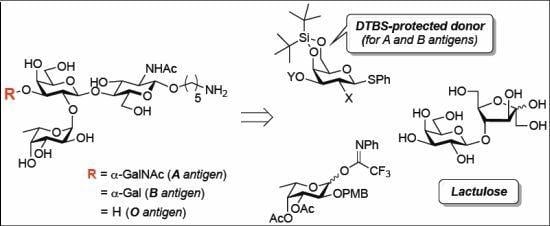
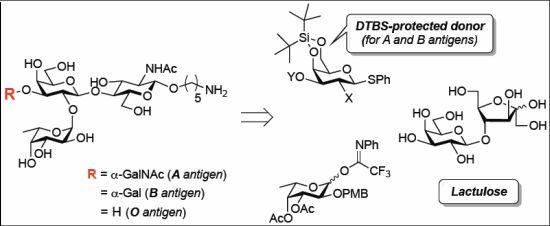
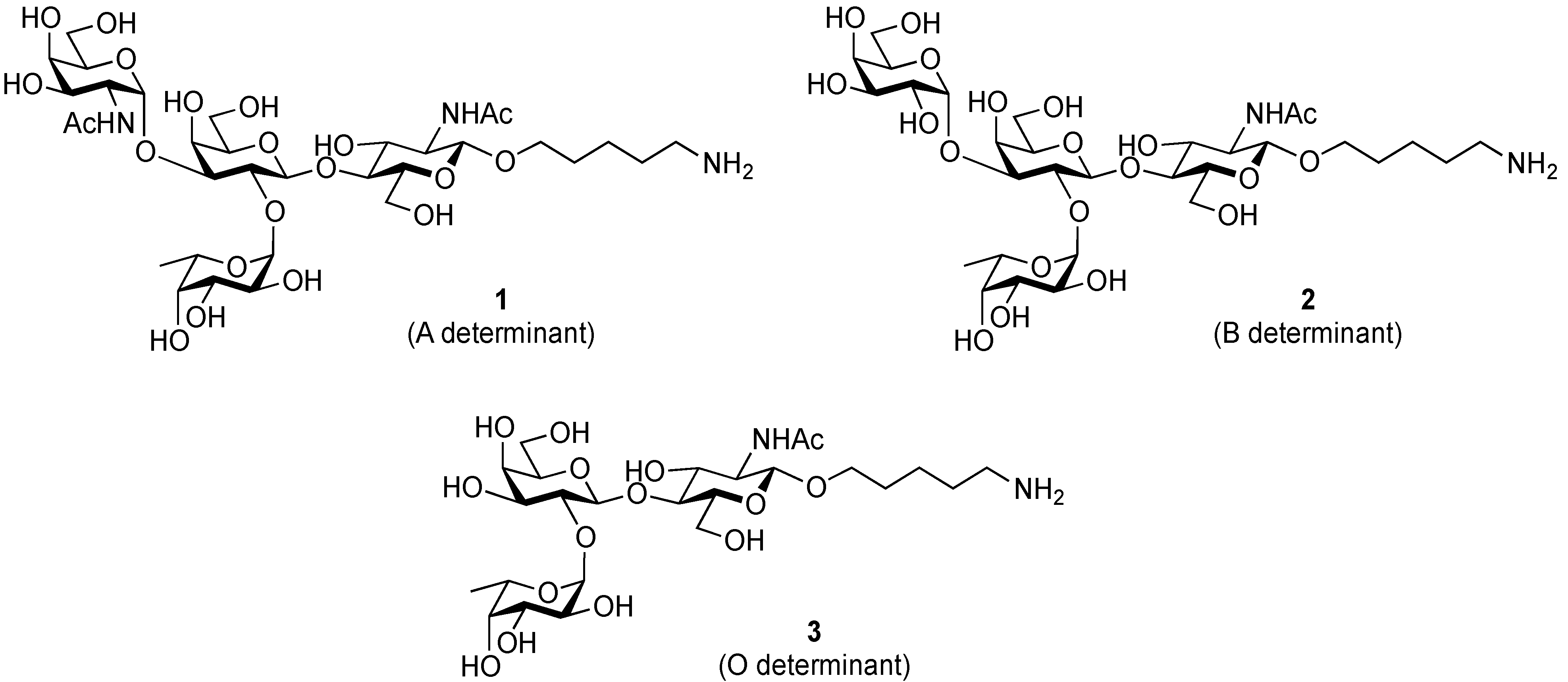
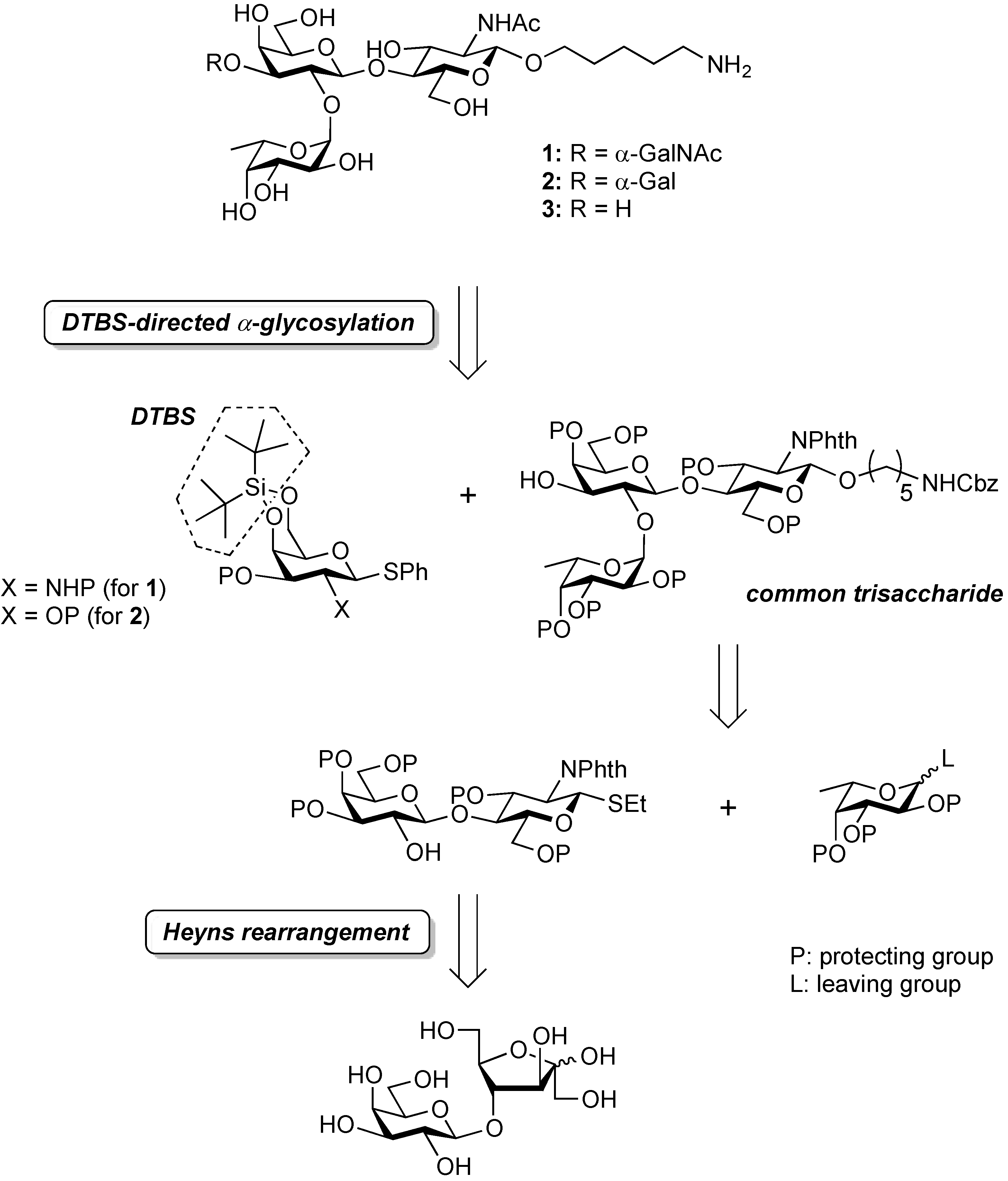
3.2. Physical Data for All New Compounds
Ethyl (2,3,4,6-tetra-O-acetyl-β-d-galactopyranosyl)-(1→4)-3,6-di-O-acetyl-2-deoxy-2-phthalimide-1-thio-β-d-glucopyranoside (
6)
. To a mixture of
5 (4.16 g, 5.44 mmol) in (CH
2Cl)
2 (27.2 mL) were added EtSH (606 µL, 8.16 mmol) and BF
3·OEt
2 (1.03 mL, 8.16 mmol) at 0 °C. After stirring for 2 h at rt as the reaction was monitored by TLC (3:2 EtOAc–hexane), the reaction was quenched by the addition of crushed ice. The solution was diluted with CHCl
3 and subsequently washed with ice-cooled H
2O, satd aq Na
2CO
3, and brine. The organic layer was then dried over Na
2SO
4, and concentrated. The resulting residue was purified by silica gel column chromatography (1:1 EtOAc–hexane) to give
6 (3.99 g, 96%). Spectroscopic data of
6 were identical to those reported in the literature [
54].
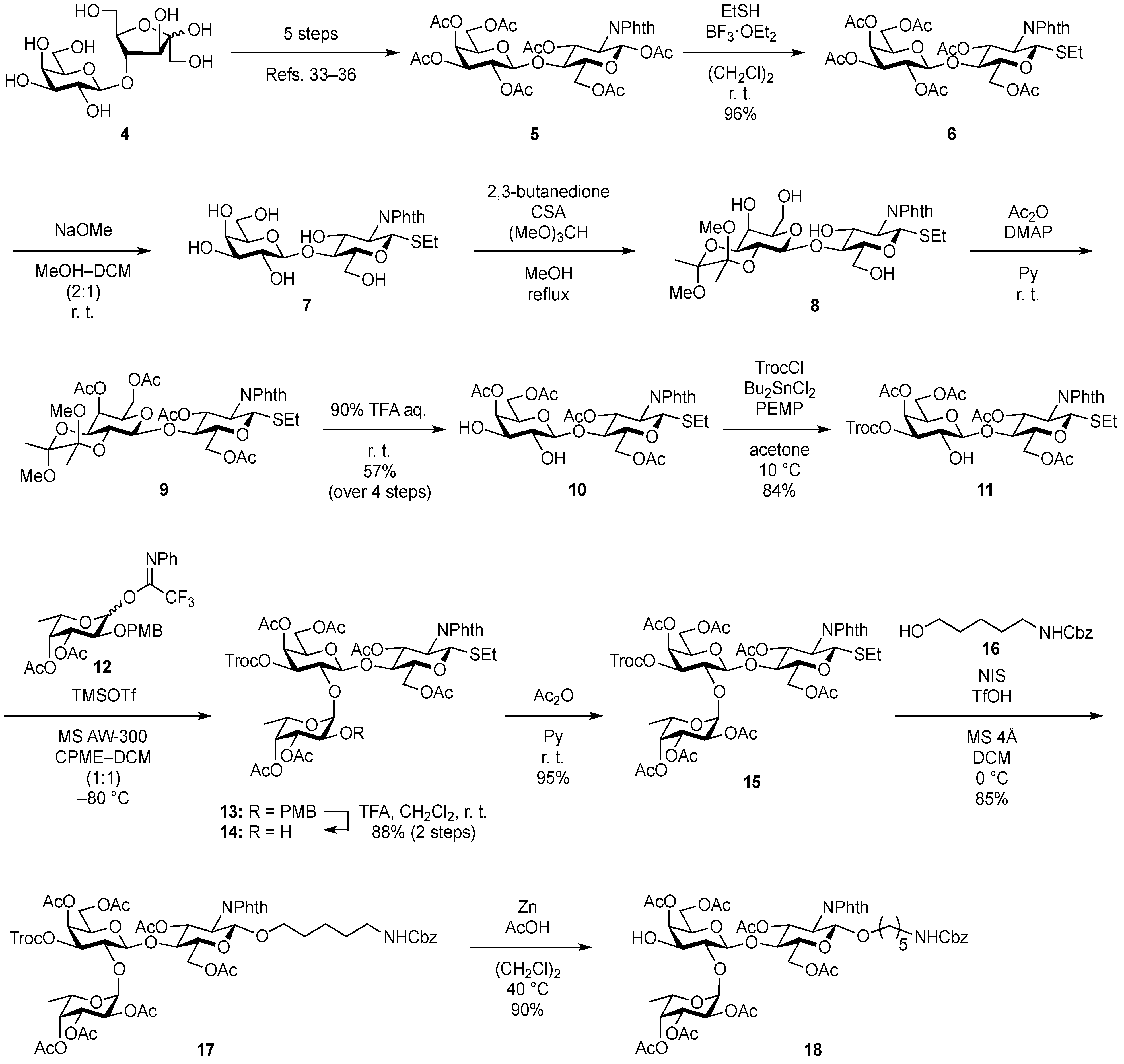
Ethyl (4,6-di-O-acetyl-β-d-galactopyranosyl)-(1→4)-3,6-di-O-acetyl-2-deoxy-2-phthalimide-1-thio-β-d-glucopyranoside (10). To a solution of 6 (1.08 g, 1.41 mmol) in MeOH/CH2Cl2 (2:1, 14.1 mL) was added NaOMe (28% solution in MeOH, 31.9 µL, 141 µmol) at 0 °C. After stirring for 2 h at rt as the reaction was monitored by TLC (3:2 EtOAc–hexane), the reaction was neutralized with AcOH. After concentration, the resulting residue was diluted with CHCl3 and subsequently washed with H2O and brine. The organic layer was dried over Na2SO4, of which solid was filtered through cotton and the filtrate was then evaporated (giving 7). The residue was subjected to next reaction without further purification. The crude product 7 was dissolved in MeOH (28.2 mL). To the solution were added 2,3-butanedione (492 µL, 5.64 mmol), trimethyl orthoformate (1.95 mL, 17.8 mmol), and (±)-10-camphorsulfonic acid (66 mg, 282 µmol) at rt. After stirring for 20 h at reflux as the reaction was monitored by TLC (10:1 CHCl3–MeOH), the reaction was quenched by the addition of triethylamine (218 µmol) and concentrated. The resulting residue was diluted with CHCl3 and subsequently washed with H2O and brine. The organic layer was dried over Na2SO4, filtered, concentrated. The resulting residue was roughly purified by silica gel column chromatography (20:1 CHCl3–MeOH) to give 2,3-O-BDA-protected product 8 along with small amounts of contaminants. The crude mixture (494 mg) was dissolved in pyridine (7.8 mL). To the solution were added Ac2O (890 µL, 9.42 mmol) and a catalytic amount of DMAP at 0 °C. After stirring for 1 h at rt as the reaction was monitored by TLC (3:2 EtOAc–hexane), the mixture was co-evaporated with toluene. The resulting residue was diluted with EtOAc and subsequently washed with 2 M HCl, H2O, satd aq NaHCO3, and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (2:3 EtOAc–hexane) to give 9 (634 mg), to which suspension in H2O (1.6 mL) was added trifluoroacetic acid (14.4 mL) at 0 °C. After stirring for 2 h at rt as the reaction was monitored by TLC (1:1 CHCl3–acetone), the mixture was diluted with toluene and concentrated. The resulting residue was purified by silica gel column chromatography (7:3 CHCl3–acetone) to give 10 (548 mg, 57% over four steps). [α]D +18.3° (c 1.0, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.88–7.74 (m, 4H, Phth), 5.84 (dd, 1H, J3,4 = 8.2 Hz, J2,3 = 11.2 Hz, H-3GlcN), 5.50 (d, 1H, J1,2 = 10.6 Hz, H-1GlcN), 5.28 (d, 1H, J3,4 = 3.3 Hz, H-4Gal), 4.63 (dd, 1H, J5,6a = 1.6 Hz, Jgem = 11.9 Hz, H-6aGlcN), 4.40–4.29 (m, 3H, H-2GlcN, H-6bGlcN, H-1Gal), 4.10–3.98 (m, 2H, H-6aGal, H-6bGal), 3.91–3.80 (m, 3H, H-4GlcN, H-5GlcN, H-5Gal), 3.75–3.73 (m, 1H, H-3Gal), 3.62 (d, 1H, J2,OH = 3.2 Hz, OH), 3.57–3.53 (m, 1H, H-2Gal), 3.21 (d, 1H, J3,OH = 3.1 Hz, OH), 2.73–2.61 (m, 2H, SCH2CH3), 2.18–1.90 (4 s, 12H, Ac), 1.22 (t, 3H, SCH2CH3); 13C-NMR (125 MHz, CDCl3) δ 171.2, 170.9, 170.5, 170.0, 167.7, 167.4, 134.4, 134.2, 131.6, 131.2, 123.7, 123.6, 103.1, 81.1, 72.0, 71.9, 71.7, 71.0, 68.7, 63.1, 61.5, 53.9, 29.7, 29.2, 24.6, 21.0, 20.7, 20.7, 20.6, 14.9. HRMS (ESI) m/z: found [M+Na]+ 706.1776, C30H37NO15S calcd for [M+Na]+ 706.1773.
Ethyl [4,6-di-O-acetyl-3-O-(2,2,2-trichloroethoxycarbonyl)-β-d-galactopyranosyl]-(1→4)-3,6-di-O-acetyl-2-deoxy-2-phthalimide-1-thio-β-d-glucopyranoside (11). A solution of 10 (103 mg, 151 µmol) and dibutyltin dichloride (4.6 mg, 15.1 µmol) in acetone (3.0 mL) was stirred for 10 min at rt. To the solution were added PEMP (55 µL, 302 µmol) and TrocCl (27 µL, 196 µmol) at 10 °C. After stirring for 20 min at the same temperature as the reaction was monitored by TLC (1:2 EtOAc–toluene, 1:1 CHCl3–acetone), the reaction was quenched by the addition of satd aq NH4Cl and concentrated. The resulting residue was diluted with EtOAc and subsequently washed with H2O and brine. The organic layer was dried over Na2SO4, filtered, concentrated. The resulting residue was purified by silica gel column chromatography (2:7 EtOAc–toluene) to give 11 (108 mg, 84%). [α]D +30.0° (c 1.0, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.88–7.74 (m, 4H, Phth), 5.70 (dd, 1H, J3,4 = 8.2 Hz, J2,3 = 10.6 Hz, H-3GlcN), 5.49 (d, 1H, J1,2 = 10.6 Hz, H-1GlcN), 5.46 (d, 1H, J3,4 = 2.9 Hz, H-4Gal), 4.79 (m, 2H, H-3Gal, OCH2CCl3), 4.65 (near dd, 1H, Jgem = 11.4 Hz, H-6aGlcN), 4.44 (d, 1H, J1,2 = 7.7 Hz, H-1Gal), 4.38 (dd, 1H, J5,6b = 4.2 Hz, H-6bGlcN), 4.31 (t, 1H, H-2GlcN), 4.11–4.03 (m, 2H, H-6aGal, H-6bGal), 3.91–3.77 (m, 4H, H-4GlcN, H-5GlcN, H-2Gal, H-5Gal), 3.47 (d, 1H, J2,OH = 5.2 Hz, OH), 2.72–2.62 (m, 2H, SCH2CH3), 2.14–1.91 (4 s, 12H, Ac), 1.22 (t, 3H, SCH2CH3); 13C-NMR (125 MHz, CDCl3) δ 171.2, 170.4, 170.2, 170.2, 167.8, 167.5, 153.2, 134.3, 134.3, 131.8, 131.3, 123.8, 103.3, 94.1, 81.2, 77.7, 77.3, 77.2, 72.2, 70.6, 69.2, 66.2, 63.1, 61.1, 54.0, 29.8, 24.6, 21.0, 20.7, 20.7, 20.6, 15.1. HRMS (ESI) m/z: found [M+Na]+ 880.0823, C33H38Cl3NO17S calcd for [M+Na]+ 880.0818.
3,4-Di-O-acetyl-2-O-p-methoxybenzyl-l-fucopyranosyl N-phenyl 2,2,2-trifluoroacetimidate (
12). To a solution of phenyl 3,4-di-
O-acetyl-2-
O-
p-methoxybenzyl-1-thio-β-
l-fucopyranoside [
42] (1.21 g, 2.63 mmol) in acetone/H
2O (13.1 mL, 96:4) was added NBS (701 mg, 3.94 mmol) at −15 °C. After stirring for 1 h at the same temperature as the reaction was monitored by TLC (1:1 EtOAc–hexane), the reaction was quenched by the addition of satd aq Na
2S
2O
3 and then diluted with EtOAc, washed with H
2O and brine. The organic layer was dried over Na
2SO
4, filtered, concentrated. The resulting residue was purified by silica gel column chromatography (2:3 EtOAc–hexane) to give the corresponding hemiacetal product (969 mg, quant.), which was then dissolved in acetone (52.6 mL). To the solution were added 2,2,2-trifluoro-
N-phenylacetimidoyl chloride (853 µL, 5.26 mmol) and K
2CO
3 (1.82 g, 13.2 mmol) at rt. After stirring for 2.5 h at rt as the reaction was monitored by TLC (1:2 EtOAc–hexane), the reaction mixture was filtered through Celite. The filtrate and washings were concentrated. The resulting residue was purified by silica gel column chromatography (1:4 EtOAc–hexane) to give 12 (1.33 g, 94%, α/β = 1/1). [α]
D −81.6° (c 1.0, CHCl
3);
13C-NMR (125 MHz, CDCl
3) δ 170.3, 170.2, 169.9, 169.8, 159.4, 159.3, 143.5, 143.2, 129.8, 129.7, 129.5, 129.3, 129.1, 128.9, 128.8, 128.6, 128.6, 128.4, 128.4, 124.3, 124.2, 119.3, 119.2, 117.2, 114.9, 114.0, 113.7, 113.7, 97.0, 93.6, 77.6, 77.2, 74.9, 74.7, 72.9, 72.6, 72.2, 70.8, 70.2, 70.0, 69.7, 67.3, 55.2, 55.1, 20.7, 20.6, 20.5, 20.5, 15.9, 15.8.
1H-NMR (500 MHz, CDCl
3)
α-isomer: δ 7.45–6.71 (m, 9H, Ar), 6.46 (br s, 1H, H-1), 5.35–5.31 (m, 2H, H-3, H-4), 4.75–4.59 (m, 2H, OC
H2Ar), 4.27 (br s, 1H, H-5), 3.95 (br d, 1H, H-2), 3.87–3.77 (m, 3H, OMe), 2.16–1.99 (m, 6H, Ac), 1.18–1.14 (m, 3H, H-6).
β-isomer: δ 7.45–6.71 (m, 9H, Ar), 5.68 (br s, 1H, H-1), 5.20 (br s, 1H, H-4), 4.98 (br s, 1H, H-3) 4.75–4.59 (m, 2H, OC
H2Ar), 3.87–3.77 (m, 5H, H-2, H-5, OMe), 2.16–1.99 (m, 6H, Ac), 1.18–1.14 (m, 3H, H-6). Possible other stereoisomers were not assigned. HRMS (ESI)
m/z: found [M+Na]
+ 562.1657, C
26H
28F
3NO
8 calcd for [M+Na]
+ 562.1659.
Ethyl (3,4-di-O-acetyl-α-l-fucopyranosyl)-(1→2)-[4,6-di-O-acetyl-3-O-(2,2,2-trichloroethoxycarbonyl)-β-d-galactopyranosyl]-(1→4)-3,6-di-O-acetyl-2-deoxy-2-phthalimide-1-thio-β-d-glucopyranoside (14). To a mixture of 11 (1.06 g, 1.24 mmol) and 12 (1.33 g, 2.47 mmol) in CPME/CH2Cl2 (1:1, 74.2 mL) was added 4 Å molecular sieves AW-300 (7.42 g) at rt. After stirring for 30 min, the mixture was cooled to −80 °C. TMSOTf (22 µL, 124 µmol) was then added to the mixture at −80 °C. After stirring for 5.5 h at the same temperature as the reaction was monitored by TLC (1:2 EtOAc–toluene, 1:2 EtOAc–hexane) and MALDI-TOF MS, the reaction was quenched by the addition of satd aq NaHCO3. The reaction mixture was diluted with CHCl3 and filtered through Celite. The filtrate was then washed with satd aq NaHCO3 and H2O. The organic layer was subsequently dried over Na2SO4, and concentrated. The resulting residue was purified by silica gel column chromatography (2:7 EtOAc–toluene) to give 13 with unidentified impurity (1.66 g). The crude mixture was then dissolved in CH2Cl2 (44.6 mL). To the solution was added trifluoroacetic acid (5.0 mL) at 0 °C. After stirring for 40 min at rt as the reaction was monitored by TLC (1:1 EtOAc–hexane), the mixture was co-evaporated with toluene. The residue was diluted with CHCl3 and subsequently washed with satd aq NaHCO3 and H2O. The organic layer was dried over Na2SO4, filtered, concentrated. The resulting residue was purified by silica gel column chromatography (1:2 EtOAc–toluene) to give 14 (1.18 g, 88% over two steps). [α]D −38.1° (c 1.0, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.88–7.73 (m, 4H, Phth), 5.80 (t, 1H, J2,3 = J3,4 = 10.6 Hz, H-3GlcN), 5.47–5.45 (m, 2H, H-1GlcN, H-4Gal), 5.29 (d, 1H, J1,2 = 2.5 Hz, H-1Fuc), 5.21 (d, 1H, J3,4 = 3.9 Hz, H-4Fuc), 4.99 (dd, 1H, J2,3 = 10.7 Hz, H-3Fuc), 4.91 (dd, 1H, J3,4 = 3.6 Hz, J2,3 = 10.1 Hz, H-3Gal), 4.75 (s, 2H, OCH2CCl3), 4.51 (dd, 1H, J5,6a = 4.2 Hz, Jgem = 12.0 Hz, H-6aGlcN), 4.33–4.31 (m, 4H, H-2GlcN, H-6bGlcN, H-1Gal, H-5Fuc), 4.17–4.09 (m, 2H, H-6aGal, H-6bGal), 3.95–3.83 (m, 5H, H-4GlcN, H-5GlcN, H-2Gal, H-5Gal, H-2Fuc), 2.74–2.62 (m, 2H, SCH2CH3), 2.16–1.91 (6 s, 18H, Ac), 1.27–1.22 (m, 6H, H-6Fuc, SCH2CH3); 13C-NMR (125 MHz, CDCl3) δ 170.6, 170.5, 170.3, 169.9, 169.8, 167.5, 167.2, 152.8, 134.3, 134.2, 131.6, 131.2, 123.6, 100.1, 99.6, 93.8, 81.4, 77.8, 77.2, 74.9, 73.2, 71.2, 70.7, 70.6, 67.0, 66.6, 65.7, 62.5, 60.9, 53.9, 29.6, 24.8, 20.8, 20.7, 20.6, 20.6, 20.5, 20.4, 15.6, 15.0. HRMS (ESI) m/z: found [M+Na]+ 1110.1609, C43H52Cl3NO23S calcd for [M+Na]+ 1110.1611.
Ethyl (2,3,4-tri-O-acetyl-α-l-fucopyranosyl)-(1→2)-[4,6-di-O-acetyl-3-O-(2,2,2-trichloroethoxycarbonyl)-β-d-galactopyranosyl]-(1→4)-3,6-di-O-acetyl-2-deoxy-2-phthalimide-1-thio-β-d-glucopyranoside (15). To a solution of 14 (1.06 g, 975 µmol) in pyridine (4.9 mL) was added acetic anhydride (4.9 mL) at 0 °C. After stirring for 2 h at rt as the reaction was monitored by TLC (1:1 EtOAc–hexane), the reaction was quenched by addition of MeOH at 0 °C and then evaporated. The residue was diluted with CHCl3, washed with 2 M HCl, H2O, satd aq NaHCO3, and brine, dried over Na2SO4, concentrated. The residue obtained was purified by silica gel column chromatography (2:3 EtOAc–hexane) to give 15 (1.05 g, 95%). [α]D −39.6° (c 1.0, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.86–7.73 (m, 4H, Phth), 5.79 (t, 1H, J2,3 = J3,4 = 10.1 Hz, H-3GlcN), 5.47 (d, 1H, J1,2 = 10.6 Hz, H-1GlcN), 5.43 (d, 1H, J3,4 = 4.3 Hz, H-4Gal), 5.39 (d, 1H, J1,2 = 3.8 Hz, H-1Fuc), 5.34 (d, 1H, J3,4 = 3.9 Hz, H-4Fuc), 5.16 (dd, 1H, J2,3 = 10.9 Hz, H-3Fuc), 5.07 (dd, 1H, H-2Fuc), 4.88 (dd, 1H, J2,3 = 9.8 Hz, H-3Gal), 4.84 (d, 1H, Jgem = 11.7 Hz, OCH2CCl3), 4.63 (d, 1H, OCH2CCl3), 4.51 (dd, 1H, J5,6a = 1.8 Hz, Jgem = 10.8 Hz, H-6aGlcN), 4.47–4.43 (m, 2H, H-1Gal, H-5Fuc), 4.39–4.30 (m, 2H, H-2GlcN, H-6bGlcN), 4.16 (dd, 1H, J5,6a = 6.6 Hz, Jgem = 11.2 Hz, H-6aGal), 4.09 (dd, 1H, H-6bGal), 3.94 (t, 1H, H-4GlcN), 3.89–3.83 (m, 3H, H-5GlcN, H-2Gal, H-5Gal), 2.74–2.62 (m, 2H, SCH2CH3), 2.17–1.91 (7 s, 21H, Ac), 1.26–1.22 (m, 6H, H-6Fuc, SCH2CH3); 13C-NMR (125 MHz, CDCl3) δ 170.6, 170.5, 170.3, 170.1, 169.9, 169.7, 169.7, 167.5, 167.2, 152.7, 134.3, 134.1, 131.6, 131.2, 123.6, 100.0, 96.2, 93.8, 81.4, 77.8, 77.2, 76.9, 74.7, 72.5, 71.1, 70.7, 70.6, 67.9, 67.7, 66.4, 65.3, 62.7, 60.9, 53.8, 29.6, 24.8, 20.8, 20.6, 20.6, 20.5, 20.3, 15.5, 15.1. HRMS (ESI) m/z: found [M+Na]+ 1152.1716, C45H54Cl3NO24S calcd for [M+Na]+ 1152.1714.
5-Benzyloxycarbonylamino-1-pentyl (2,3,4-tri-O-acetyl-α-l-fucopyranosyl)-(1→2)-[4,6-di-O-acetyl-3-O-(2,2,2-trichloroethoxycarbonyl)-β-d-galactopyranosyl]-(1→4)-3,6-di-O-acetyl-2-deoxy-2-phthalimide-β-d-glucopyranoside (17). A mixture of 15 (372 mg, 329 µmol) and 16 (234 mg, 988 µmol), and NIS (148 mg, 658 µmol) was exposed to high vacuum for 1 h. The mixture was dissolved in CH2Cl2 (13.2 mL), to which 4 Å molecular sieves (1.32 g) was added at rt. After stirring for 30 min at rt and then for 10 min at 0 °C, TfOH (7.1 µL, 65.8 µmol) was added to the mixture. After stirring for 1 h at 0 °C as the reaction was monitored by TLC (1:1 EtOAc–hexane, 2:1 EtOAc–hexane), additional portions of NIS (148 mg, 658 µmol) and TfOH (7.1 µL, 65.8 µmol) were added to the mixture. After 8 h and 16 h, further portions of TfOH (7.1 µL of each) were added to the mixture and the stirring was continued. After stirring for total 30 h, the reaction was quenched by the addition of satd aq NaHCO3. The precipitate was filtered through Celite. The filtrate was diluted with CHCl3, washed with satd aq Na2S2O3 and brine. The organic layer was subsequently dried over Na2SO4, concentrated and the residue was then purified by silica gel column chromatography (1:1 EtOAc–hexane) and gel filtration column chromatography (LH-20, 1:1 CHCl3–MeOH) to give 17 (375 mg, 87%). [α]D −41.1° (c 1.0, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.85–7.69 (m, 4H, Phth), 7.47–7.30 (m, 5H, Ph), 5.74 (dd, 1H, J3,4 = 9.0 Hz, J2,3 = 10.8 Hz, H-3GlcN), 5.42 (d, 1H, J3,4 = 3.1 Hz, H-4Gal), 5.40 (d, 1H, J1,2 = 3.8 Hz, H-1Fuc), 5.33 (d, 1H, J3,4 = 3.8 Hz, H-4Fuc), 5.31 (d, 1H, J1,2 = 8.5 Hz, H-1GlcN), 5.17 (dd, 1H, J2,3 = 10.9 Hz, H-3Fuc), 5.07–5.04 (m, 3H, H-2Fuc, OCH2), 4.88 (dd, 1H, J2,3 = 9.8 Hz, H-3Gal), 4.84 (d, 1H, Jgem = 11.6 Hz, OCH2), 4.64–4.62 (m, 2H, OCH2(CH2)3CH2NH, OCH2), 4.55 (dd, 1H, J5,6a = 1.8 Hz, Jgem = 12.1 Hz, H-6aGlcN), 4.47–4.43 (m, 2H, H-1Gal, H-5Fuc), 4.37 (dd, 1H, J5,6b = 5.2 Hz, H-6bGlcN), 4.24 (dd, 1H, H-2GlcN), 4.16 (dd, 1H, J5,6a = 6.7 Hz, Jgem = 11.2 Hz, H-6aGal), 4.09 (dd, 1H, H-6bGal), 3.94 (t, 1H, H-4GlcN), 3.88–3.79 (m, 4H, H-5GlcN, H-2Gal, H-5Gal, OCH2(CH2)3CH2NH), 3.46–3.44 (m, 1H, OCH2(CH2)3CH2NH), 2.95–2.91 (m, 2H, OCH2(CH2)3CH2NH), 2.17–1.91 (7 s, 21H, Ac), 1.51–1.11 (m, 9H, H-6Fuc, OCH2(CH2)3CH2NH); 13C-NMR (125 MHz, CDCl3) δ 170.7, 170.6, 170.3, 170.2, 170.0, 169.8, 156.2, 152.8, 136.7, 134.3, 128.5, 128.1, 128.1, 123.6, 100.1, 98.1, 96.2, 93.8, 77.6, 74.8, 72.9, 72.5, 71.1, 70.6, 70.6, 70.0, 69.8, 67.9, 67.8, 66.5, 66.4, 65.3, 62.3, 61.0, 54.7, 40.8, 29.3, 28.8, 23.0, 20.9, 20.7, 20.6, 20.6, 20.4, 15.5. HRMS (ESI) m/z: found [M+Na]+ 1327.2890, C56H67Cl3N2O27 calcd for [M+Na]+ 1327.2889.
5-Benzyloxycarbonylamino-1-pentyl (2,3,4-tri-O-acetyl-α-l-fucopyranosyl)-(1→2)-(4,6-di-O-acetyl-β-d-galactopyranosyl)-(1→4)-3,6-di-O-acetyl-2-deoxy-2-phthalimide-β-d-glucopyranoside (18). To a solution of 17 (289 mg, 215 µmol) in AcOH/(CH2Cl)2 (3:1, 14.3 mL) was added Zn powder (2.89 g) at rt. The reaction mixture was stirred for 1 h at 40 °C as the reaction was monitored by TLC (3:1 EtOAc–hexane). The precipitate was filtered through Celite and the filtrate was co-evaporated with toluene. The residue obtained was purified by silica gel column chromatography (3:1 EtOAc–hexane) to give 18 (233 mg, 97%). [α]D −56.6° (c 1.0, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.85–7.69 (m, 4H, Phth), 7.38–7.30 (m, 5H, Ph), 5.74 (dd, 1H, J3,4 = 9.0 Hz, J2,3 = 10.8 Hz, H-3GlcN), 5.39 (d, 1H, J3,4 = 3.6 Hz, H-4Gal), 5.33 (d, 1H, J3,4 = 3.8 Hz, H-4Fuc), 5.31 (d, 1H, J1,2 = 8.5 Hz, H-1GlcN), 5.26–5.23 (m, 2H, H-3Fuc, OCH2Ph), 5.16 (dd, 1H, J1,2 = 3.6 Hz, J2,3 = 9.9 Hz, H-2Fuc), 5.07 (m, 2H, H-1Fuc, OCH2Ph), 4.65 (s, 1H, OCH2(CH2)3CH2NH), 4.51 (dd, 1H, J5,6a = 1.8 Hz, Jgem = 12.0 Hz, H-6aGlcN), 4.42–4.37 (m, 2H, H-6bGlcN, H-5Fuc), 4.31 (d, 1H, J1,2 = 7.7 Hz, H-1Gal), 4.23 (dd, 1H, H-2GlcN), 4.11 (m, 2H, H-6aGal, H-6bGal), 3.92 (t, 1H, J4,5 = 9.0 Hz, H-4GlcN), 3.86–3.79 (m, 4H, H-5GlcN, H-3Gal, H-5Gal, OCH2(CH2)3CH2NH), 3.54 (dd, 1H, J2,3 = 9.5 Hz, H-2Gal), 3.47–3.43 (m, 1H, OCH2(CH2)3CH2NH), 2.94–2.90 (m, 2H, OCH2(CH2)3CH2NH), 2.18–1.91 (7 s, 21H, Ac), 1.49–1.11 (m, 9H, H-6Fuc, OCH2(CH2)3CH2NH); 13C-NMR (125 MHz, CDCl3) δ 171.0, 170.7, 170.7, 170.4, 170.1, 170.1, 169.9, 156.2, 136.6, 134.3, 131.4, 128.5, 128.0, 123.5, 100.1, 98.1, 97.8, 74.9, 73.0, 72.4, 71.1, 71.0, 69.9, 69.7, 69.6, 68.2, 67.7, 66.5, 65.2, 62.4, 61.5, 54.8, 40.8, 29.6, 29.3, 28.8, 23.0, 20.8, 20.7, 20.6, 20.6, 20.5, 15.7. HRMS (ESI) m/z: found [M+Na]+ 1153.3847, C53H66N2O25 calcd for [M+Na]+ 1153.3851.
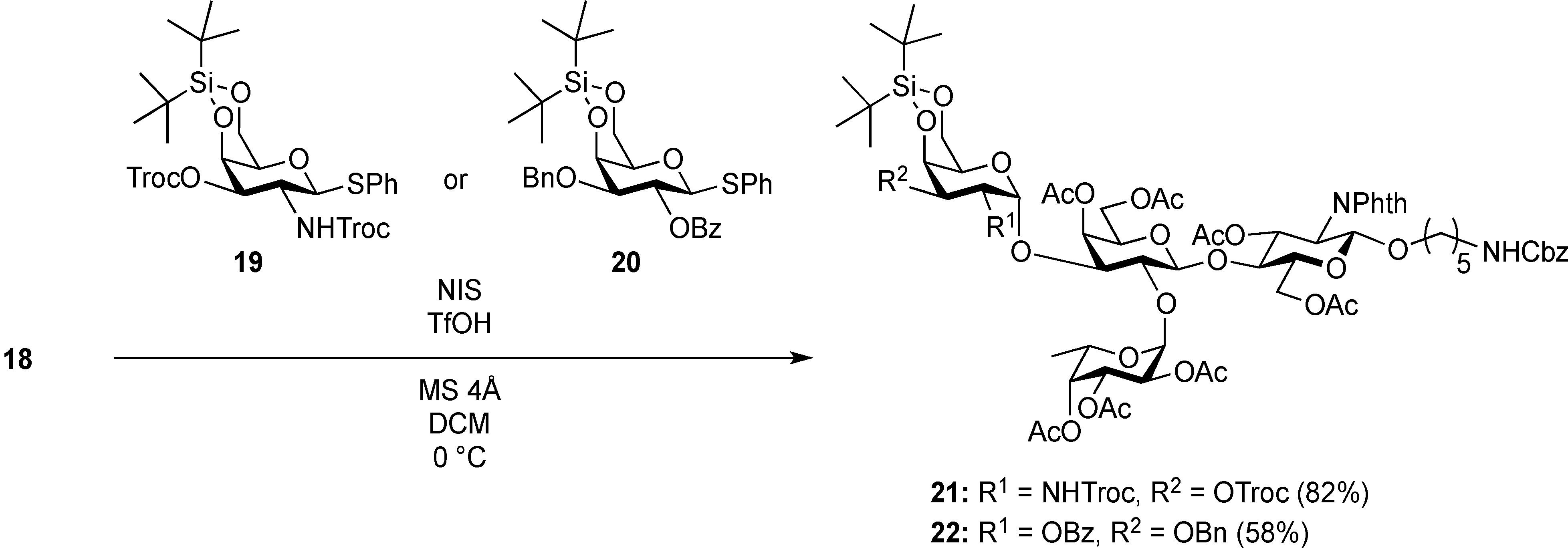
Phenyl 2-O-benzoyl-3-O-benzyl-4,6-O-di-tert-butylsilylene-1-thio-β-d-galactopyranoside (20). To a solution of Phenyl 3-O-benzyl-1-thio-β-d-galactopyranoside (262 mg, 724 µmol) in pyridine (7.2 mL) was added di-tert-butylsilyl bis(trifluoromethanesulfonate) (260 µL, 796 µmol) at 0 °C. After stirring for 3.5 h at 0 °C as the reaction was monitored by TLC (1:1 EtOAc–hexane), benzoic anhydride (328 mg, 1.45 mmol) was added to the mixture at 0 °C. After stirring for 22 h at rt as the reaction was monitored by TLC (1:3 EtOAc–hexane), the reaction was quenched by the addition of MeOH at 0 °C. The mixture was co-evaporated with toluene. The residue obtained was diluted with EtOAc, washed with 2 M HCl, H2O, satd aq NaHCO3, and brine, dried over Na2SO4, concentrated. The resulting residue was purified by silica gel column chromatography (1:7 EtOAc–hexane) to give 20 (324 mg, 74%). [α]D +66.9° (c 0.6, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 8.06–7.16 (m, 15H, Ph), 5.70 (t, 1H, J1,2 = J2,3 = 9.8 Hz, H-2), 4.79 (d, 1H, H-1), 4.72 (d, 1H, Jgem = 12.8 Hz, OCH2Ph), 4.60–4.57 (m, 2H, H-4, OCH2Ph), 4.30–4.23 (m, 2H, H-6a, H-6b), 3.57 (dd, 1H, H-3), 3.40 (s, 1H, H-5), 1.16–1.08 (2 s, 18H, 2 t-Bu); 13C-NMR (125 MHz, CDCl3) δ 165.4, 137.9, 134.4, 133.0, 132.1, 130.1, 129.9, 128.8, 128.3, 127.6, 127.5, 87.6, 79.3, 75.1, 70.0, 69.8, 69.4, 67.3, 27.6, 23.4, 20.7. HRMS (ESI) m/z: found [M+Na]+ 629.2363, C34H42O6SSi calcd for [M+Na]+ 629.2364.
5-Benzyloxycarbonylamino-1-pentyl [2-deoxy-4,6-O-di-tert-butylsilylene-2-(2,2,2-trichloroethoxycarbamoyl)-3-O-(2,2,2-trichloroethoxycarbonyl)-α-d-galactopyranosyl]-(1→3)-[2,3,4-tri-O-acetyl-α-l-fucopyranosyl-(1→2)]-(4,6-di-O-acetyl-β-d-galactopyranosyl)-(1→4)-3,6-di-O-acetyl-2-deoxy-2-phthalimide-β-d-glucopyranoside (21). A mixture of 18 (103 mg, 91.1 µmol) and 19 (138 mg, 182 µmol), and NIS (46 mg, 364 µmol) was exposed to high vacuum for 1 h. The mixture was dissolved in CH2Cl2 (2.7 mL), to which 4 Å molecular sieves (273 mg) was added at rt. After stirring for 30 min at rt and then for 10 min at 0 °C, TfOH (1.9 µL, 18.2 µmol) was added to the mixture. After stirring for 3 h at 0 °C as the reaction was monitored by TLC (3:1 EtOAc–hexane, 1:1 EtOAc–hexane, 1:3 EtOAc–hexane), additional portions of NIS (23 mg) and TfOH (1.0 µL) were added to the mixture and the stirring was continued. After stirring for total 5 h, the reaction was quenched by the addition of satd aq NaHCO3. The precipitate was filtered through Celite. The filtrate was diluted with CHCl3, washed with satd aq Na2S2O3 and brine. The organic layer was subsequently dried over Na2SO4, concentrated and the residue was then purified by silica gel column chromatography (1:1 EtOAc–hexane) to give 21 (132 mg, 82%), and 9.5 mg (9%) of 18 was recovered. [α]D +23.8° (c 1.7, CHCl3); 1H-NMR (500 MHz, CD3CN) δ 7.77–7.70 (m, 4H, Phth), 7.30–7.22 (m, 5H, Ph), 5.74 (d, 1H, JNH,2 = 9.7 Hz, NHGalN), 5.64 (dd, 1H, J3,4 = 9.0 Hz, J2,3 = 11.9 Hz, H-3GlcN), 5.35 (d, 1H, J3,4 = 2.8 Hz, H-4Gal), 5.30 (m, 2H, H-1Fuc, OCH2(CH2)3CH2NH), 5.24 (d, 1H, J3,4 = 2.3 Hz, H-4Fuc), 5.20 (d, 1H, J1,2 = 10.8 Hz, H-1GlcN), 5.08 (d, 1H, J1,2 = 4.0 Hz, H-1GalN), 5.07 (dd, 1H, J1,2 = 3.5 Hz, J2,3 = 11.0 Hz, H-2Fuc), 4.97 (dd, 1H, H-3Fuc), 4.93 (s, 2H, OCH2), 4.86–4.79 (m, 2H, OCH2), 4.78–4.68 (m, 3H, H-3GalN, H-4GalN, OCH2), 4.59 (d, 1H, Jgem = 12.3 Hz, OCH2), 4.41–4.28 (m, 6H, H-6aGlcN, H-1Gal, H-5Fuc, H-2GalN, H-6aGalN, H-6bGalN), 4.09–3.96 (m, 5H, H-2GlcN, H-4GlcN, H-6bGlcN, H-3Gal, H-6aGal), 3.92 (dd, 1H, J5,6b = 6.1 Hz, Jgem = 11.3 Hz, H-6bGal), 3.80–3.75 (m, 3H, H-5GlcN, H-5Gal, H-5GalN), 3.65–3.61 (m, 2H, H-2Gal, OCH2(CH2)3CH2NH), 3.39–3.34 (m, 1H, OCH2(CH2)3CH2NH), 2.71–2.65 (m, 2H, OCH2(CH2)3CH2NH), 2.18–1.80 (7 s, 21H, Ac), 1.31–0.96 (m, 27H, H-6Fuc, 2 t-Bu, OCH2(CH2)3CH2NH); 13C-NMR (125 MHz, CD3CN) δ 171.6, 171.5, 171.3, 171.2, 171.1, 155.5, 154.2, 135.7, 132.3, 129.4, 128.8, 128.7, 118.6, 118.3, 101.2, 98.9, 97.5, 96.6, 95.5, 94.4, 94.4, 79.1, 77.5, 76.6, 75.2, 74.4, 74.1, 73.5, 71.9, 71.5, 71.3, 70.8, 70.4, 69.0, 68.9, 68.6, 67.3, 66.6, 66.5, 65.7, 63.2, 62.2, 55.5, 49.4, 41.3, 30.0, 29.5, 27.9, 27.8, 23.7, 23.7, 21.5, 21.3, 21.2, 21.1, 21.0, 21.0, 20.8, 16.1. HRMS (ESI) m/z: found [M+Na]+ 1802.3642, C73H95Cl6N3O33Si calcd for [M+Na]+ 1802.3640.
5-Benzyloxycarbonylamino-1-pentyl (2-O-benzoyl-3-O-benzyl-4,6-O-di-tert-butylsilylene-α-d-galactopyranosyl)-(1→3)-[2,3,4-tri-O-acetyl-α-l-fucopyranosyl-(1→2)]-(4,6-di-O-acetyl-β-d-galactopyranosyl)-(1→4)-3,6-di-O-acetyl-2-deoxy-2-phthalimide-β-d-glucopyranoside (22). A mixture of 18 (49.7 mg, 44.0 µmol) and 20 (53.3 mg, 87.9 µmol), and NIS (22.0 mg, 176 µmol) was exposed to high vacuum for 1 h. The mixture was dissolved in CH2Cl2 (1.3 mL), to which 4 Å molecular sieves (132 mg) was added at rt. After stirring for 30 min at rt and then for 10 min at 0 °C, TfOH (1.0 µL, 8.79 µmol) was added to the mixture. After stirring for 1.5 h at 0 °C as the reaction was monitored by TLC (3:1 EtOAc–hexane, 1:1 EtOAc–hexane, 1:3 EtOAc–hexane), additional portion of TfOH (1.0 µL) was added to the mixture and the stirring was continued. After stirring for total 2 h, the reaction was quenched by the addition of satd aq NaHCO3. The precipitate was filtered through Celite. The filtrate was diluted with CHCl3, washed with satd aq Na2S2O3 and brine. The organic layer was subsequently dried over Na2SO4, concentrated and the residue was then purified by silica gel column chromatography (7:8 EtOAc–hexane) to give 22 (41.2 mg, 58%), and 10.8 mg (22%) of 18 was recovered. [α]D +43.5° (c 1.3, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.96–7.21 (m, 19H, Ar), 5.70 (dd, 1H, J3,4 = 8.7 Hz, J2,3 = 10.9 Hz, H-3GlcN), 5.64 (dd, 1H, J1,2 = 3.6 Hz, J2,3 = 10.4 Hz, H-2GalII), 5.52 (d, 1H, J3,4 = 2.3 Hz, H-4Fuc), 5.41–5.40 (m, 2H, H-4GalI, H-1GalII), 5.34–5.33 (m, 2H, H-1GlcN, H-1Fuc), 5.13–5.07 (m, 4H, H-2Fuc, H-3Fuc, OCH2Ph), 4.83 (d, 1H, J3,4 = 2.1 Hz, H-4GalII), 4.75 (d, 1H, Jgem = 12.0 Hz, OCH2Ph), 4.63 (br s, 1H, OCH2(CH2)3CH2NH), 4.54 (d, 1H, OCH2Ph), 4.47 (dd, 1H, J5,6a = 3.9 Hz, Jgem = 12.2 Hz, H-6aGlcN), 4.41 (d, 1H, H-6bGlcN), 4.37–4.19 (m, 5H, H-2GlcN, H-1GalI, H-6aGalI, H-6bGalI, H-5Fuc), 4.13 (dd, 1H, J5,6a = 6.7 Hz, Jgem = 11.3 Hz, H-6aGalII), 3.98–3.90 (m, 3H, H-4GlcN, H-3GalII, H-6bGalII), 3.86–3.78 (m, 3H, H-5GlcN, H-3GalI, OCH2(CH2)3CH2NH), 3.69–3.63 (m, 3H, H-2GalI, H-5GalI, H-5GalII), 3.47–3.45 (m, 1H, OCH2(CH2)3CH2NH), 2.93–2.89 (m, 2H, OCH2(CH2)3CH2NH), 2.25–1.81 (6 s, 18H, Ac), 1.47–1.11 (m, 30H, H-6Fuc, 2 t-Bu, Ac, OCH2(CH2)3CH2NH); 13C-NMR (125 MHz, CDCl3) δ 170.6, 170.6, 170.4, 170.1, 170.0, 169.8, 169.2, 169.3, 165.7, 138.4, 136.6, 134.3, 133.0, 131.4, 130.2, 129.8, 128.5, 128.2, 128.1, 128.1, 127.5, 127.4, 123.5, 100.9, 98.1, 95.9, 92.7, 77.6, 74.3, 72.6, 71.1, 70.9, 70.3, 69.9, 69.7, 69.6, 68.6, 68.4, 68.1, 67.8, 66.8, 66.5, 65.4, 64.4, 62.3, 61.2, 54.6, 40.8, 29.7, 29.3, 28.8, 27.7, 27.3, 23.4, 23.0, 20.9, 20.8, 20.7, 20.6, 20.6, 19.5, 15.9. HRMS (ESI) m/z: found [M+Na]+ 1649.6129, C81H102N2O31Si calcd for [M+Na]+ 1649.6128.
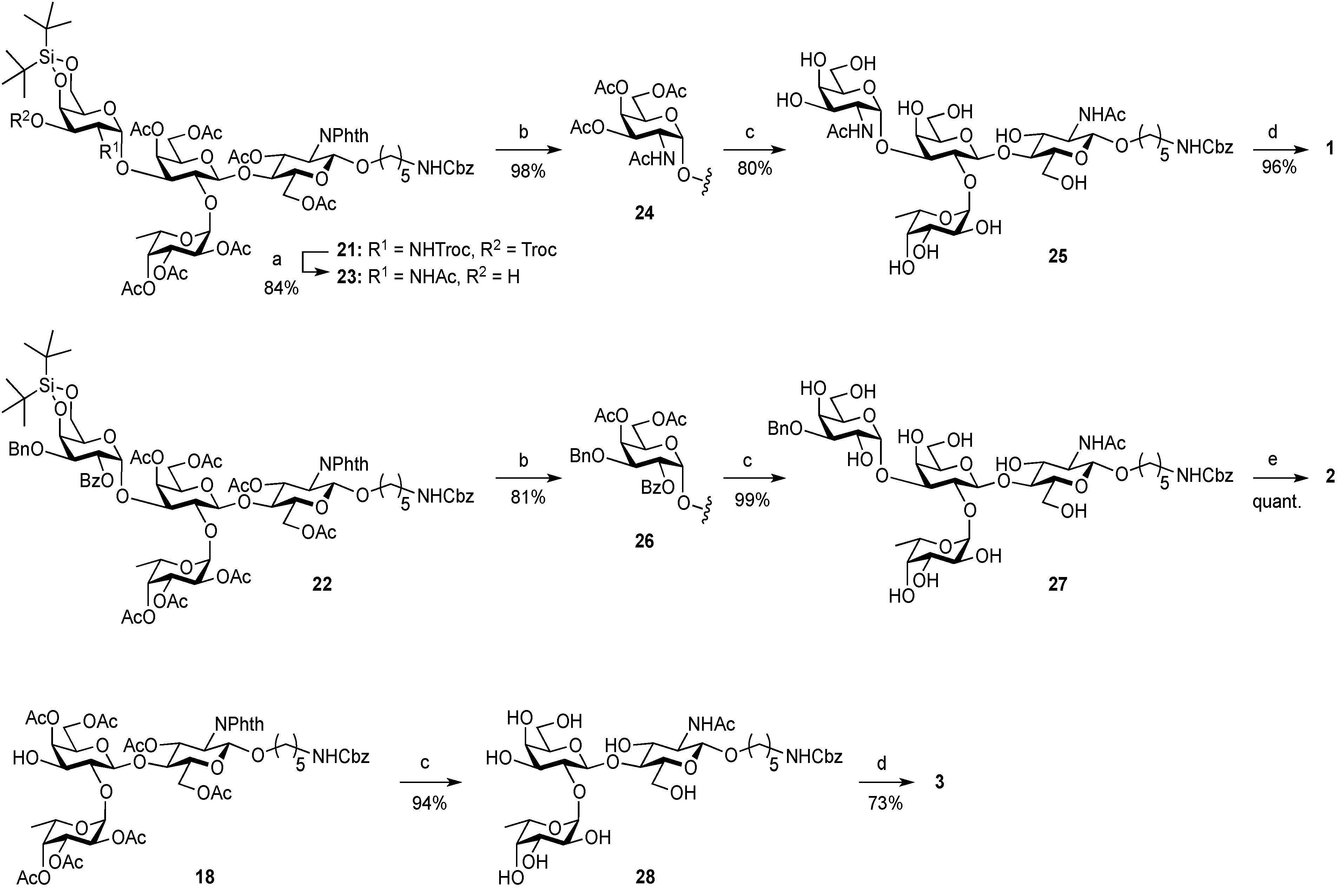
5-Benzyloxycarbonylamino-1-pentyl (2-acetamido-2-deoxy-4,6-O-di-tert-butylsilylene-α-d-galactopyranosyl)-(1→3)-[2,3,4-tri-O-acetyl-α-l-fucopyranosyl-(1→2)]-(4,6-di-O-acetyl-β-d-galactopyranosyl)-(1→4)-3,6-di-O-acetyl-2-deoxy-2-phthalimide-β-d-glucopyranoside (23). To a solution of 21 (45 mg, 25.2 µmol) in CH2Cl2 (1.7 mL) were added AcOH (288 µL, 5.04 mmol) and Zn powder (225 mg, 3.44 mmol) at rt. After stirring for 20 min at rt as the reaction was monitored by TLC (20:1 CHCl3–MeOH), another portion of Zn powder (225 mg) was added to the mixture and the stirring was continued. After 30 min, AcOH (288 µL) and CH2Cl2 (1.7 mL) were added to the mixture. After stirring for total 4 h, the precipitate was filtered through Celite and the filtrate was washed with satd aq NaHCO3. The organic layer was subsequently dried over Na2SO4, concentrated and the residue obtained was then dissolved in CH2Cl2 (2.5 mL). To the mixture was added acetic anhydride (48 µL, 252 µmol) at 0 °C. After stirring for 1 h at rt as the reaction was monitored by TLC (2:1 CHCl3–acetone), the reaction mixture was concentrated. The resulting residue was purified by silica gel column chromatography (2:1 CHCl3–acetone) to give 23 (31 mg, 84%). [α]D +6.3° (c 0.6, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.85–7.70 (m, 4H, Phth), 7.38–7.26 (m, 5H, Ph), 5.76–5.69 (m, 2H, H-3GlcN, NHGalN), 5.45–5.43 (m, 2H, H-4Gal, H-4Fuc), 5.36–5.31 (m, 2H, H-1GlcN, H-2Fuc), 5.15–5.07 (m, 5H, H-1Fuc, H-3Fuc, H-1GalN, OCH2), 4.63 (br s, 1H, OCH2(CH2)3CH2NH), 4.50–4.43 (m, 4H, H-2GlcN, H-6aGlcN, H-6bGlcN, H-4GalN), 4.41–4.35 (m, 2H, H-1Gal, H-5Fuc), 4.29 (d, 1H, Jgem = 11.2 Hz, H-6aGalN), 4.25–4.18 (m, 2H, H-2GalN, H-6bGalN), 4.10–4.04 (m, 2H, H-6aGal, H-6bGal), 3.95–3.92 (t, 1H, J3,4 = J4,5 = 9.9 Hz, H-4GlcN), 3.89–3.72 (m, 5H, H-5GlcN, H-2Gal, H-3Gal, H-5Gal, OCH2(CH2)3CH2NH), 3.56–3.45 (m, 3H, H-3GalN, H-5GalN, OCH2(CH2)3CH2NH), 2.94–2.90 (m, 2H, OCH2(CH2)3CH2NH), 2.60 (d, 1H, J3,OH = 11.5 Hz, OHGalN), 2.18–1.88 (8 s, 24H, Ac), 1.51–1.05 (m, 27H, H-6Fuc, 2 t-Bu, OCH2(CH2)3CH2NH); 13C-NMR (125 MHz, CD3CN) δ 170.2, 170.2, 169.9, 169.9, 169.8, 169.7, 169.6, 155.9, 137.3, 134.4, 131.0, 128.1, 127.5, 127.4, 123.1, 117.0, 99.5, 97.6, 95.6, 93.4, 73.8, 73.6, 73.1, 72.6, 72.2, 70.8, 70.4, 69.8, 69.1, 67.8, 67.7, 67.6, 67.5, 66.3, 65.3, 64.8, 64.7, 61.7, 61.1, 54.2, 53.9, 48.6, 40.0, 30.9, 29.0, 28.7, 28.4, 28.3, 26.7, 26.4, 22.5, 22.4, 21.8, 20.2, 19.9, 19.9, 19.8, 19.7, 19.7, 19.6, 19.5, 14.7. HRMS (ESI) m/z: found [M+Na]+ 1496.5661, C69H95N3O30Si calcd for [M+Na]+ 1496.5662.
5-Benzyloxycarbonylamino-1-pentyl (2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-α-d-galactopyranosyl)-(1→3)-[2,3,4-tri-O-acetyl-α-l-fucopyranosyl-(1→2)]-(4,6-di-O-acetyl-β-d-galactopyranosyl)-(1→4)-3,6-di-O-acetyl-2-deoxy-2-phthalimide-β-d-glucopyranoside (24). To a solution of 23 (31 mg, 21.2 µmol) in THF (1.1 mL) was added TBAHF 1.0 m solution (212 µL) at rt. After stirring for 40 min at rt as the reaction was monitored by TLC (10:1 CHCl3–MeOH), the reaction mixture was diluted with EtOAc and washed with 2 M HCl, H2O, satd aq NaHCO3, and brine. The organic layer was then dried over Na2SO4 and concentrated. The residue obtained was dissolved in pyridine (1.0 mL). To the mixture was added acetic anhydride (1.0 mL) at 0 °C. After stirring for 21 h at rt as the reaction was monitored by TLC (10:1 CHCl3–MeOH), the reaction was quenched by the addition of MeOH at 0 °C. The mixture was co-evaporated with toluene. The residue was diluted with EtOAc and washed with 2 M HCl, H2O, satd aq NaHCO3, and brine. The organic layer was then dried over Na2SO4 and concentrated. The resulting residue was purified by silica gel column chromatography (10:10:1 CHCl3-toluene-MeOH) and gel filtration column chromatography (LH-20, 1:1 CHCl3–MeOH) to give 24 (30 mg, 98% over two steps). [α]D +2.6° (c 1.0, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.85–7.70 (m, 4H, Phth), 7.38–7.26 (m, 5H, Ph), 6.32 (br d, 1H, J2,NH = 6.9 Hz, NHGalN), 5.76 (dd, 1H, J2,3 = 10.8 Hz, J3,4 = 9.2 Hz, H-3GlcN), 5.50 (d, 1H, J1,2 = 3.7 Hz, H-1Fuc), 5.42 (d, 1H, J3,4 = 1.5 Hz, H-4GalN), 5.37–5.34 (m, 3H, H-4Gal, H-2Fuc, H-4Fuc), 5.31 (d, 1H, J1,2 = 8.5 Hz, H-1GlcN), 5.22 (d, 1H, J1,2 = 3.2 Hz, H-1GalN), 5.13 (dd, 1H, J3,4 = 3.2 Hz, J2,3 = 11.0 Hz, H-3Fuc), 5.07 (s, 2H, OCH2Ph), 4.97 (dd, 1H, J2,3 = 11.3 Hz, H-3GalN), 4.66 (br s, 1H, OCH2(CH2)3CH2NH), 4.56–4.51 (m, 2H, H-6aGlcN, H-5Fuc), 4.48–4.43 (m, 1H, H-2GalN), 4.40–4.37 (m, 2H, H-6bGlcN, H-1Gal), 4.24 (dd, 1H, H-2GlcN), 4.17–4.14 (m, 2H, H-6aGal, H-6aGalN), 4.11–4.07 (m, 2H, H-6bGal, H-6bGalN), 4.03 (br d, 1H, H-5GalN), 3.96 (t, 1H, J4,5 = 9.2 Hz, H-4GlcN), 3.86–3.74 (m, 5H, H-5GlcN, H-2Gal, H-3Gal, H-5Gal, OCH2(CH2)3CH2NH), 3.48–3.44 (m, 1H, OCH2(CH2)3CH2NH), 2.94–2.90 (m, 2H, OCH2(CH2)3CH2NH), 2.20–1.92 (11 s, 33H, Ac), 1.49–1.05 (m, 9H, H-6Fuc, OCH2(CH2)3CH2NH); 13C-NMR (125 MHz, CDCl3) δ 170.9, 170.6, 170.5, 170.4, 170.3, 170.0, 170.0, 169.9, 136.6, 134.3, 128.5, 128.1, 128.1, 123.6, 100.3, 98.1, 96.6, 77.6, 74.6, 74.5, 72.8, 71.2, 70.6, 69.9, 69.8, 68.6, 67.9, 67.4, 66.9, 66.7, 66.5, 65.3, 62.7, 62.2, 61.0, 54.7, 48.1, 40.8, 29.7, 29.3, 28.8, 23.0, 20.9, 20.7, 20.6, 20.6, 15.6. HRMS (ESI) m/z: found [M+Na]+ 1482.4956, C67H85N3O33Si calcd for [M+Na]+ 1482.4958.
5-Benzyloxycarbonylamino-1-pentyl (2-acetamido-2-deoxy-α-d-galactopyranosyl)-(1→3)-[α-l-fucopyranosyl-(1→2)]-β-d-galactopyranosyl-(1→4)-2-acetamide-2-deoxy-β-d-glucopyranoside (25). To a solution of 24 (19.8 mg, 13.6 µmol) in MeOH (1.4 mL) was added NaOMe (1m solution in MeOH, 6.8 µL, 6.78 µmol) at 0 °C. After stirring for 4 h at rt as the reaction was monitored by TLC (20:12:1 CHCl3–MeOH–H2O), the reaction was neutralized with Muromac (H+) resin. The resin was filtered out and the filtrate was concentrated. The residue obtained was then dissolved in EtOH (2.8 mL). To the solution was added NH2NH2·H2O (1.0 µL, 27.2 µmol) at rt. The reaction mixture was stirred at reflux as monitored by TLC (5:4:1 CHCl3–MeOH–H2O). Additional portions of NH2NH2·H2O (2.0 µL) was added to the mixture every 15 min (total amounts of NH2NH2·H2O added was 32 µL). After 6.5 h, the reaction mixture was concentrated and exposed to high vacuum for 1 h. The resulting residue was then dissolved in MeOH/CH2Cl2 (3:1, 4.4 mL). To the mixture was added acetic anhydride (26 µL, 272 µmol) at 0 °C. After stirring for 1.5 h at rt as the reaction was monitored by TLC (5:4:1 CHCl3-MeOH-H2O), the reaction mixture was concentrated. The residue obtained was purified by silica gel column chromatography (Iatrobeads, 9:5:0.5 CHCl3–MeOH–H2O) to give 25 (10.3 mg, 80% over three steps). [α]D +4.4° (c 0.3, MeOH); 1H-NMR (500 MHz, CD3OD) δ 7.45–7.43 (m, 5H, Ph), 5.36 (d, 1H, J1,2 = 3.9 Hz, α-anomer H), 5.15 (d, 1H, J1,2 = 3.7 Hz, α-anomer H), 5.06 (s, 2H, OCH2Ph), 4.52 (d, 1H, J1,2 = 7.7 Hz, β-anomer H), 4.39 (d, 1H, J1,2 = 8.4 Hz, β-anomer H), 4.34–4.31 (m, 1H, H-5Fuc), 4.18–3.46 (m, 27H, ring H, OCH2(CH2)3CH2NH), 3.11–3.08 (m, 2H, OCH2(CH2)3CH2NH), 2.00–1.96 (2 s, 6H, Ac), 1.57–1.20 (m, 9H, H-6Fuc, OCH2(CH2)3CH2NH); 13C-NMR (125 MHz, CD3OD) δ 174.5, 173.5, 158.9, 138.5, 129.4, 128.9, 128.8, 102.8, 102.2, 100.3, 93.6, 78.5, 77.9, 77.2, 76.9, 74.2, 73.6, 73.5, 72.7, 71.9, 70.5, 70.5, 70.1, 69.9, 67.7, 67.3, 64.9, 63.4, 62.6, 61.8, 56.9, 51.3, 41.8, 30.5, 30.2, 24.3, 23.0, 22.7, 16.6. HRMS (ESI) m/z: found [M+Na]+ 974.3954, C41H65N3O22 calcd for [M+Na]+ 974.3952.
5-Amino-1-pentyl 2-acetamido-2-deoxy-α-d-galactopyranosyl-(1→3)-[α-l-fucopyranosyl-(1→2)]-β-d-galactopyranosyl-(1→4)-2-acetamide-2-deoxy-β-d-glucopyranoside (1). To a solution of 25 (3.2 mg, 3.36 µmol) in MeOH/H2O (1:1, 3.2 mL) was added Pd/C (5 wt. %, 0.5 mg). After stirring for 3.5 h at rt under a hydrogen atmosphere as the reaction was monitored by TLC (5:4:1:1 CHCl3–MeOH–H2O–AcOH), additional portion of Pd/C (0.5 mg) was added to the mixture and the stirring was continued. After 12.5 h, further portion of Pd/C (0.5 mg) was added to the mixture. After stirring for total 21 h, the mixture was filtered through membrane filter. The filtrate was concentrated and the residue obtained was purified by gel filtration column chromatography (LH-20, MeOH) to give 1 (2.2 mg, 96%). [α]D +4.4° (c 0.3, MeOH); 1H-NMR (500 MHz, D2O) δ 5.36 (d, 1H, J1,2 = 4.1 Hz, α-anomer H), 5.16 (d, 1H, J1,2 = 3.9 Hz, α-anomer H), 4.58 (d, 1H, J1,2 = 7.7 Hz, β-anomer H), 4.47 (d, 1H, J1,2 = 8.4 Hz, β-anomer H), 4.31–4.29 (m, 1H, H-5Fuc), 4.23–3.56 (m, 27H, ring H, OCH2(CH2)3CH2NH), 2.98–2.95 (m, 2H, OCH2(CH2)3CH2NH), 2.02 (2 s, 6H, Ac), 1.67–1.22 (m, 9H, H-6Fuc, OCH2(CH2)3CH2NH); 13C-NMR (200 MHz, CD3OD) δ 174.4, 173.6, 103.0, 102.2, 100.3, 93.5, 78.3, 77.8, 77.1, 77.0, 74.1, 73.6, 73.5, 72.7, 71.9, 70.5, 70.3, 70.0, 69.9, 67.7, 64.8, 63.4, 62.6, 61.6, 56.8, 51.2, 40.7, 33.1, 30.8, 30.5, 29.8, 28.3, 24.2, 23.8, 23.0, 22.7, 16.6, 14.5. HRMS (ESI) m/z: found [M+Na]+ 840.3584, C33H59N3O20 calcd for [M+Na]+ 840.3584.
5-Benzyloxycarbonylamino-1-pentyl (4,6-di-O-acetyl-2-O-benzoyl-3-O-benzyl-α-d-galactopyranosyl)-(1→3)-[2,3,4-tri-O-acetyl-α-l-fucopyranosyl-(1→2)]-(4,6-di-O-acetyl-β-d-galactopyranosyl)-(1→4)-3,6-di-O-acetyl-2-deoxy-2-phthalimide-β-d-glucopyranoside (26). Compound 22 (30.1 mg, 18.5 µmol) was converted into 26 (23.6 mg, 81%) according to the procedure described for 24. [α]D +75.3° (c 0.2, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.96–7.16 (m, 19H, Ar), 5.75 (d, 1H, J3,4 = 1.9 Hz, H-4GalII), 5.72 (dd, 1H, J3,4 = 8.2 Hz, J2,3 = 8.8 Hz, H-3GlcN), 5.56 (d, 1H, J3,4 = 2.6 Hz, H-4Fuc), 5.42 (d, 1H, J1,2 = 3.9 Hz, H-1Fuc) 5.41 (d, 1H, J3,4 = 2.3 Hz, H-4GalI), 5.38–5.35 (m, 2H, H-1GalII, H-2GalII), 5.31 (d, 1H, J1,2 = 8.4 Hz, H-1GlcN), 5.22–5.17 (m, 2H, H-2Fuc, H-3Fuc), 5.07 (s, 2H, OCH2Ph), 4.70 (d, 1H, Jgem = 11.8 Hz, OCH2Ph), 4.64 (br s, 1H, OCH2(CH2)3CH2NH), 4.49–4.40 (m, 4H, H-6aGlcN, H-6bGlcN, H-5Fuc, OCH2Ph), 4.30 (d, 1H, J1,2 = 7.4 Hz, H-1GalI), 4.23–4.13 (m, 4H, H-2GlcN, H-5GalII, H-6aGalII, H-6bGalII), 4.08 (dd, 1H, J3,4 = 3.2 Hz, J2,3 = 7.2 Hz, H-3GalII), 4.00 (dd, 1H, J5,6a = 6.7 Hz, Jgem = 11.3 Hz, H-6aGalI), 3.94–3.90 (m, 2H, H-4GlcN, H-6bGalI), 3.86–3.79 (m, 2H, H-5GlcN, OCH2(CH2)3CH2NH), 3.76 (dd, 1H, J2,3 = 7.4 Hz, J3,4 = 2.9 Hz, H-3GalI), 3.65 (t, 1H, H-2GalI), 3.57 (t, 1H, J5,6b = 6.7 Hz, H-5GalI), 3.48–3.43 (m, 1H, OCH2(CH2)3CH2NH), 2.93–2.89 (m, 2H, OCH2(CH2)3CH2NH), 2.24–1.83 (9 s, 27H, Ac), 1.47–1.09 (m, 9H, H-6Fuc, OCH2(CH2)3CH2NH); 13C-NMR (125 MHz, CDCl3) δ 170.6, 170.6, 170.5, 17.3, 170.2, 170.1, 169.9, 169.8, 169.4, 165.6, 156.2, 137.8, 136.6, 134.3, 133.3, 131.4, 129.9, 129.6, 128.5, 128.4, 128.2, 128.1, 128.1, 127.9, 127.5, 123.5, 100.5, 98.1, 96.1, 77.6, 74.2, 72.7, 71.4, 71.3, 70.9, 70.1, 69.8, 69.6, 68.0, 67.8, 67.7, 67.2, 66.5, 65.2, 62.5, 62.3, 61.2, 54.6, 40.8, 29.7, 29.3, 28.8, 23.0, 20.8, 20.8, 20.7, 20.7, 20.7, 20.6, 19.8, 15.8. HRMS (ESI) m/z: found [M+Na]+ 1593.4316, C81H102N2O31Si calcd for [M+Na]+ 1593.4318.
5-Benzyloxycarbonylamino-1-pentyl (3-O-benzyl-α-d-galactopyranosyl)-(1→3)-[α-l-fucopyranosyl-(1→2)]-β-d-galactopyranosyl-(1→4)-2-acetamide-2-deoxy-β-d-glucopyranoside (27). Compound 26 (23.3 mg, 14.8 µmol) was converted into 27 (14.7 mg, 99%) according to the procedure described for 25. [α]D −6.2° (c 0.3, MeOH); 1H-NMR (500 MHz, CD3OD) δ 7.45–7.26 (m, 10H, Ph), 5.30 (near s, 1H, α-anomer H), 5.15 (d, 1H, J1,2 = 3.9 Hz, α-anomer H), 5.05 (s, 2H, OCH2Ph), 4.75 (d, 1H, Jgem = 11.7 Hz, OCH2Ph), 4.64 (d, 1H, OCH2Ph), 4.53 (d, 1H, J1,2 = 7.5 Hz, β-anomer H), 4.38 (d, 1H, J1,2 = 8.4 Hz, β-anomer H), 4.29–4.28 (m, 1H, H-5Fuc), 4.12–3.45 (m, 27H, ring H, OCH2(CH2)3CH2NH), 3.11–3.08 (m, 2H, OCH2(CH2)3CH2NH), 1.96 (s, 3H, Ac), 1.56–1.21 (m, 9H, H-6Fuc, OCH2(CH2)3CH2NH); 13C-NMR (125 MHz, CD3OD) δ 173.5, 158.9, 139.9, 138.5, 129.4, 129.3, 129.1, 128.9, 128.8, 128.7, 102.8, 102.2, 100.2, 95.9, 79.5, 79.3, 78.6, 77.1, 76.7, 74.1, 73.6, 73.0, 72.6, 71.9, 70.5, 69.9, 69.1, 68.1, 67.6, 67.3, 65.6, 63.3, 62.6, 61.7, 56.7, 41.8, 30.5, 30.2, 24.3, 23.0, 16.6. HRMS (ESI) m/z: found [M+Na]+ 1023.4156, C46H68N2O22 calcd for [M+Na]+ 1023.4156.
5-Amino-1-pentyl α-d-galactopyranosyl-(1→3)-[α-l-fucopyranosyl-(1→2)]-β-d-galactopyranosyl-(1→4)-2-acetamide-2-deoxy-β-d-glucopyranoside (2). Compound 27 (2.1 mg, 2.10 µmol) was converted into 2 (1.6 mg, quant.) according to the procedure described for 1, except for the use of a mixed solvent (1:1, 1,4-dioxane–2% aq formic acid) as reaction media. [α]D +6.3° (c 0.3, MeOH); 1H-NMR (500 MHz, D2O) δ 5.31 (d, 1H, J1,2 = 4.1 Hz, α-anomer H), 5.22 (d, 1H, J1,2 = 2.5 Hz, α-anomer H), 4.59 (d, 1H, J1,2 = 7.6 Hz, β-anomer H), 4.46 (d, 1H, J1,2 = 8.4 Hz, β-anomer H), 4.30–3.42 (m, 28H, ring H, OCH2(CH2)3CH2NH), 2.98–2.95 (m, 2H, OCH2(CH2)3CH2NH), 2.01 (s, 3H, Ac), 1.67–1.21 (m, 9H, H-6Fuc, OCH2(CH2)3CH2NH); 13C-NMR (200 MHz, CD3OD) δ 173.5, 103.0, 102.2, 100.3, 96.2, 79.9, 78.5, 77.1, 76.7, 74.1, 73.8, 73.6, 73.2, 71.8, 71.4, 71.3, 70.3, 70.0, 69.9, 67.6, 65.8, 63.3, 62.6, 61.7, 56.7, 40.7, 29.8, 28.4, 24.2, 23.0, 16.5. HRMS (ESI) m/z: found [M+Na]+ 779.3320, C31H56N2O20 calcd for [M+Na]+ 779.3319.
5-Benzyloxycarbonylamino-1-pentyl α-l-fucopyranosyl-(1→2)-β-d-galactopyranosyl-(1→4)-2-acetamide-2-deoxy-β-d-glucopyranoside (28). Compound 18 (19.3 mg, 17.0 µmol) was converted into 28 (12.0 mg, 94%) according to the procedure described for 25. [α]D −110.0° (c 0.2, MeOH); 1H-NMR (500 MHz, CD3OD) δ 7.34–7.28 (m, 5H, Ph), 5.22 (d, 1H, J1,2 = 3.1 Hz, α-anomer H), 5.05 (s, 2H, OCH2Ph), 4.48 (d, 1H, J1,2 = 6.1 Hz, β-anomer H), 4.37 (d, 1H, J1,2 = 8.3 Hz, β-anomer H), 4.18–4.17 (m, 1H, H-5Fuc), 3.96–3.45 (m, 20H, ring H, OCH2(CH2)3CH2NH), 3.11–3.08 (m, 2H, OCH2(CH2)3CH2NH), 1.96 (s, 3H, Ac), 1.57–1.20 (m, 9H, H-6Fuc, OCH2(CH2)3CH2NH); 13C-NMR (125 MHz, CD3OD) δ 173.5, 158.9, 138.5, 129.4, 128.9, 128.8, 102.8, 102.5, 101.8, 79.0, 78.2, 77.1, 77.0, 76.9, 75.3, 74.1, 73.6, 71.7, 70.7, 70.5, 68.3, 67.3, 62.6, 61.6, 56.7, 41.8, 30.5, 30.2, 24.3, 23.0, 16.7. HRMS (ESI) m/z: found [M+Na]+ 771.3156, C33H52N2O17 calcd for [M+Na]+ 771.3158.
5-Amino-1-pentyl α-l-fucopyranosyl-(1→2)-β-d-galactopyranosyl-(1→4)-2-acetamide-2-deoxy-β-d-glucopyranoside (3). Compound 28 (5.9 mg, 7.88 µmol) was converted into 3 (3.5 mg, 73%) according to the procedure described for 1. [α]D −76.3° (c 0.2, MeOH); 1H-NMR (500 MHz, D2O) δ 5.29 (d, 1H, J1,2 = 3.1 Hz, α-anomer H), 4.52 (d, 1H, J1,2 = 7.8 Hz, β-anomer H), 4.48 (d, 1H, J1,2 = 8.2 Hz, β-anomer H), 4.22–4.20 (m, 1H, H-5Fuc), 3.98–3.42 (m, 18H, ring H, OCH2(CH2)3CH2NH), 2.98–2.95 (m, 2H, OCH2(CH2)3CH2NH), 2.02 (s, 3H, Ac), 1.69–1.21 (m, 9H, H-6Fuc, OCH2(CH2)3CH2NH); 13C-NMR (200 MHz, CD3OD) δ 173.6, 103.0, 102.5, 101.8, 79.0, 78.0, 77.1, 76.9, 75.2, 74.1, 73.6, 71.7, 70.7, 70.7, 70.2, 68.3, 62.7, 61.5, 56.6, 40.6, 39.5, 29.8, 28.2, 24.1, 23.0, 16.8. HRMS (ESI) m/z: found [M+Na]+ 637.2791, C25H46N2O15 calcd for [M+Na]+ 637.2790.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
























