Development of a Supercritical Fluid Chromatography-Tandem Mass Spectrometry Method for the Determination of Azacitidine in Rat Plasma and Its Application to a Bioavailability Study

Abstract
:1. Introduction

2. Results and Discussion
2.1. Method Development
2.2. Method Validation


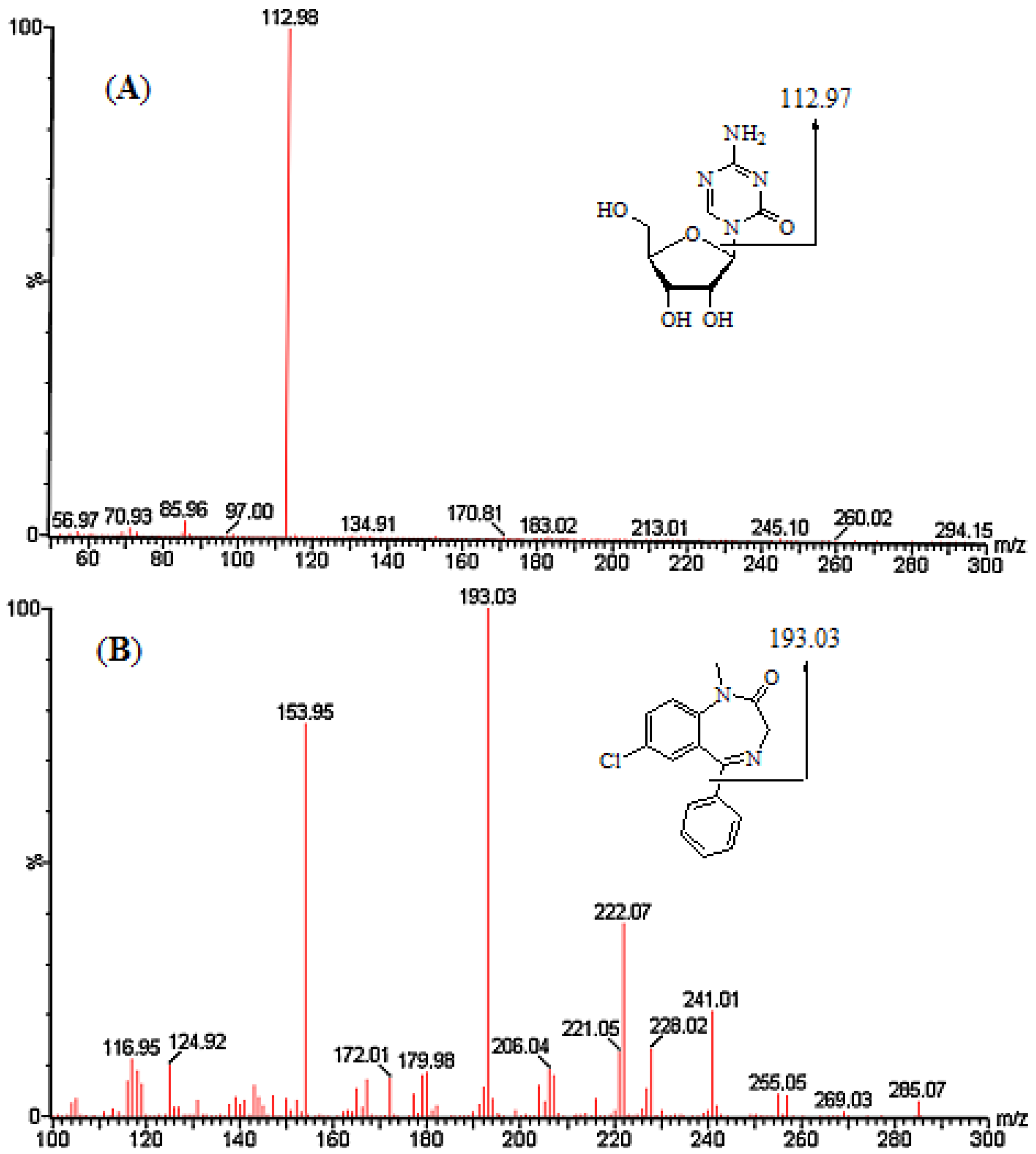{kind=link}
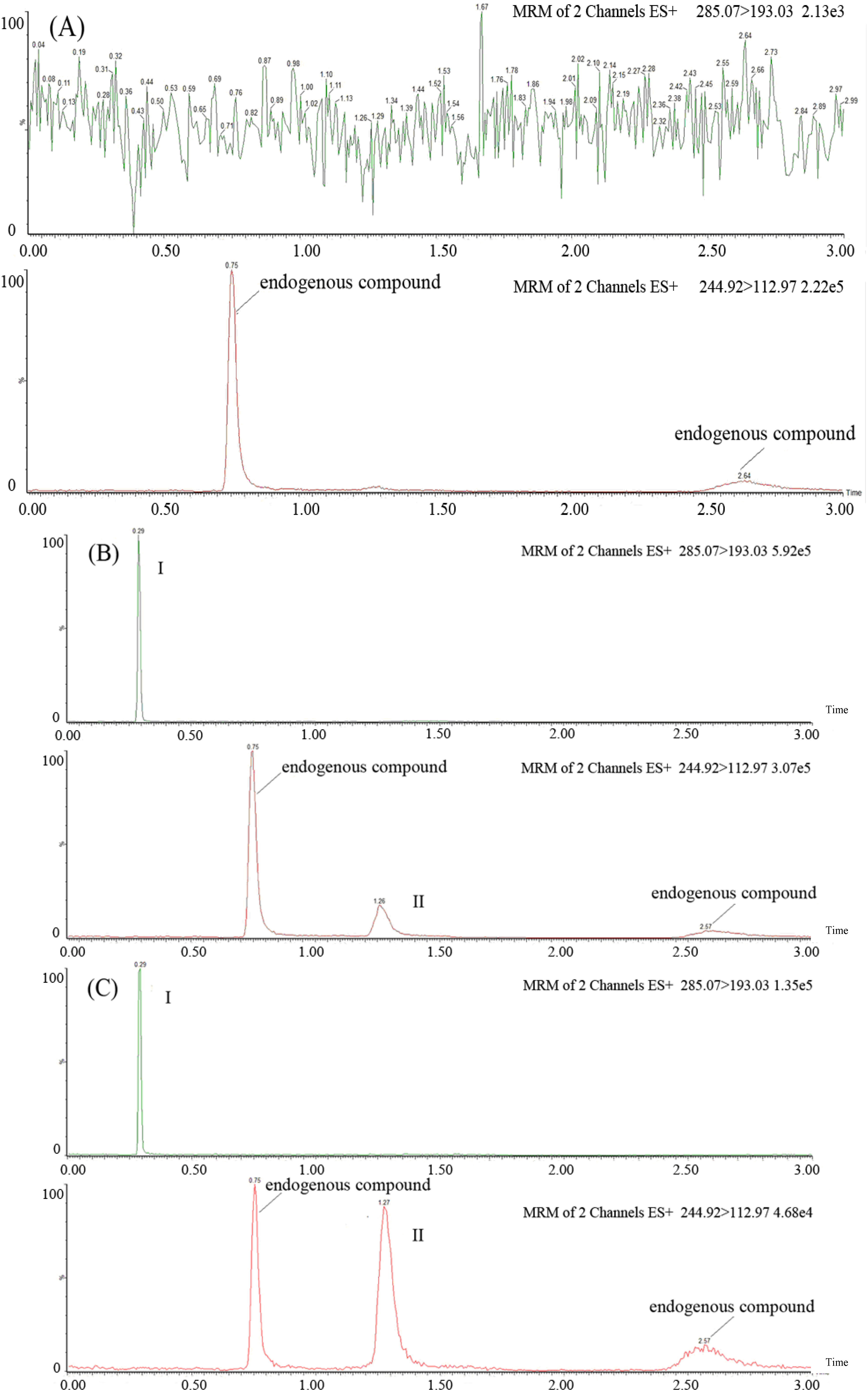{kind=link}
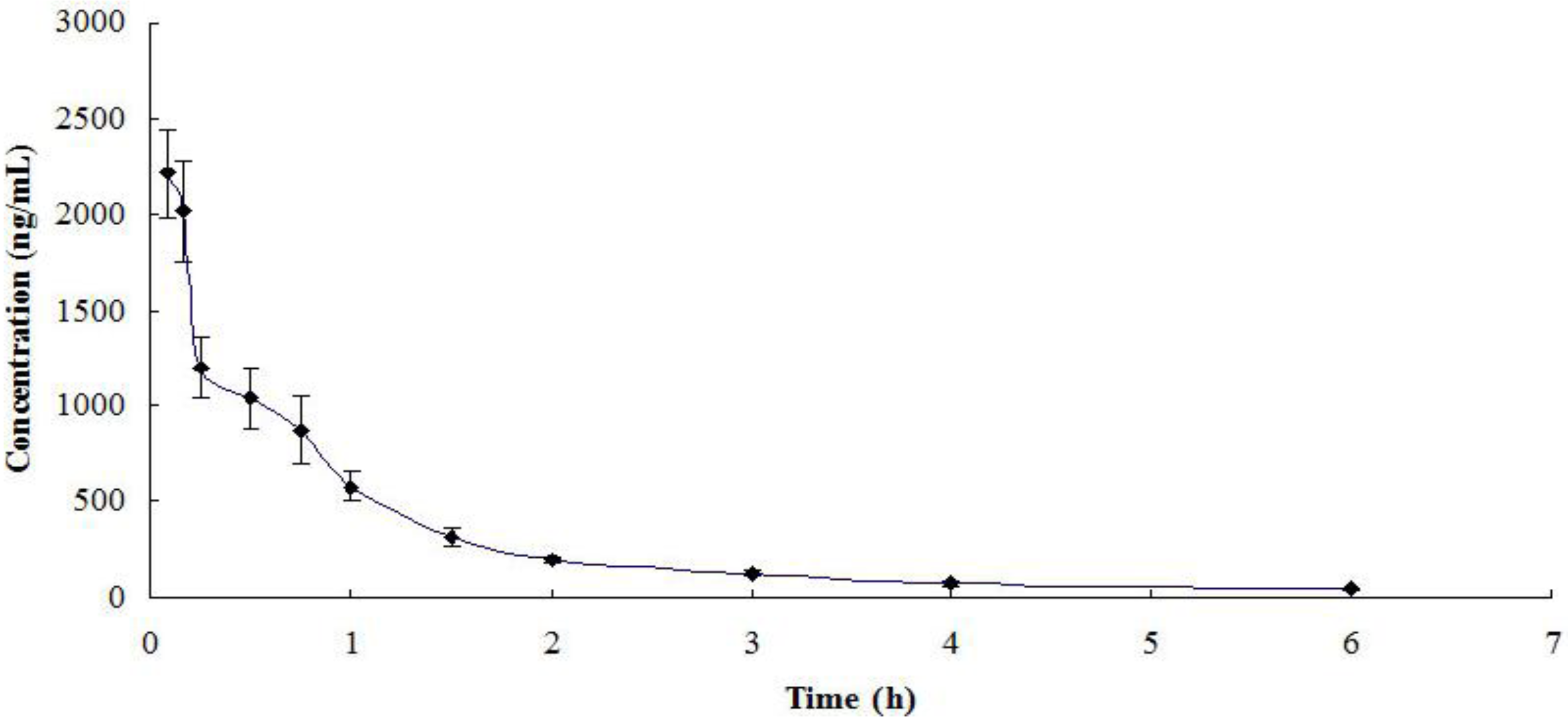{kind=link}
| Nominal Concentration (ng/mL) | Peak area | Matrix effect (%) | Recovery (%) | |||
|---|---|---|---|---|---|---|
| SET 1 | SET 2 | SET 3 | ||||
| Plasma azacitidine | 20 | 337 ± 21 | 270 ± 22 | 214 ± 9 | 80.3 | 79.2 |
| 2,000 | 53,115 ± 1,275 | 43,340 ± 2,915 | 36,507 ± 1,394 | 81.6 | 84.2 | |
| 10,000 | 256,686 ± 2,592 | 235,618 ± 13,942 | 202,066 ± 10,854 | 91.8 | 85.8 | |
| Diazepam (IS) | 2000 | 62,114 ± 1,497 | 55,308 ± 3,698 | 47,741 ± 2,797 | 89.0 | 86.3 |
| Concentration (ng/mL) | RSD (%) | RE (%) | ||
|---|---|---|---|---|
| Added | Found | Intra-day | Inter-day | |
| 20 | 18.6 | 5.9 | 18.3 | −7.0 |
| 2,000 | 2,074.5 | 6.8 | 10.6 | 3.7 |
| 10,000 | 10,014.8 | 8.8 | 9.0 | 0.2 |
| Storage conditions | Concentration (ng/mL) | RSD (%) | RE (%) | |
|---|---|---|---|---|
| Added | Found | |||
| Ambient, 2 h | 20 | 24.0 | 3.4 | 20.0 |
| 10,000 | 10,388.5 | 6.0 | 3.9 | |
| −80 °C, 30 days | 20 | 24.0 | 1.8 | 20.0 |
| 10,000 | 9,519.1 | 6.7 | −4.8 | |
| Three freeze–thaw cycles | 20 | 24.0 | 2.5 | 20.0 |
| 10,000 | 10,073.4 | 10.6 | 0.7 | |
| Sample rack for 24 h at 4 °C | 20 | 23.8 | 1.8 | 19.0 |
| 10,000 | 9,966.3 | 8.7 | −0.3 | |
2.3. Pharmacokinetic Application of the Method

| Parameters | Azacitidine |
|---|---|
| t1/2β (h) | 0.506 ± 0.198 |
| AUC 0-∞ (μg·h/mL) | 1996.0 ± 198.7 |
| Vd (L/kg) | 2.07 ± 0.17 |
| CL (L/h/kg) | 1.253 ± 0.085 |
3. Experimental
3.1. Chemicals and Reagents
3.2. Instrumentation
3.3. SFC/MS/MS Condition
3.4. Preparation of Standard and QC Samples
3.5. Plasma Sample Preparation
3.6. Method Validation
3.7. Pharmacokinetic (PK) Study in Rats
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Aul, C.; Gatterman, N.; Schneider, W. Age-related incidence and other epidemiological aspects of myelodysplastic syndromes. Br. J. Haematol. 1992, 82, 358–367. [Google Scholar] [CrossRef]
- Pui, C.H.; Robison, L.L.; Look, A.T. Acute lymphoblastic leukaemia. Lancet 2008, 371, 1030–1043. [Google Scholar] [CrossRef]
- Yoo, C.B.; Jones, P.A. Epigenetic therapy of cancer: Past, present and future. Nat. Rev. Drug Discov. 2006, 5, 37–50. [Google Scholar] [CrossRef]
- Anna, S.K.; Tadeusz, R. Hypomethylating agents in the treatment of myelodysplastic syndromes and myeloid leukemia. Curr. Cancer Drug Targets 2011, 11, 837–848. [Google Scholar] [CrossRef]
- Bryan, J.; Kantarjian, H.; Garcia-Manero, G.; Jabbour, E. Pharmacokinetic evaluation of decitabine for the treatment of leukemia. Expert Opin. Drug Metab. Toxicol. 2011, 7, 661–672. [Google Scholar] [CrossRef]
- Lin, K.T.; Momparler, R.L.; Rivard, G.E. High-performance liquid chromatographic analysis of chemical stability of 5-aza-2'-deoxycytidine. J. Pharm. Sci. 1981, 70, 1228–1232. [Google Scholar] [CrossRef]
- Notari, R.E.; Deyoung, J.L. Kinetics and mechanisms of degradation of the antileukemic agent 5-azacytidine in aqueous solutions. J. Pharm. Sci. 1975, 64, 1148–1157. [Google Scholar]
- Chan, K.K.; Giannini, D.D.; Staroscik, J.A.; Sadee, W. 5-azacytidine hydrolysis kinetics measured by high-pressure liquid chromatography and 13C-NMR spectroscopy. J. Pharm. Sci. 1979, 68, 807–812. [Google Scholar] [CrossRef]
- Pittillo, R.F.; Woolley, C. 5-Azacytidine: Microbiological assay in mouse blood. Appl. Microbiol. 1969, 18, 284–286. [Google Scholar]
- Weiss, A.J.; Stambaugh, J.E.; Mastrangelo, M.J.; Laucius, J.F.; Bellet, R.E. Phase I study of 5-azacytidine (NSC-102816). Cancer Chemother. Rep. 1972, 56, 413–419. [Google Scholar]
- Kissinger, L.D.; Stemm, N.L. Determination of the antileukemia angents cytarabine and azacitidine and their respective degradation products by high-performance liquid chromatography. J. Chromatogr. 1986, 353, 309–318. [Google Scholar] [CrossRef]
- Cheung, Y.W.; Vishnuvajjala, B.R.; Morris, N.L.; Flora, K.P. Stability of azacitidine in infusion fluids. Am. J. Hosp. Pharm. 1984, 41, 1156–1159. [Google Scholar]
- Rustum, A.M.; Hoffman, N.E. High-performance liquid chromatographic determination of 5-azacytidine in plasma. J. Chromatogr. B 1987, 421, 387–391. [Google Scholar] [CrossRef]
- Zhao, M.; Rudek, M.A.; He, P.; Hartke, C.; Gore, S.; Carducci, M.A.; Baker, S.D. Quantification of 5-azacytidine in plasma by electrospray tandem mass spectrometry coupled with high-performance liquid chromatography. J. Chromatogr. B 2004, 813, 81–88. [Google Scholar] [CrossRef]
- Sun, Q.; Olesik, S.V. Chiral separations performed by enhanced-fluidity liquid chromatography on a macrocyclic antibiotic chiral stationary phase. Anal. Chem. 1999, 71, 2139–2145. [Google Scholar] [CrossRef]
- Cashen, A.F.; Shah, A.K.; Todt, L.; Fisher, N.; DiPersio, J. Pharmacokinetics of decitabine administered as a 3-h infusion to patients with acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS). Cancer Chemother. Pharmacol. 2008, 61, 759–766. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds, azacitidine and diazepam are available from the authors.
© 2013 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, D.; Zhang, T.; Kou, L.; Zhang, Y.; Sun, J.; He, Z. Development of a Supercritical Fluid Chromatography-Tandem Mass Spectrometry Method for the Determination of Azacitidine in Rat Plasma and Its Application to a Bioavailability Study. Molecules 2014, 19, 342-351. https://doi.org/10.3390/molecules19010342
Li D, Zhang T, Kou L, Zhang Y, Sun J, He Z. Development of a Supercritical Fluid Chromatography-Tandem Mass Spectrometry Method for the Determination of Azacitidine in Rat Plasma and Its Application to a Bioavailability Study. Molecules. 2014; 19(1):342-351. https://doi.org/10.3390/molecules19010342
Chicago/Turabian StyleLi, Dongpo, Tianhong Zhang, Longfa Kou, Youxi Zhang, Jin Sun, and Zhonggui He. 2014. "Development of a Supercritical Fluid Chromatography-Tandem Mass Spectrometry Method for the Determination of Azacitidine in Rat Plasma and Its Application to a Bioavailability Study" Molecules 19, no. 1: 342-351. https://doi.org/10.3390/molecules19010342