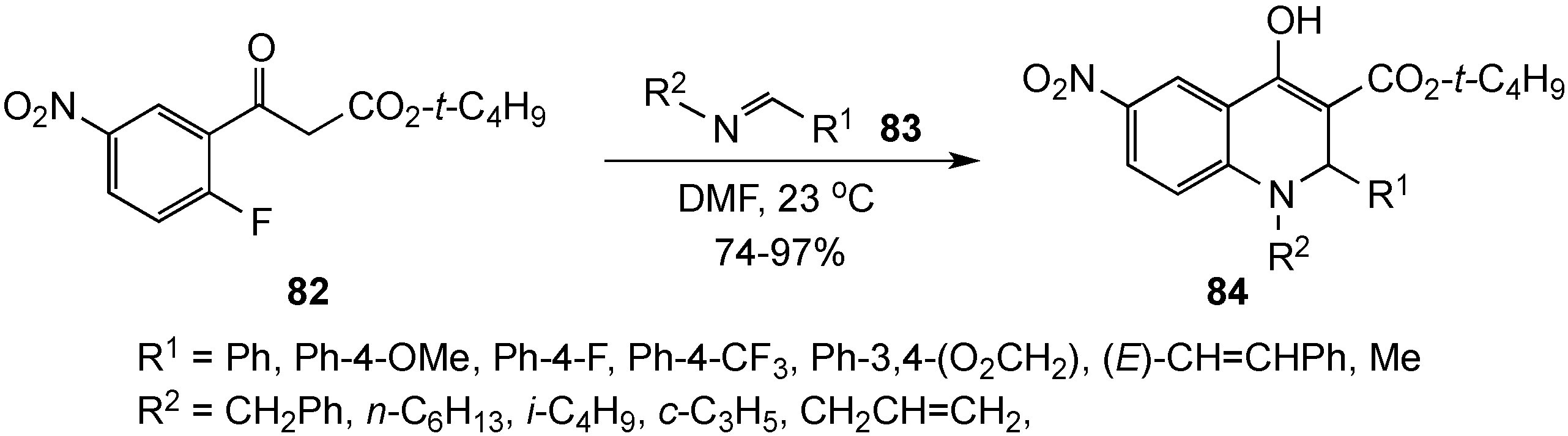{kind=link}
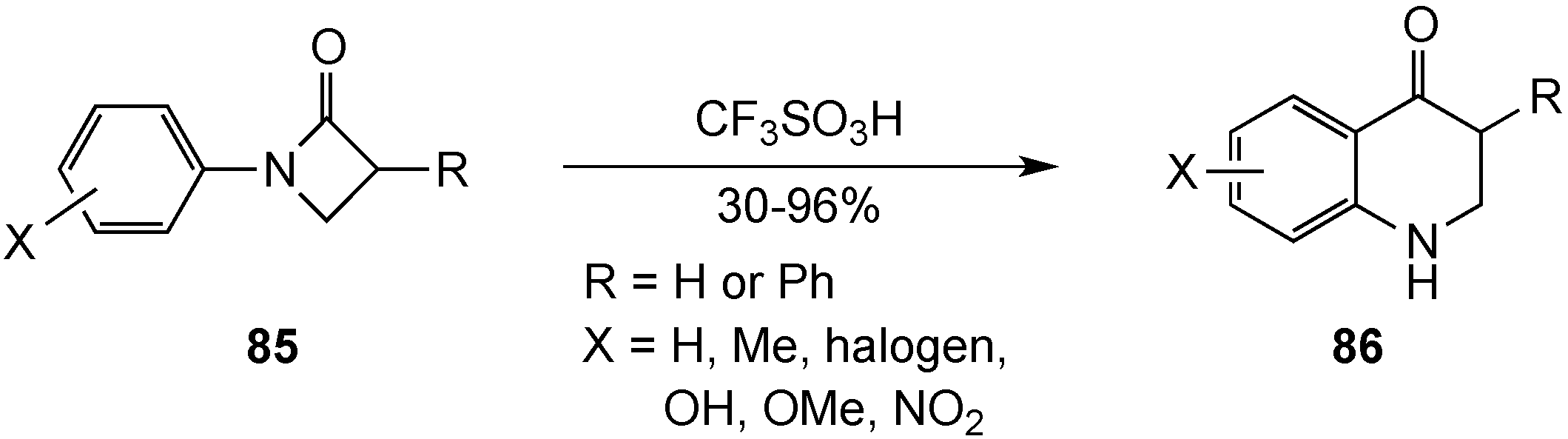{kind=link}
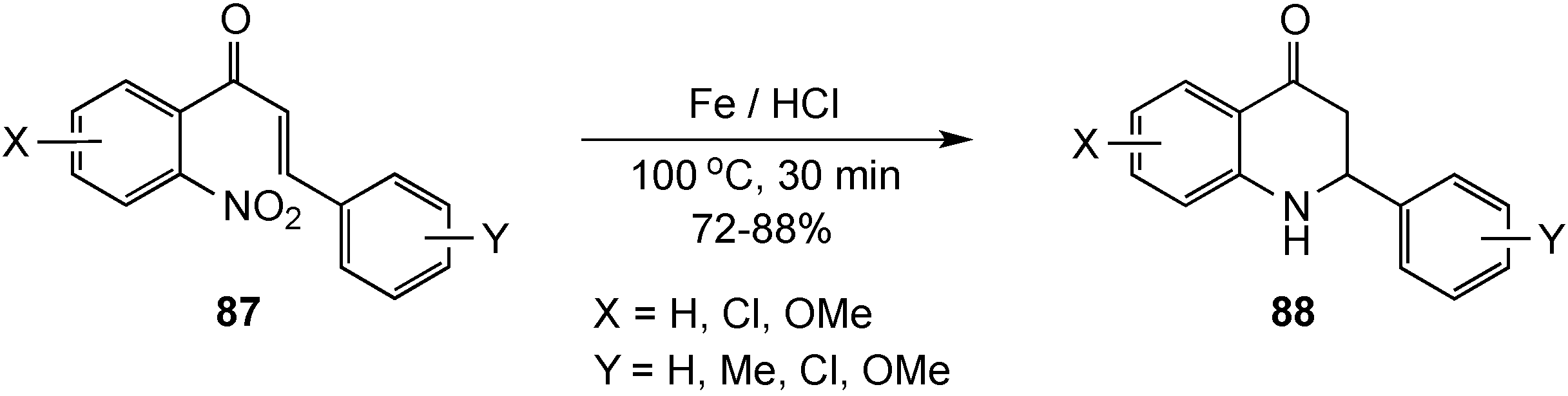{kind=link}
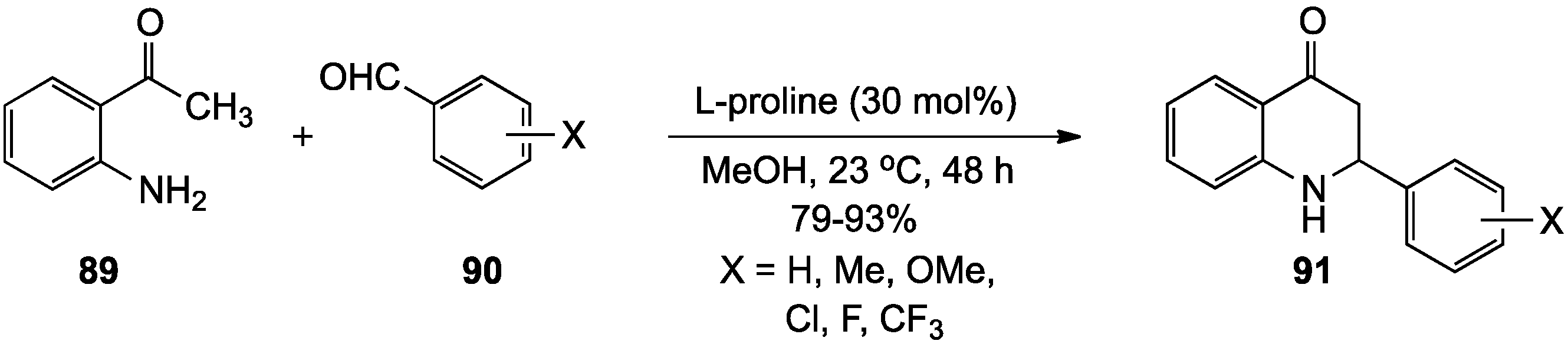{kind=link}
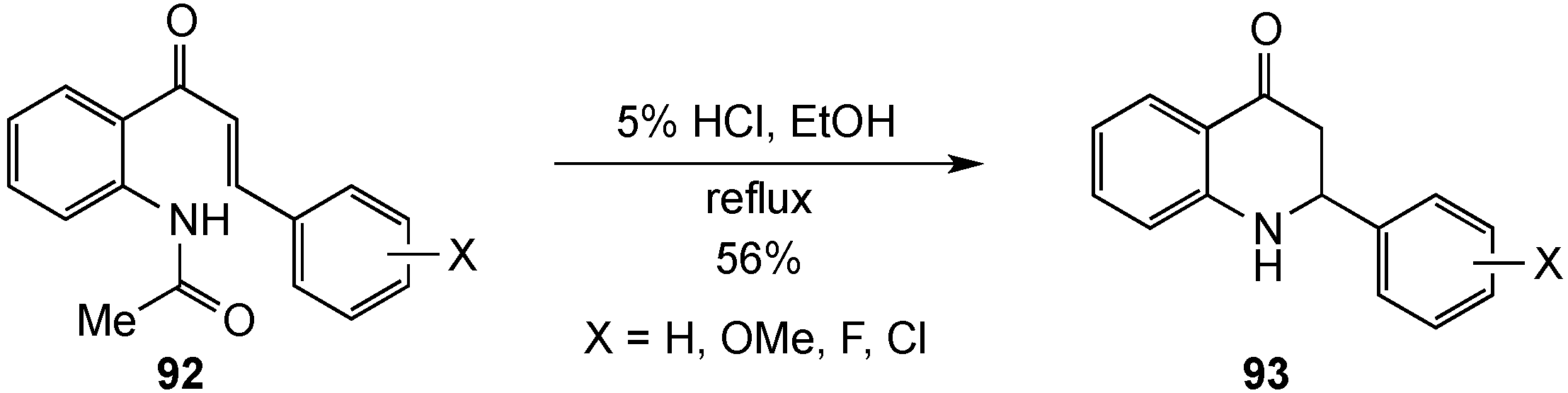{kind=link}
{kind=link}
{kind=link}
{kind=link}
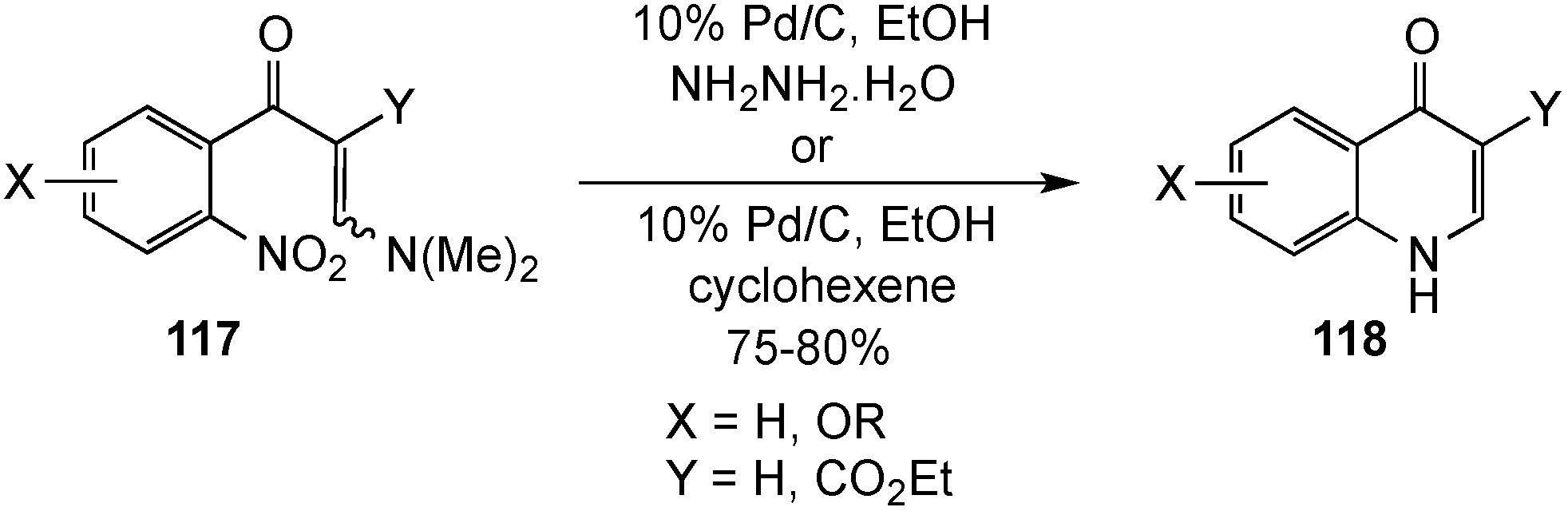{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
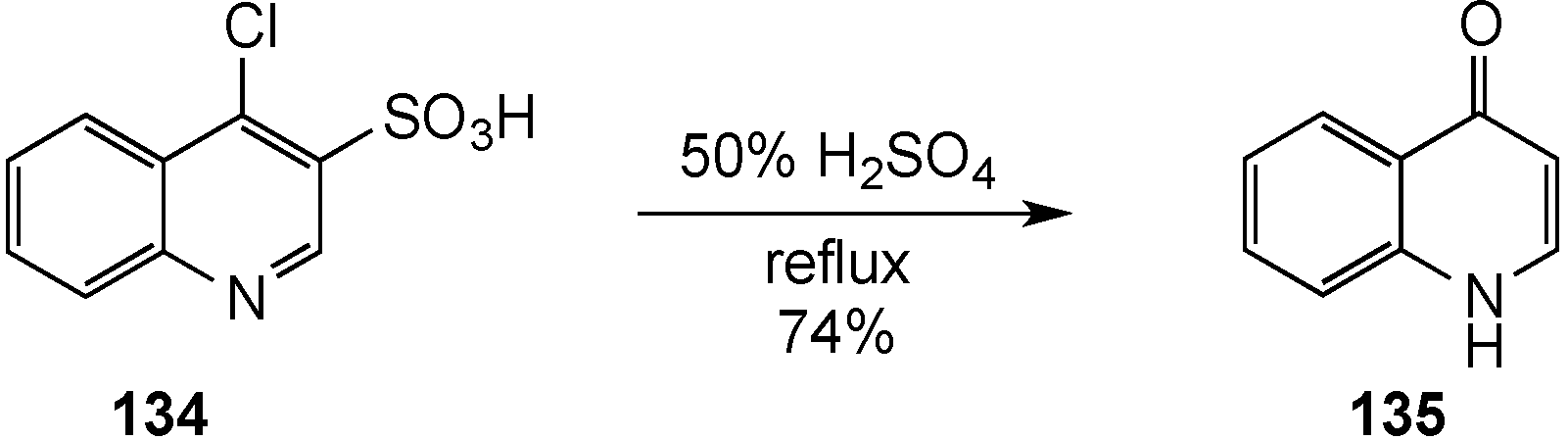{kind=link}
Abstract
:1. Introduction
2. Survey of New Methodology
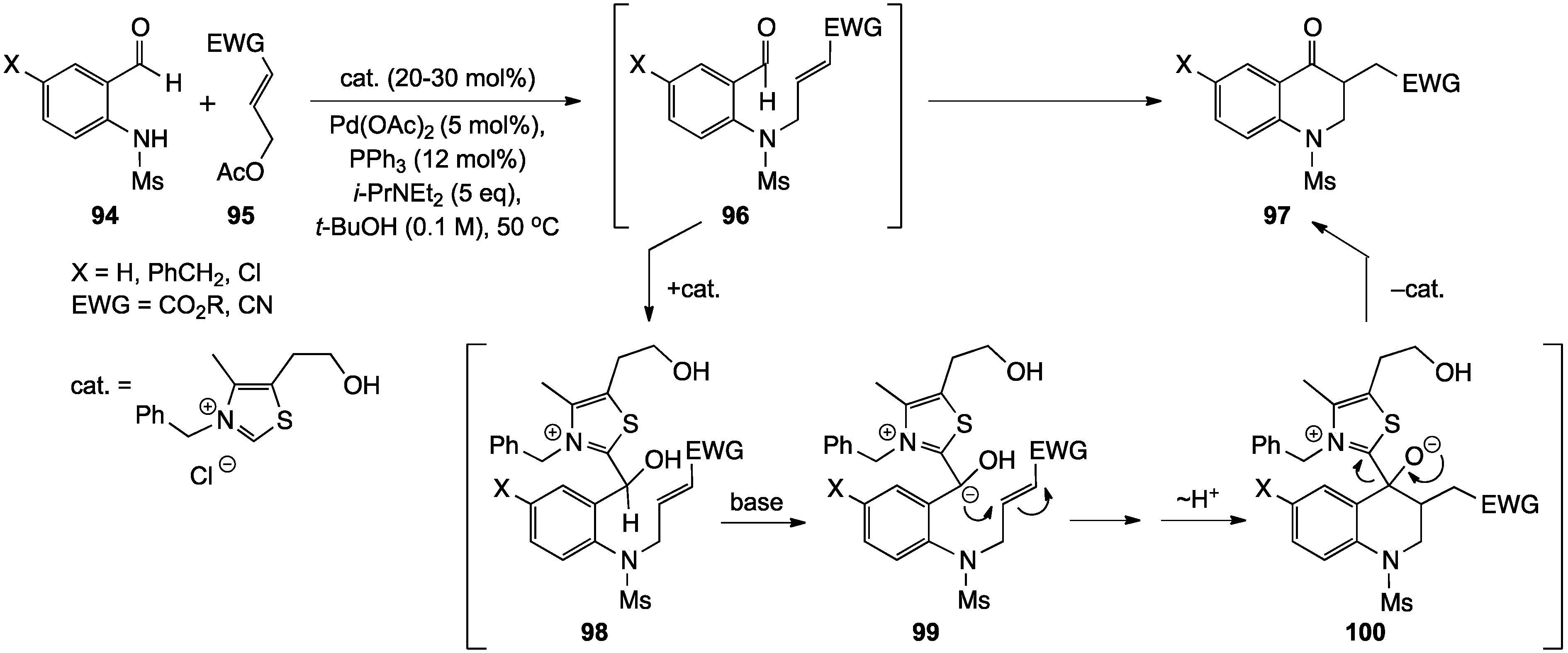
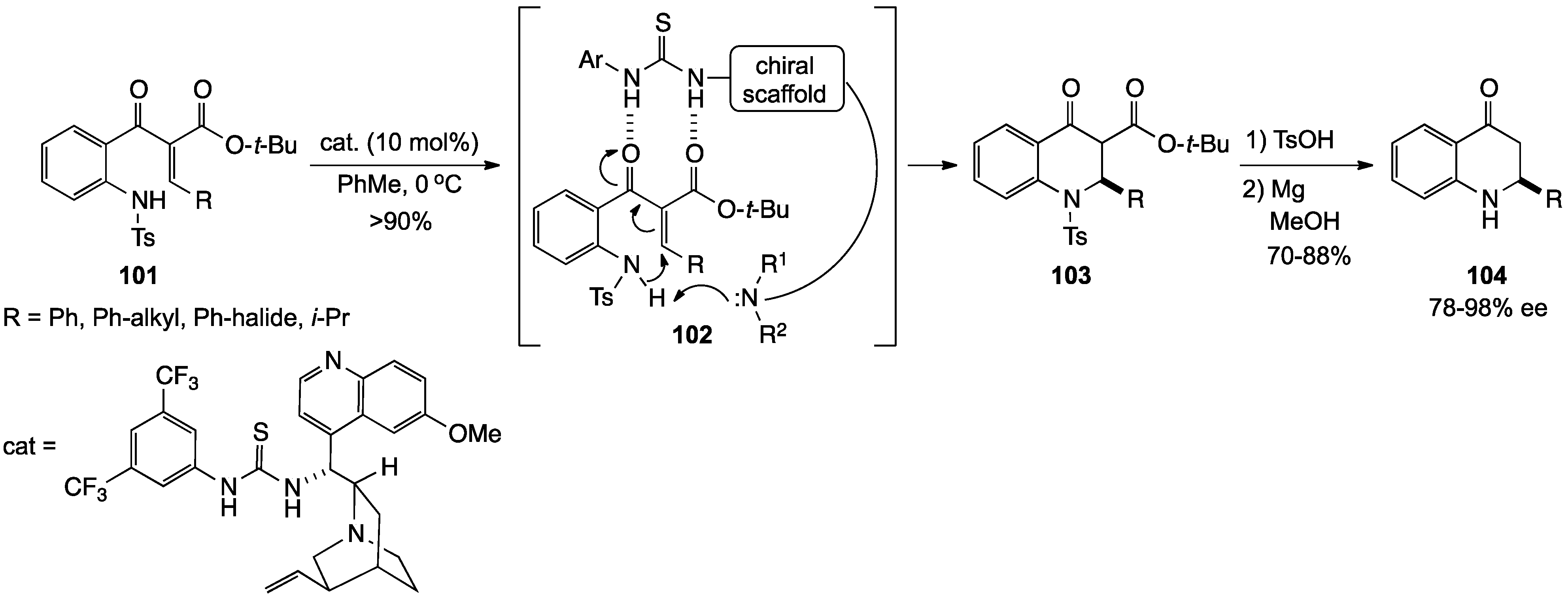
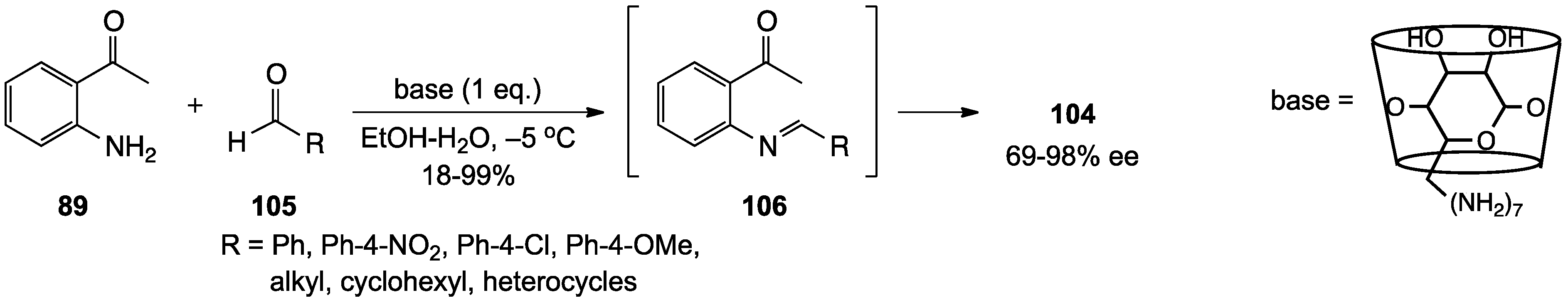
2.1. 1,2,3,4-Tetrahydroquinolines





















2.2. 2,3-Dihydro-4(1H)-quinolinones










2.3. 4(1H)-Quinolinones









3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Nicolaou, K.C.; Edmonds, D.J.; Bulger, P.G. Cascade reactions in total synthesis. Angew. Chem. Int. Ed. 2006, 45, 7134–7186. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Montagnon, T.; Snyder, S.A. Tandem reactions, cascade sequences, and biomimetic strategies in total synthesis. Chem. Commun. 2003, 551–564. [Google Scholar] [CrossRef]
- Trost, B.M. Atom economy—a challenge for organic-synthesis: Homogeneous catalysis leads the way. Angew. Chem. Int. Ed. 1995, 34, 259–281. [Google Scholar] [CrossRef]
- Trost, B.M. The atom economy–a search for synthetic efficiency. Science 1991, 254, 1471–1477. [Google Scholar]
- Katritzky, A.R.; Rachwal, S.; Rachwal, B. Recent progress in the synthesis of 1,2,3,4-tetrahydroquinolines. Tetrahedron 1996, 52, 15031–15070. [Google Scholar] [CrossRef]
- Sridharan, V.; Suryavanshi, P.A.; Menéndez, J.C. Advances in the chemistry of tetrahydroquinolines. Chem. Rev. 2011, 111, 7157–7259. [Google Scholar] [CrossRef]
- Barluenga, J.; Rodriguez, F.; Fananas, F.J. Recent advances in the synthesis of indole and quinoline derivatives through cascade reactions. Chem.-Asian J. 2009, 4, 1036–1048. [Google Scholar] [CrossRef]
- Bunce, R.A.; Herron, D.M.; Johnson, L.B.; Kotturi, S.V. Diastereoselective synthesis of substituted tetrahydroquinoline-4-carboxylic esters by a tandem reduction−reductive amination reaction. J. Org. Chem. 2001, 66, 2822–2827. [Google Scholar] [CrossRef]
- Fujita, K.; Yamamoto, K.; Yamaguchi, R. Oxidative cyclization of amino alcohols catalyzed by a Cp*Ir complex. Synthesis of indoles, 1,2,3,4-tetrahydroquinolines, and 2,3,4,5-tetrahydro-1-benzazepine. Org. Lett. 2002, 4, 2691–2694. [Google Scholar] [CrossRef]
- Bunce, R.A.; Nago, T.; Sonobe, N.; Slaughter, L.M. Benzo-fused heterocycles and carbocycles by intramolecular SNAr and tandem SN2-SNAr reactions. J. Heterocycl. Chem. 2008, 45, 551–557. [Google Scholar] [CrossRef]
- Lange, J.; Bissember, A.C.; Banwell, M.G.; Cade, I.A. Synthesis of 2,3-dihydro-4(1H)-quinolones and the corresponding 4(1H)-quinolones via low-temperature Fries rearrangement of N-arylazetidin-2-ones. Aust. J. Chem. 2011, 64, 454–470. [Google Scholar]
- Gigant, N.; Gillaizeau, I. Construction of nitrogen-fused tetrahydroquinolines via a domino reaction. Org. Lett. 2012, 14, 4622–4625. [Google Scholar] [CrossRef]
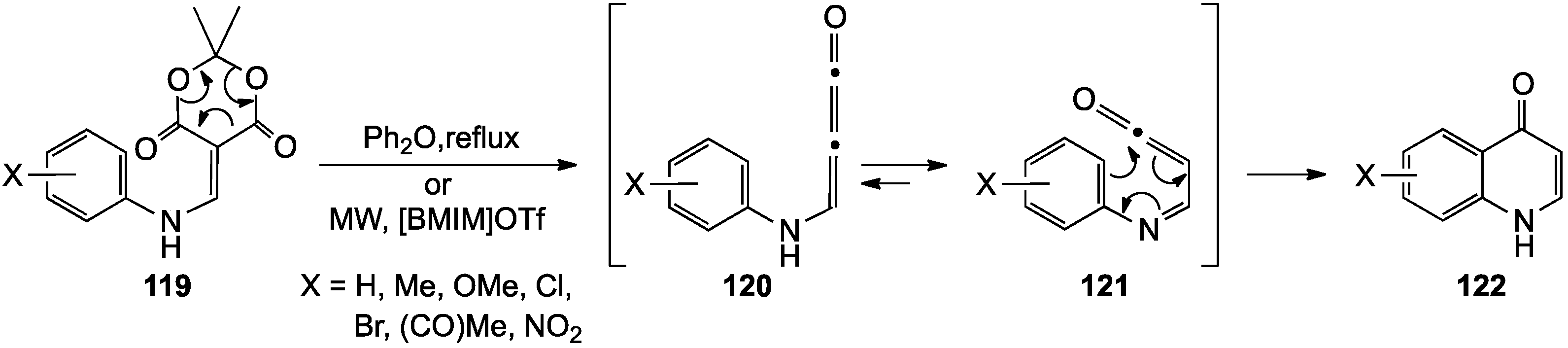
- Cassis, R.; Tapia, R.; Valderrama, J.A. Synthesis of 4(1H)-quinolones by thermolysis of arylaminomethylene Meldrum’s acid derivatives. Synth. Commun. 1985, 15, 125–133. [Google Scholar] [CrossRef]
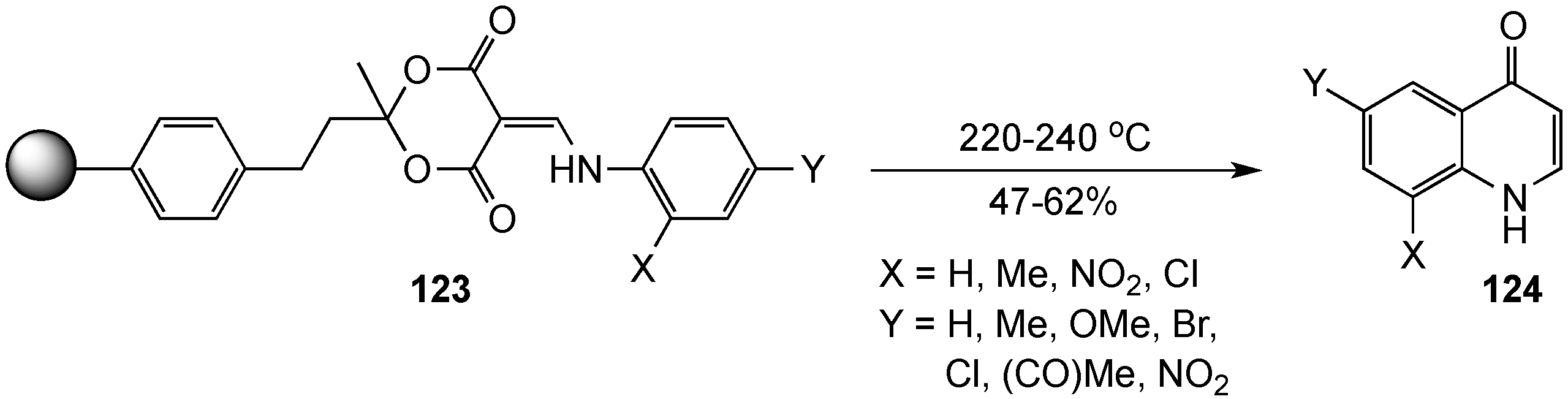
- Tang, J.; Huang, X. Preparation of resin-bound bismethylene cyclic malonic acid ester and facile solid-phase synthesis of 2-alkylthio-4(1H)-quinolone and 2-alkyl-4(1H)-quinolone. Synth. Commun. 2003, 33, 3953–3960. [Google Scholar] [CrossRef]
- Chen, B.; Huang, X.; Wang, J. A versatile synthesis of 2-alkyl and 2-aryl-4-quinolones. Synthesis 1987, 482–483. [Google Scholar] [CrossRef]
- Ueda, M.; Kawai, S.; Hayashi, M.; Naito, T.; Miyata, O. Efficient entry into 2-substituted tetrahydroquinoline systems through alkylative ring expansion: Stereoselective formal synthesis of (±)-martinellic acid. J. Org. Chem. 2010, 75, 914–921. [Google Scholar] [CrossRef]
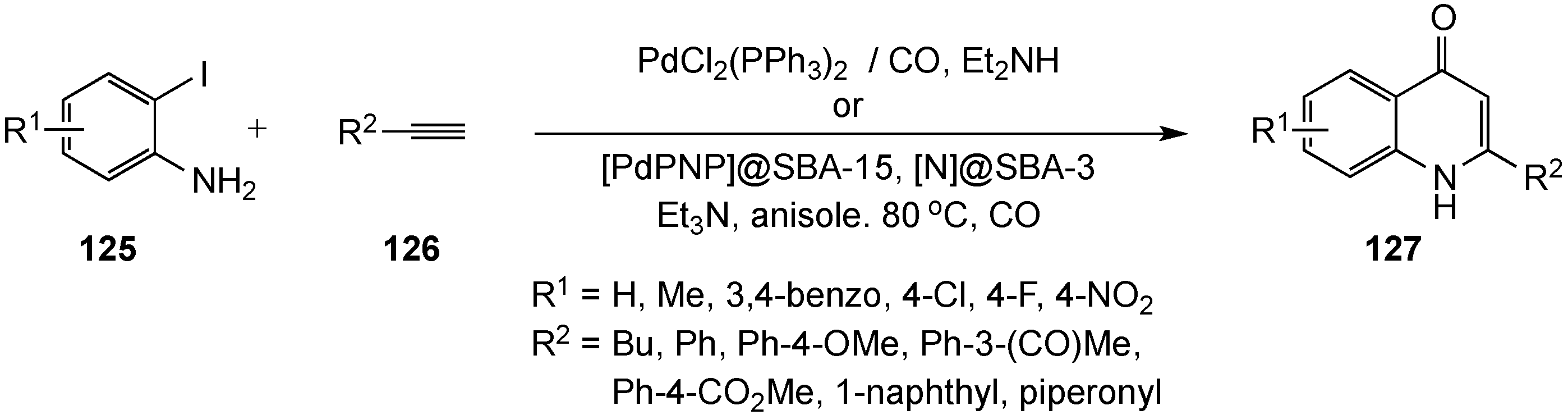
- Kalinin, V.N.; Shostakovsky, M.V.; Ponomaryov, A.B. A new route to 2-aryl-4-quinolones via palladium-catalyzed carbonylative coupling of o-iodoanilines with terminal arylacetylenes. Tetrahedron Lett. 1992, 33, 373–376. [Google Scholar] [CrossRef]
- Ward, T.R.; Turunen, B.J.; Haack, T.; Neuenswander, B.; Shadrick, W.; Georg, G.I. Synthesis of a quinolone library from ynones. Tetrahedron Lett. 2009, 50, 6494–6497. [Google Scholar] [CrossRef]
- Perry, N.B.; Blunt, J.W.; Munro, M.H.G.; Higa, T.; Sakai, R. Discorhabdin-D, an antitumor alkaloid from the sponges Latrunculia brevis and Prianos sp. J. Org. Chem. 1988, 53, 4127–4128. [Google Scholar] [CrossRef]
- Perry, N.B.; Blunt, J.W.; Munro, M.H.G. Cytotoxic pigments from New Zealand sponges of the genus Latrunculia–discorhabdin-A, discorhabdin-B and discorhabdin-C. Tetrahedron 1988, 44, 1727–1734. [Google Scholar] [CrossRef]
- Perry, N.B.; Blunt, J.W.; McCombs, J.D.; Munro, M.H.G. Discorhabdin-C, a highly cytotoxic pigment from a sponge of the genus Latrunculia. J. Org. Chem. 1986, 51, 5476–5478. [Google Scholar] [CrossRef]
- Nishiyama, S.; Cheng, J.F.; Tao, X.L.; Yamamura, S. Synthetic studies on novel sulfur-containing alkaloids, prianosins and discorhabdins—Total synthesis of discorhabdin-C. Tetrahedron Lett. 1991, 32, 4151–4154. [Google Scholar] [CrossRef]
- Asolkar, R.N.; Schroeder, D.; Heckmann, R.; Lang, S.; Wagner-Doebler, I.; Laatsch, H. Marine bacteria XXVII. Helquinoline, a new tetrahydroquinoline antibiotic from Janibacter limosus Hel 1. J. Antibiot. 2004, 57, 17–23. [Google Scholar] [CrossRef]
- Satyanarayana, G.; Pflaesterer, D.; Helmchen, G. Enantioselective syntheses of tetrahydroquinolines based on iridium-catalyzed allylic substitutions: Total syntheses of (+)-angustureine and (−)-cuspareine. Eur. J. Org. Chem. 2011, 6877–6886. [Google Scholar] [CrossRef] [Green Version]
- Staub, G.M.; Gloer, J.B.; Wicklow, D.T.; Dowd, P.F. Aspernomine: A cytotoxic antiinsectan metabolite with a novel ring system from the sclerotia of Aspergillus nomius. J. Am. Chem. Soc. 1992, 114, 1015–1017. [Google Scholar] [CrossRef]
- Magomedov, N.A. Efficient construction of cyclopenta[b]quinoline core of isoschizozygane alkaloids via intramolecular formal hetero-Diels-Alder reaction. Org. Lett. 2003, 5, 2509–2512. [Google Scholar] [CrossRef]
- Snider, B.B.; Ahn, Y.; O’Hare, S.M. Total synthesis of (±)-martinellic acid. Org. Lett. 2001, 3, 4217–4220. [Google Scholar] [CrossRef]
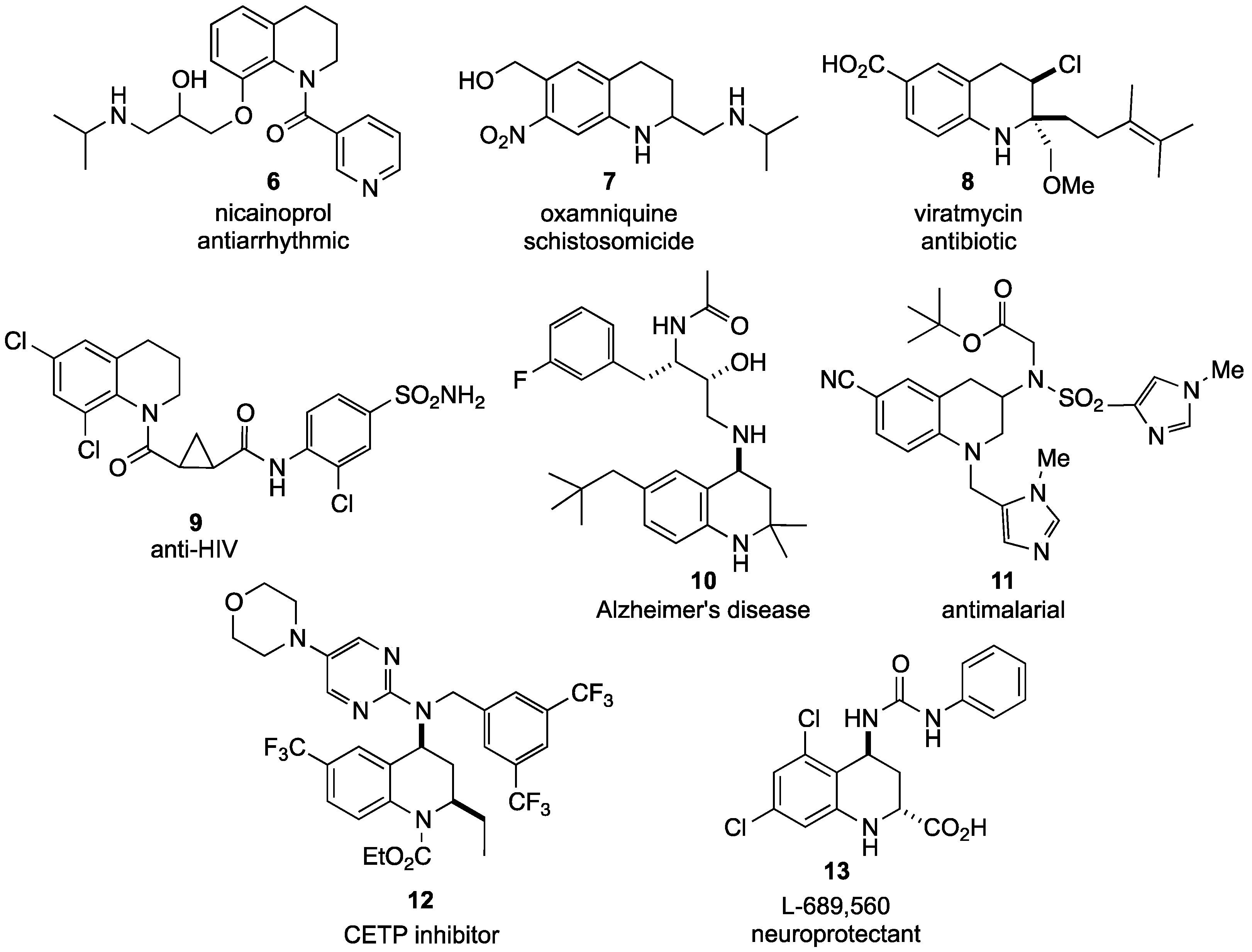
- Imanishi, S.; Kimura, T.; Arita, M. Nicainoprol. Cardiovasc. Drug Rev. 1991, 9, 223–236. [Google Scholar] [CrossRef]
- Galdino, D.R.P.M.; Barreto, D.M.R.M.J.; Galdino, S.L. The evolution of drugs on schistosoma treatment: Looking to the past to improve the future. Mini-Rev. Med. Chem. 2013, 13, 493–508. [Google Scholar] [CrossRef]
- Keck, D.; Vanderheiden, S.; Brase, S. A formal total synthesis of virantmycin: A modular approach towards tetrahydroquinoline natural products. Eur. J. Org. Chem. 2006, 2006, 4916–4923. [Google Scholar]
- Ori, M.; Toda, N.; Takami, K.; Tago, K.; Kogen, H. Stereospecific synthesis of 2,2,3-trisubstituted tetrahydroquinolines: Application to the total syntheses of benzastatin E and natural virantmycin. Tetrahedron 2005, 61, 2075–2104. [Google Scholar] [CrossRef]
- Steinhagen, H.; Corey, E.J. A Simple convergent approach to the synthesis of the antiviral agent virantmycin. Org. Lett. 1999, 1, 823–824. [Google Scholar] [CrossRef]
- Back, T.G.; Wulff, J.E. A stereodivergent synthesis of virantmycin by an enzyme-mediated diester desymmetrization and a highly hindered aryl amination. Angew. Chem. Int. Ed. 2004, 43, 6493–6496. [Google Scholar] [CrossRef]
- Su, D.-S.; Lim, J.J.; Tinney, E.; Wan, B.-L.; Young, M.B.; Anderson, K.D.; Rudd, D.; Munshi, V.; Bahnck, C.; Felock, P.J.; et al. Substituted tetrahydroquinolines as potent allosteric inhibitors of reverse transcriptase and its key mutants. Bioorg. Med. Chem. Lett. 2009, 19, 5119–5123. [Google Scholar] [CrossRef]
- Zhong, W.; Hitchcock, S.; Albrecht, B.K.; Bartberger, M.; Brown, J.; Brown, R.; Chaffee, S.C.; Cheng, Y.; Croghan, M.; Graceffa, R.; et al. Preparation of 2-Hydroxy-1,3-diaminoalkanes including Spiro Substituted Chroman Derivatives as β-Secretase Modulators and their use for Treatment Alzheimer’s Disease and Related Conditions. WO2007061670A1, 2007. Chem. Abstr. 2007, 147, 52808. [Google Scholar]
- Guo, T.; Gu, H.; Hobbs, D.W.; Rokosz, L.L.; Stauffer, T.M.; Jacob, B.; Clader, J.W. Design, synthesis, and evaluation of tetrahydroquinoline and pyrrolidine sulfonamide carbamates as γ-secretase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 3010–3013. [Google Scholar] [CrossRef]
- Asberom, T.; Bara, T.A.; Clader, J.W.; Greenlee, W.J.; Guzik, H.S.; Josien, H.B.; Li, W.; Parker, E.M.; Pissarnitski, D.A.; Song, L.; et al. Tetrahydroquinoline sulfonamides as γ-secretase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 205–207. [Google Scholar] [CrossRef]
- Nallan, L.; Bauer, K.D.; Bendale, P.; Rivas, K.; Yokoyama, K.; Horney, C.P.; Rao, P.P.; Floyd, D.; Lombardo, L.J.; Williams, D.K.; et al. Protein farnesyltransferase inhibitors exhibit potent antimalarial activity. J. Med. Chem. 2005, 48, 3704–3713. [Google Scholar] [CrossRef]
- Kubota, H.; Sugahara, M.; Furukawa, M.; Takano, M.; Motomura, D. Tetrahydroquinoline Derivatives as Cholesteryl Ester Transfer Protein Inhibitors and a Process for Preparing the Same. US20070082896A1, 2007. Chem. Abstr. 2007, 146, 500906. [Google Scholar]
- Escribano, A.; Mateo, A.I.; Martin de la Nava, E.M.; Mayhugh, D.R.; Cockerham, S.L.; Beyer, T.P.; Schmidt, R.J.; Cao, G.; Zhang, Y.; Jones, T.M.; et al. Design and synthesis of new tetrahydroquinolines derivatives as CETP inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 3671–3675. [Google Scholar] [CrossRef]
- Leeson, P.D.; Carling, R.W.; Moore, K.W.; Moseley, A.M.; Smith, J.D.; Stevenson, G.; Chan, T.; Baker, R.; Foster, A.C.; Grimwood, S.; et al. 4-Amido-2-carboxytetrahydroquinolines. Structure-activity relationships for antagonism at the glycine site of the NMDA receptor. J. Med. Chem. 1992, 35, 1954–1968. [Google Scholar] [CrossRef]
- Ferranti, A.; Garuti, L.; Giovanninetti, G.; Gaggi, R.; Roncada, P.; Nardi, P. Preparation and analgesic activity of some tetrahydroquinolines and tetrahydroisoquinolines. Farmaco 1987, 42, 237–249. [Google Scholar]
- Carling, R.W.; Leeson, P.D.; Moseley, A.M.; Smith, J.D.; Saywell, K.; Tricklebank, M.D.; Kemp, J.A.; Marshall, G.R.; Foster, A.C.; Grimwood, S. Anticonvulsant activity of glycine-site NMDA antagonists. 2. Trans 2-carboxy-4-substituted tetrahydroquinolines. Bioorg. Med. Chem. Lett. 1993, 3, 65–70. [Google Scholar] [CrossRef]
- Smirnova, T.A.; Gavrilov, M.Y.; Nazmetdinov, F.Y.; Kolla, V.E.; Kon’shin, M.E. Synthesis and antidepressant activity of acylhydrazides of 2-chloro- and 2-anilino-5,6,7,8-tetrahydroquinoline-4-carboxylic acids. Pharm. Chem. J. 1999, 33, 370–371. [Google Scholar] [CrossRef]
- Oshiro, Y.; Sakurai, Y.; Sato, S.; Kurahashi, N.; Tanaka, T.; Kikuchi, T.; Tottori, K.; Uwahodo, Y.; Miwa, T.; Nishi, T. 3,4-Dihydro-2(1H)-quinolinone as a novel antidepressant drug: Synthesis and pharmacology of 1-[3-[4-(3-chlorophenyl)-1-piperazinyl]propyl]-3,4-dihydro-5-methoxy-2(1H)-quinolinone and its derivatives. J. Med. Chem. 2000, 43, 177–189. [Google Scholar] [CrossRef]
- Scott, J.D.; Miller, M.W.; Li, S.W.; Lin, S.-I.; Vaccaro, H.A.; Hong, L.; Mullins, D.E.; Guzzi, M.; Weinstein, J.; Hodgson, R.A.; et al. Tetrahydroquinoline sulfonamides as vasopressin 1b receptor anatgonists. Bioorg. Med. Chem. Lett. 2009, 19, 6018–6022. [Google Scholar] [CrossRef]
- Moon, M.W.; Morris, J.K.; Heier, R.F.; Chidester, C.G.; Hoffmann, W.E.; Piercey, M.F.; Althaus, J.S.; von Voigtlander, P.F.; Evans, D.L.; Figur, L.M.; et al. Dopaminergic and serotonergic activities of imidazoquinolinones and related compounds. J. Med. Chem. 1992, 35, 1076–1092. [Google Scholar] [CrossRef]
- Holsworth, D.D.; Cai, C.; Cheng, X.-M.; Cody, W.L.; Downing, D.M.; Erasga, N.; Lee, C.; Powell, N.A.; Edmunds, J.J.; Stier, M.; et al. Ketopiperazine-based renin inhibitors: Optimization of the “C” ring. Bioorg. Med. Chem. Lett. 2006, 16, 2500–2504. [Google Scholar] [CrossRef]
- Holsworth, D.D.; Jalaie, M.; Belliotti, T.; Cai, C.; Collard, W.; Ferreira, S.; Powell, N.A.; Stier, M.; Zhang, E.; McConnell, P.; et al. Discovery of 6-ethyl-2,4-diaminopyrimidine-based small molecule renin inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 3575–3580. [Google Scholar] [CrossRef]
- Powell, N.A.; Ciske, F.L.; Cai, C.; Holsworth, D.D.; Mennen, K.; van Huis, C.A.; Jalaie, M.; Day, J.; Mastronardi, M.; McConnell, P.; et al. Rational design of 6-(2,4-diaminopyrimidinyl)-1,4-benzoxazin-3-ones as small molecule renin inhibitors. Bioorg. Med. Chem. 2007, 15, 5912–5949. [Google Scholar] [CrossRef]
- Güller, R.; Binggeli, A.; Breu, V.; Bur, D.; Fischli, W.; Hirth, G.; Jenny, C.; Kansy, M.; Montavon, F.; Müller, M.; et al. Piperidine-renin inhibitors compounds with improved physicochemical properties. Bioorg. Med. Chem. Lett. 1999, 9, 1403–1408. [Google Scholar] [CrossRef]
- Baumgarth, M.; Lues, I.; Minck, K.-O.; Beier, N. Preparation of (Benzocyclylethyl)arylpiperidines and -Piperazines for Treating Arrhythmia and Tachycardia. DE4321366A1, 1995. Chem. Abstr. 1995, 122, 187620. [Google Scholar]
- Huang, X.; Brubaker, J.; Peterson, S.L.; Butcher, J.W.; Close, J.T.; Martinez, M.; Maccoss, R.N.; Jung, J.O.; Siliphaivanh, P.; Zhang, H.; et al. Preparation of Cycloalkyl-fused Tetrahydroquinolines as CRTH2 Receptor Modulators. WO2012174176A1, 2012. Chem. Abstr. 2013, 158, 104992. [Google Scholar]
- Sun, Y.; Zhang, Y.; Liu, M. Advanced process in research of quinoline-and related heterocycle-like antimalarial agents. Guowai Yiyao Kangshengsu Fence 2012, 33, 6–21, Chem. Abstr. 2012, 158, 690082. [Google Scholar]
- Wang, X.-F.; Wang, S.-B.; Ohkoshi, E.; Wang, L.-T.; Hamel, E.; Qian, K.; Morris-Natschke, S.L.; Lee, K.-H.; Xie, L. N-Aryl-6-methoxy-1,2,3,4-tetrahydroquinolines: A novel class of antitumor agents targeting the colchicine site on tubulin. Eur. J. Med. Chem. 2013, 67, 196–207. [Google Scholar] [CrossRef]
- Madacsi, R.; Kanizsai, I.; Feher, L.Z.; Gyuris, M.; Ozsvari, B.; Erdelyi, A.; Wolfling, J.; Puskas, L.G. Aromatic sulfonamides containing a condensed piperidine moiety as potential oxidative stress-inducing anticancer agents. Med. Chem. 2013, 9, 911–919. [Google Scholar] [CrossRef]
- Gutierrez, M.; Carmona, U.; Vallejos, G.; Astudillo, L. Antifungal activity of tetrahydroquinolines against some phytopathogenic fungi. Z. Naturforsch. C: J. Biosci. 2012, 67, 551–556. [Google Scholar] [CrossRef]
- Fonseca-Berzal, C.; Merchan, A.D.R.; Romero, B.A.R.; Escario, J.A.; Kouznetsov, V.V.; Gomez-Barrio, A. Selective activity of 2,4-diaryl-1,2,3,4-tetrahydroquinolines on Trypanosoma cruzi epimastigotes and amastigotes expressing β-galactosidase. Bioorg. Med. Chem. Lett. 2013, 23, 4851–4856. [Google Scholar] [CrossRef]
- Wallace, O.B. Preparation of tetrahydroquinolines as selective estrogen receptor modulators. WO2002094788A1, 2002. Chem. Abstr. 2002, 137, 384759. [Google Scholar]
- Kohno, Y.; Kojima, E. Preparation of 1,2,3,4-tetrahydroquinoline-4,8-dicarboxylates as drugs. EP403980A1, 1990. Chem. Abstr. 1991, 114, 207056. [Google Scholar]
- Kohno, Y.; Awano, K.; Ishizaki, T.; Kojima, E.; Kudoh, S.; Sakoe, Y.; Saito, K. Preparation of Tetrahydroquinolineacetic Acid Derivatives as Immunosuppressants. WO9218482A1, 1992. Chem. Abstr. 1993, 119, 117135. [Google Scholar]
- Feng, L.; Huang, M.; Liu, Y.; Wu, G.; Yan, S.; Yun, H.; Zhou, M. Preparation of Novel Tetrahydroquinoline Derivatives as AMP-activated Protein Kinase Activators. US20120190677A1, 2012. Chem. Abstr. 2012, 157, 294945. [Google Scholar]
- Bunce, R.A.; Schammerhorn, J.E.; Slaughter, L.M. Catalyst and pressure dependent reductive cyclizations for the diastereoselective synthesis of hexahydropyrrolo[1,2-a]quinoline-5-carboxylic esters. J. Heterocycl. Chem. 2006, 43, 1505–1511. [Google Scholar] [CrossRef]
- Bunce, R.A.; Herron, D.M.; Lewis, J.R.; Kotturi, S.V.; Holt, E.M. Diastereoselective synthesis of linear-fused tricyclic nitrogen heterocycles by a tandem reduction-reductive amination reaction. J. Heterocycl. Chem. 2003, 40, 101–106. [Google Scholar] [CrossRef]
- Bunce, R.A.; Schammerhorn, J.E.; Slaughter, L.M. (±)-2,3-Dialkyl-1,2,3,4-tetrahydroquinoline-3-carboxylic esters by a tandem reduction-reductive amination reaction. J. Heterocycl. Chem. 2007, 44, 1051–1057. [Google Scholar] [CrossRef]
- Bunce, R.A.; Nago, T.; Sonobe, N. (±)-2-Alkyl-1,2,3,4-tetrahydroquinoline-3-carboxylic esters by a catalyst and pressure dependent tandem reduction-reductive amination reaction. J. Heterocycl. Chem. 2007, 44, 1059–1064. [Google Scholar] [CrossRef]
- Bunce, R.A.; Herron, D.M.; Ackerman, M.L. Aryl-fused nitrogen heterocycles by a tandem reduction−Michael addition reaction. J. Org. Chem. 2000, 65, 2847–2850. [Google Scholar] [CrossRef]
- Bunce, R.A.; Nammalwar, B.; Slaughter, L.M. Divergent reactivity in tandem reduction–Michael ring closures of five- and six-membered cyclic enones. J. Heterocycl. Chem. 2009, 46, 854–860. [Google Scholar] [CrossRef]
- Bunce, R.A.; Nago, T. 6-Nitro-1,2,3,4-tetrahydroquinolines by a tandem reductive amination-SNAr reaction. J. Heterocycl. Chem. 2008, 45, 1155–1160. [Google Scholar] [CrossRef]
- Bunce, R.A.; Schammerhorn, J.E. 6-Nitro-1,2,3,4-tetrahydroquinoline-4-carboxylic Esters and 7-Nitro-3,4-dihydroquinoxaline-1(2H)-carboxylic Esters by a Tandem Reductive Amination-SNAr Reaction. Org. Prep. Proced. Int. 2010, 42, 71–82. [Google Scholar] [CrossRef]
- Bunce, R.A.; Lee, E.J. Ester- and ketone-substituted (±)-1-alkyl-6-nitro-1,2,3,4-tetrahydroquinolines by a tandem SNAr-Michael reaction. J. Heterocycl. Chem. 2010, 47, 1176–1182. [Google Scholar] [CrossRef]
- Patti, A.; Pedotti, S. Hydrogenation of ortho-nitrochalcones over Pd/C as a simple access to 2-substituted 1,2,3,4-tetrahydroquinolines. Tetrahedron 2010, 66, 5607–5611. [Google Scholar] [CrossRef]
- Liu, Y.; Wei, J.; Che, C.-M. [Fe(F20TPP)Cl] catalyzed intramolecular C-N bond formation for alkaloid synthesis using aryl azides as nitrogen source. Chem. Commun. 2010, 46, 6926–6928. [Google Scholar] [CrossRef]
- Söderberg, B.C.G.; Shriver, J.A.; Cooper, S.H.; Shrout, T.L.; Scott Helton, E.; Austin, L.R.; Odens, H.H.; Hearn, B.R.; Jones, P.C.; Kouadio, T.N.; et al. Intramolecular cyclization reactions of unsaturated amino Fischer chromium carbenes forming indoles and quinolines. Tetrahedron 2003, 59, 8775–8791. [Google Scholar] [CrossRef]
- Ortiz-Marciales, M.; Figueroa, D.; López, J.A.; de Jesús, M.; Vega, R. Steric and electronic effects on the reduction of O-silylated aromatic ketoximes with borane. Tetrahedron Lett. 2000, 41, 6567–6570. [Google Scholar] [CrossRef]
- Park, K.H.; Joo, H.S.; Ahn, K.I.; Jun, K. One step synthesis of 4-ethoxy-1,2,3,4-tetrahydroquinoline from nitroarene and ethanol: A TiO2 mediated photocatalytic reaction. Tetrahedron Lett. 1995, 36, 5943–5946. [Google Scholar] [CrossRef]
- Dhiman, R.; Sharma, S.; Singh, G.; Nepali, K.; Bedi, P.M.S. Design and synthesis of aza-flavones as a new class of xanthine oxidase inhibitors. Arch. Pharm. 2013, 346, 7–16. [Google Scholar] [CrossRef]
- Hradil, P.; Hlavac, J.; Soural, M.; Hajduch, M.; Kolar, M.; Vecerova, R. 3-Hydroxy-2-phenyl-4(1H)-quinolinones as promising biologically active compounds. Mini-Rev. Med. Chem. 2009, 9, 696–702. [Google Scholar] [CrossRef]
- Gill, N.S.; Kaur, A.; Arora, R.; Dhawan, V.; Bali, M. Synthetic studies of novel azaflavanone derivatives and its biological activities. Curr. Res. Chem. 2012, 4, 88–98. [Google Scholar] [CrossRef]
- Chandrasekhar, S.; Pushpavalli, S.N.C.V.L.; Chatla, S.; Mukhopadhyay, D.; Ganganna, B.; Vijeender, K.; Srihari, P.; Reddy, C.R.; Janaki, R.M.; Bhadra, U. Aza-flavanones as potent cross-species microRNA inhibitors that arrest cell cycle. Bioorg. Med. Chem. Lett. 2012, 22, 645–648. [Google Scholar] [CrossRef]
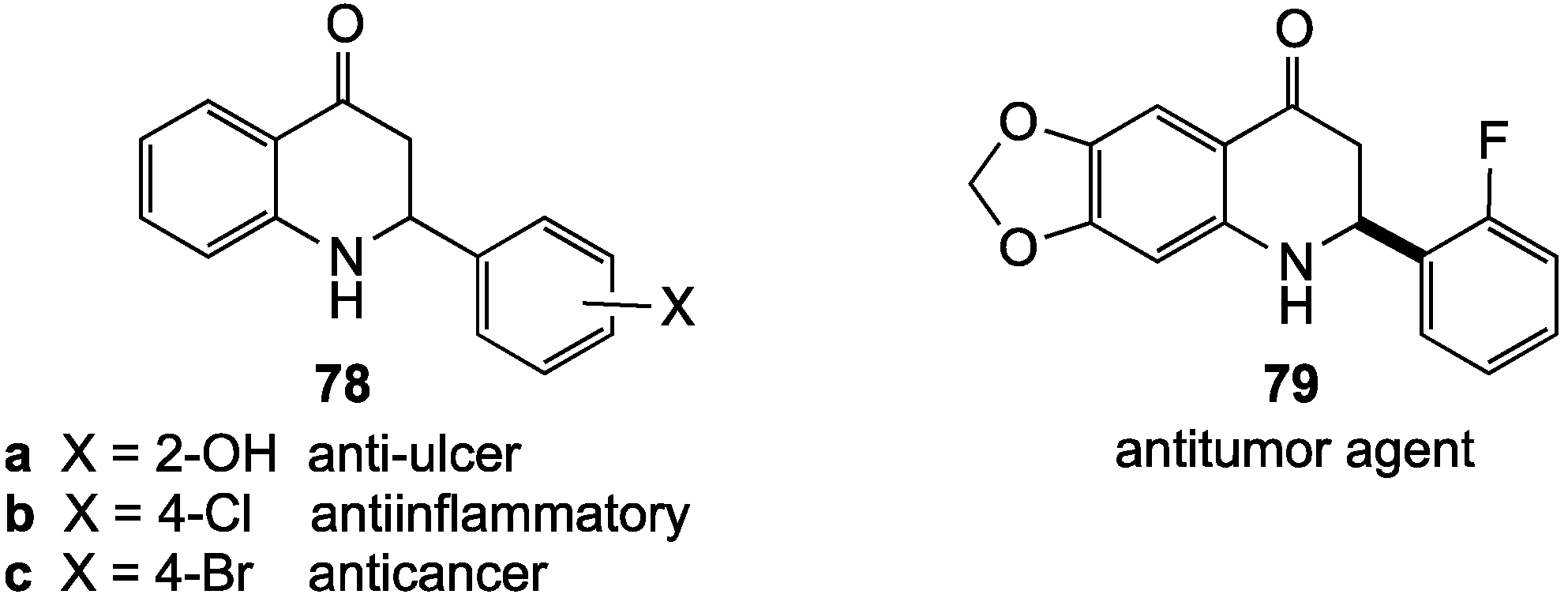
- Xia, Y.; Yang, Z.-Y.; Xia, P.; Bastow, K.F.; Tachibana, Y.; Kuo, S.-C.; Hamel, E.; Hackl, T.; Lee, K.-H. Antitumor agents. 181. Synthesis and biological evaluation of 6,7,2',3',4'-substituted-1,2,3,4-tetrahydro-2-phenyl-4-quinolones as a new class of antimitotic antitumor agents. J. Med. Chem. 1998, 41, 1155–1162. [Google Scholar] [CrossRef]
- Stern, E.; Muccioli, G.G.; Bosier, B.; Hamtiaux, L.; Millet, R.; Poupaert, J.H.; Hénichart, J.-P.; Depreux, P.; Goossens, J.-F.; Lambert, D.M. Pharmacomodulations around the 4-oxo-1,4-dihydroquinoline-3-carboxamides, a class of potent CB2-selective cannabinoid receptor ligands: Consequences in receptor affinity and functionality. J. Med. Chem. 2007, 50, 5471–5484. [Google Scholar] [CrossRef]
- Rasetti, V.; Rueeger, H.; Maibaum, J.K.; Mah, R.; Gruetter, M.; Cohen, N.C. Preparation of 2,9-Diamino- and 2-Amino-8-carbamoyl-4-hydroxyalkanoic Acid Amides as Renin Inhibitors. EP702004A2, 1996. Chem. Abstr. 1996, 125, 10631. [Google Scholar]
- Ibrahim, M.M.; Deng, H.; Zvonok, A.; Cockayne, D.A.; Kwan, J.; Mata, H.P.; Vanderah, T.W.; Lai, J.; Porreca, F.; Makriyannis, A.; Malan, T.P. Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: Pain inhibition by receptors not present in the CNS. Proc. Natl. Acad. Sci. USA 2003, 100, 10529–10533. [Google Scholar] [CrossRef]
- Mines, M.A.; Beurel, E.; Jope, R.S. Regulation of cell survival mechanisms in Alzheimer’s disease by glycogen synthase kinase-3. Int. J. Alzheimers Dis. 2011, 861072. [Google Scholar] [CrossRef]
- Cociorva, O.; Li, B.; Szardenings, K.; Fukuda, Y.; Nomura, M.; Seto, S.; Yumoto, K.; Okada, K.; Nakamura, A. Preparation of Aminoquinolones as GSK-3 Inhibitors. WO2007106537A2, 2007. Chem. Abstr. 2007, 147, 385853. [Google Scholar]
- Yamada, M.; Maehara, A.; Kawai, Y. Preparation of 1,2,3,4-Tetrahydro-4-oxo-3-quinolinecarboxylic Acid Esters and 2,3-Dihydro-4(1H)-quinolinones. JP2006083079A, 2006. Chem. Abstr. 2006, 144, 331280. [Google Scholar]
- Bunce, R.A.; Nago, T. 1-Alkyl-2,3-dihydro-4(1H)-quinolinones by a tandem Michael-SNAr annulation reaction. J. Heterocycl. Chem. 2009, 46, 623–628. [Google Scholar] [CrossRef]
- Bunce, R.A.; Schammerhorn, J.E.; Sigle, J. Substituted 4-oxo-1,2,3,4-tetrahydroquinoline-3-carboxylic esters by a tandem imine addition-SNAr reaction. J. Heterocycl. Chem. 2013, 50, 373–380. [Google Scholar] [CrossRef]
- Kano, S.; Ebata, T.; Shibuya, S. Formation of 2,3-dihydro-4(1H)-quinolones and related-compounds via Fries-type acid-catalyzed rearrangement of 1-arylazetidin-2-ones. J. Chem. Soc. Perk. Trans. 1 1980, 2105–2111. [Google Scholar] [CrossRef]
- Anderson, K.W.; Tepe, J.J. Trifluoromethanesulfonic acid catalyzed Friedel-Crafts acylation of aromatics with beta-lactams. Tetrahedron 2002, 58, 8475–8481. [Google Scholar] [CrossRef]
- Anderson, K.W.; Tepe, J.J. The first intermolecular Friedel-Crafts acylation with beta-lactams. Org. Lett. 2002, 4, 459–461. [Google Scholar] [CrossRef]
- Schmidt, R.G.; Bayburt, E.K.; Latshaw, S.P.; Koenig, J.R.; Daanen, J.F.; McDonald, H.A.; Bianchi, B.R.; Zhong, C.; Joshi, S.; Honore, P.; et al. Chroman and tetrahydroquinoline ureas as potent TRPV1 antagonists. Bioorg. Med. Chem. Lett. 2011, 21, 1338–1341. [Google Scholar] [CrossRef]
- Bunce, R.A.; Nammalwar, B. (±)-2-Aryl-2,3-dihydro-4(1H)-quinolinones by a tandem reduction–Michael addition reaction. J. Heterocycl. Chem. 2011, 48, 613–619. [Google Scholar]
- Chandrasekhar, S.; Vijeender, K.; Sridhar, C. L-Proline-catalyzed one-pot synthesis of 2-aryl-2,3-dihydroquinolin-4(1H)-ones. Tetrahedron Lett. 2007, 48, 4935–4937. [Google Scholar] [CrossRef]
- Nemoto, T.; Fukuda, T.; Hamada, Y. Efficient synthesis of 3-substituted 2,3-dihydro-4-quinolinones using a one-pot sequential multi-catalytic process: Pd-catalyzed allylic amination-thiazolium salt-catalyzed Stetter reaction cascade. Tetrahedron Lett. 2006, 47, 4365–4368. [Google Scholar] [CrossRef]
- Liu, X.; Lu, Y. Asymmetric synthesis of 2-aryl-2,3-dihydro-4-quinolones via bifunctional thiourea-mediated intramolecular cyclization. Org. Lett. 2010, 12, 5592–5595. [Google Scholar] [CrossRef]
- Kanagaraj, K.; Pitchumani, K. Per-6-amino-β-cyclodextrin as a chiral base catalyst promoting one-pot asymmetric synthesis of 2-aryl-2,3-dihydro-4-quinolones. J. Org. Chem. 2013, 78, 744–751. [Google Scholar] [CrossRef]
- Boteva, A.A.; Krasnykh, O.P. The methods of synthesis, modification, and biological activity of 4-quinolones (review). Chem. Heterocycl. Compd. 2009, 45, 757–785. [Google Scholar] [CrossRef]
- Basuri, T.S.; Modi, V.; Thakar, P.M. Quinolones in 2011: An update. J. Pharm. Res. 2011, 4, 1294–1297. [Google Scholar]
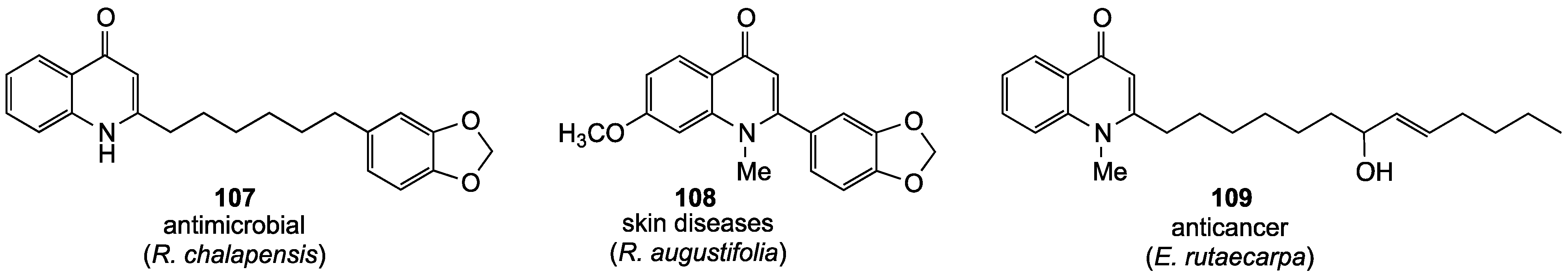
- El, S.K.; Al-Said, M.S.; El-Feraly, F.S.; Ross, S.A. New quinoline alkaloids from Ruta chalepensis. J. Nat. Prod. 2000, 63, 995–997. [Google Scholar] [CrossRef]
- Subehan; Takahashi, N.; Kadota, S.; Tezuka, Y. Cytochrome P450 2D6 inhibitory constituents of Lunasia amara. Phytochem. Lett. 2011, 4, 30–33. [Google Scholar] [CrossRef]
- Adams, M.; Kunert, O.; Haslinger, E.; Bauer, R. Inhibition of leukotriene biosynthesis by quinolone alkaloids from the fruits of Evodia rutaecarpa. Planta Med. 2004, 70, 904–908. [Google Scholar] [CrossRef]
- Adams, M.; Wube, A.A.; Bucar, F.; Bauer, R.; Kunert, O.; Haslinger, E. Quinolone alkaloids from Evodia rutaecarpa: A potent new group of antimycobacterial compounds. Int. J. Antimicrob. Agents 2005, 26, 262–264. [Google Scholar] [CrossRef]
- Huang, X.; Li, W.; Yang, X.-W. New cytotoxic quinolone alkaloids from fruits of Evodia rutaecarpa. Fitoterapia 2012, 83, 709–714. [Google Scholar] [CrossRef]
- Wang, X.-X.; Zan, K.; Shi, S.-P.; Zeng, K.-W.; Jiang, Y.; Guan, Y.; Xiao, C.-L.; Gao, H.-Y.; Wu, L.-J.; Tu, P.-F. Quinolone alkaloids with antibacterial and cytotoxic activities from the fruits of Evodia rutaecarpa. Fitoterapia 2013, 89, 1–7. [Google Scholar] [CrossRef]
- Tang, Y.; Ding, Y.; Liu, R.; Zhu, X. Progress in synthesis of quinolones as antibacterial agents. Yaoxue Jinzhan 2012, 36, 433–444, Chem. Abstr. 2013, 158, 246149. [Google Scholar]
- Nilsen, A.; LaCrue, A.N.; White, K.L.; Forquer, I.P.; Cross, R.M.; Marfurt, J.; Mather, M.W.; Delves, M.J.; Shackleford, D.M.; Saenz, F.E.; et al. Quinolone-3-diarylethers: A new class of antimalarial drug. Sci. Transl. Med. 2013, 5, 177ra37. [Google Scholar]
- Davies, R.V.; Robinson, K. Preparation of 1-alkyl-3-(alkylthio)-4-quinolones and analogs as antihypertensive agents. WO9102724A1, 1991. Chem. Abstr. 1991, 115, 92095. [Google Scholar]
- Mugnaini, C.; Pasquini, S.; Corelli, F. The 4-quinolone-3-carboxylic acid motif as a multivalent scaffold in medicinal chemistry. Curr. Med. Chem. 2009, 16, 1746–1767. [Google Scholar] [CrossRef]
- Wang, H.; Sun, Y.; Zhou, Y.; You, Q. Advances in privileged structure 4-quinolones as antitumor agents research. Zhongguo Yaowu Huaxue Zazhi 2012, 22, 59–67, Chem. Abstr. 2012, 157, 165392. [Google Scholar]
- Sato, M.; Kawakami, H.; Motomura, T.; Aramaki, H.; Matsuda, T.; Yamashita, M.; Ito, Y.; Matsuzaki, Y.; Yamataka, K.; Ikeda, S.; et al. Quinolone carboxylic acids as a novel monoketo acid class of human immunodeficiency virus type 1 integrase inhibitors. J. Med. Chem. 2009, 52, 4869–4882. [Google Scholar]
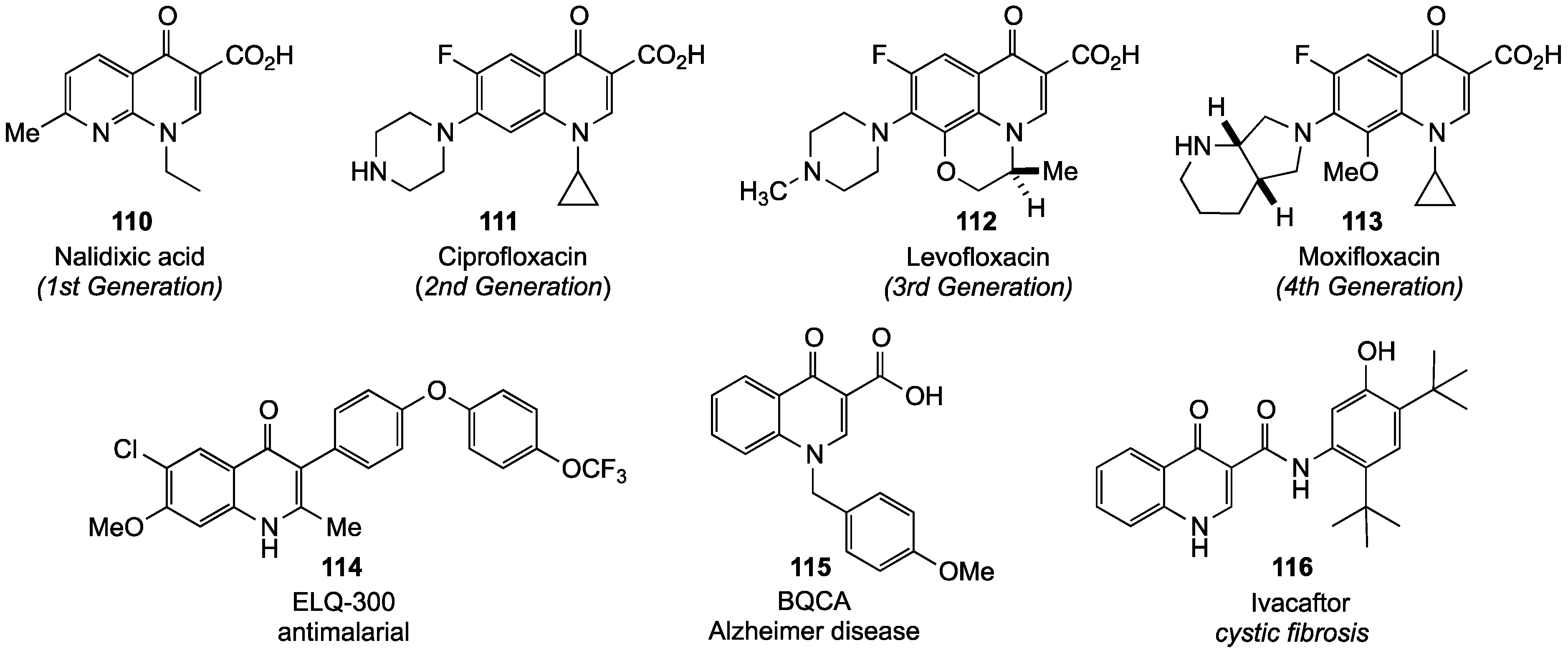
- Mistry, S.N.; Valant, C.; Sexton, P.M.; Capuano, B.; Christopoulos, A.; Scammells, P.J. Synthesis and pharmacological profiling of analogues of benzyl quinolone carboxylic acid (BQCA) as allosteric modulators of the M1 muscarinic receptor. J. Med. Chem. 2013, 56, 5151–5172. [Google Scholar] [CrossRef]
- Otsubo, K.; Yamauchi, T.; Ochi, Y. Preparation of Quinolone Compounds for Treating Neurological Diseases, Diseases Induced by Mitochondrial Dysfunction, Cardiovascular Disease, and Other Disorders. WO2010064735A1, 2010. Chem. Abstr. 2010, 153, 37063. [Google Scholar]
- Oliphant, C.M.; Green, G.M. Quinolones: A comprehensive review. Am. Fam. Physician 2002, 65, 455–464. [Google Scholar]
- Sanchez, J.P.; Domagala, J.M.; Hagen, S.E.; Heifetz, C.L.; Hutt, M.P.; Nichols, J.B.; Trehan, A.K. Quinolone antibacterial agents. Synthesis and structure-activity relationships of 8-substituted quinoline-3-carboxylic acids and 1,8-naphthyridine-3-carboxylic acids. J. Med. Chem. 1988, 31, 983–991. [Google Scholar] [CrossRef]
- Sharma, P.C.; Jain, A.; Jain, S.; Pahwa, R.; Yar, M.S. Ciprofloxacin: Review on developments in synthetic, analytical, and medicinal aspects. J. Enzyme Inhib. Med. Chem. 2010, 25, 577–589. [Google Scholar] [CrossRef]
- Hermecz, I. Recent development in the chemistry of bicyclic 6–6 systems containing one bridgehead nitrogen atom and one extra heteroatom and their benzologs: An update. Adv. Heterocycl. Chem. 2011, 104, 1–126. [Google Scholar]
- Barrett, J.F. Moxifloxacin (Bayer). Curr. Opin. Invest. Drugs 2000, 1, 45–51. [Google Scholar]
- Lees, P.; Shojaee, A.F. Rational dosing of antimicrobial drugs: Animals versus humans. Int. J. Antimicrob. Agents 2002, 19, 269–284. [Google Scholar] [CrossRef]
- Raju, S.V.; Rowe, S.M. Ivacaftor: CFTR potentiator treatment of cystic fibrosis. Drugs Future 2012, 37, 167–174. [Google Scholar]
- Schwarz, C. Drug therapy for cystic fibrosis. Arzneimitteltherapie 2013, 31, 80–88. [Google Scholar]
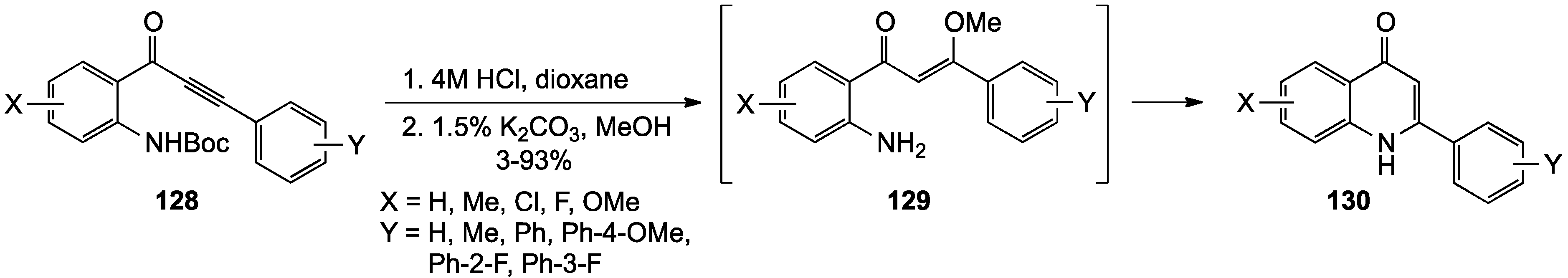
- Bunce, R.A.; Nammalwar, B. 4(1H)-Quinolinones by a tandem reduction-addition-elimination reaction. Org. Prep. Proced. Int. 2010, 42, 557–563. [Google Scholar] [CrossRef]
- Tois, J.; Vahermo, M.; Koskinen, A. Novel and convenient synthesis of 4(1H)-quinolones. Tetrahedron Lett. 2005, 46, 735–737. [Google Scholar] [CrossRef]
- Gordon, H.J.; Martin, J.C.; McNab, H. Thermal functionalization of nitrogen substituents: Formation of dihydropyrrol-3-ones, quinolin-4-ones, and enaminoenaminones by gas-phase hydrogen transfer reactions. J. Chem. Soc. Chem. Commun. 1983, 957–958. [Google Scholar] [CrossRef]
- Al-Awadi, N.A.; Abdelhamid, I.A.; Al-Etaibi, A.M.; Elnagdi, M.H. Gas-phase pyrolysis in organic synthesis: Rapid green synthesis of 4-quinolinones. Synlett 2007, 2205–2208. [Google Scholar] [CrossRef]
- Yadav, A.K.; Sharma, G.R.; Dhakad, P.; Yadav, T. A novel ionic liquid mediated synthesis of 4(1H)-quinolones, 5H-thiazolo[3,2-a]pyrimidin-5-one and 4H-pyrimido[2,1-b]benzothiazol-4-ones. Tetrahedron Lett. 2012, 53, 859–862. [Google Scholar] [CrossRef]
- Huang, X.; Liu, Z. Preparation of a resin-bound cyclic malonic ester and a facile solid-phase synthesis of 4(1H)quinolones. Tetrahedron Lett. 2001, 42, 7655–7657. [Google Scholar] [CrossRef]
- Huang, X.; Liu, Z. Solid-phase synthesis of 4(1H)-quinolone and pyrimidine derivatives based on a new scaffold polymer-bound cyclic malonic acid ester. J. Org. Chem. 2002, 67, 6731–6737. [Google Scholar] [CrossRef]
- Torii, S.; Okumoto, H.; Xu, L.H.; Sadakane, M.; Shostakovsky, M.V.; Ponomaryov, A.B.; Kalinin, V.N. Syntheses of chromones and quinolones via Pd-catalyzed carbonylation of o-iodophenols and anilines in the presence of acetylenes. Tetrahedron 1993, 49, 6773–6784. [Google Scholar] [CrossRef]
- Genelot, M.; Dufaud, V.; Djakovitch, L. Heterogeneous metallo-organocatalysis for the selective one-pot synthesis of 2-benzylidene-indoxyl and 2-phenyl-4-quinolone. Tetrahedron 2011, 67, 976–981. [Google Scholar] [CrossRef]
- Genelot, M.; Bendjeriou, A.; Dufaud, V.; Djakovitch, L. Optimized procedures for the one-pot selective syntheses of indoxyls and 4-quinolones by a carbonylative Sonogashira/cyclization sequence. Appl. Catal. A 2009, 369, 125–132. [Google Scholar] [CrossRef]
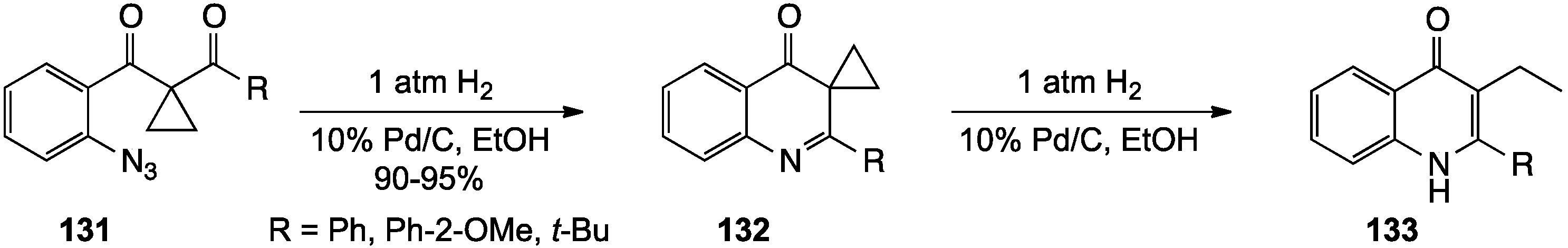
- Yang, W.; Xu, L.; Chen, Z.; Zhang, L.; Miao, M.; Ren, H. Ru-catalyzed synthesis of dihydrofuroquinolines from azido-cyclopropyl ketones. Org. Lett. 2013, 15, 1282–1285. [Google Scholar] [CrossRef]
- Skrzypek, L. Azinyl sulfides. XLIX. Synthesis of 4-amino-3-quinolinesulfonic acids and 4-aminoquinolines. Heterocycles 1998, 48, 71–78. [Google Scholar] [CrossRef]
© 2013 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nammalwar, B.; Bunce, R.A. Recent Syntheses of 1,2,3,4-Tetrahydroquinolines, 2,3-Dihydro-4(1H)-quinolinones and 4(1H)-Quinolinones using Domino Reactions. Molecules 2014, 19, 204-232. https://doi.org/10.3390/molecules19010204
Nammalwar B, Bunce RA. Recent Syntheses of 1,2,3,4-Tetrahydroquinolines, 2,3-Dihydro-4(1H)-quinolinones and 4(1H)-Quinolinones using Domino Reactions. Molecules. 2014; 19(1):204-232. https://doi.org/10.3390/molecules19010204
Chicago/Turabian StyleNammalwar, Baskar, and Richard A. Bunce. 2014. "Recent Syntheses of 1,2,3,4-Tetrahydroquinolines, 2,3-Dihydro-4(1H)-quinolinones and 4(1H)-Quinolinones using Domino Reactions" Molecules 19, no. 1: 204-232. https://doi.org/10.3390/molecules19010204