Lessons from Chlorophylls: Modifications of Porphyrinoids Towards Optimized Solar Energy Conversion

 ,
,
Abstract
:
1. Introduction

2. Results and Discussion
2.1. Results
2.1.1. Ground- and Excited-State Properties




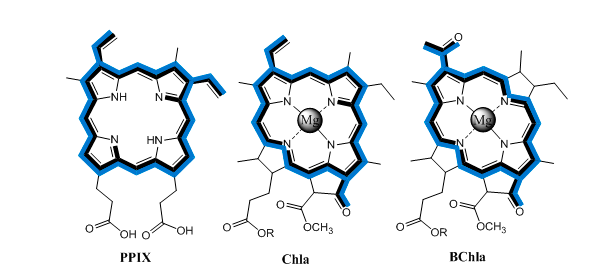
{kind=link}
{kind=link}
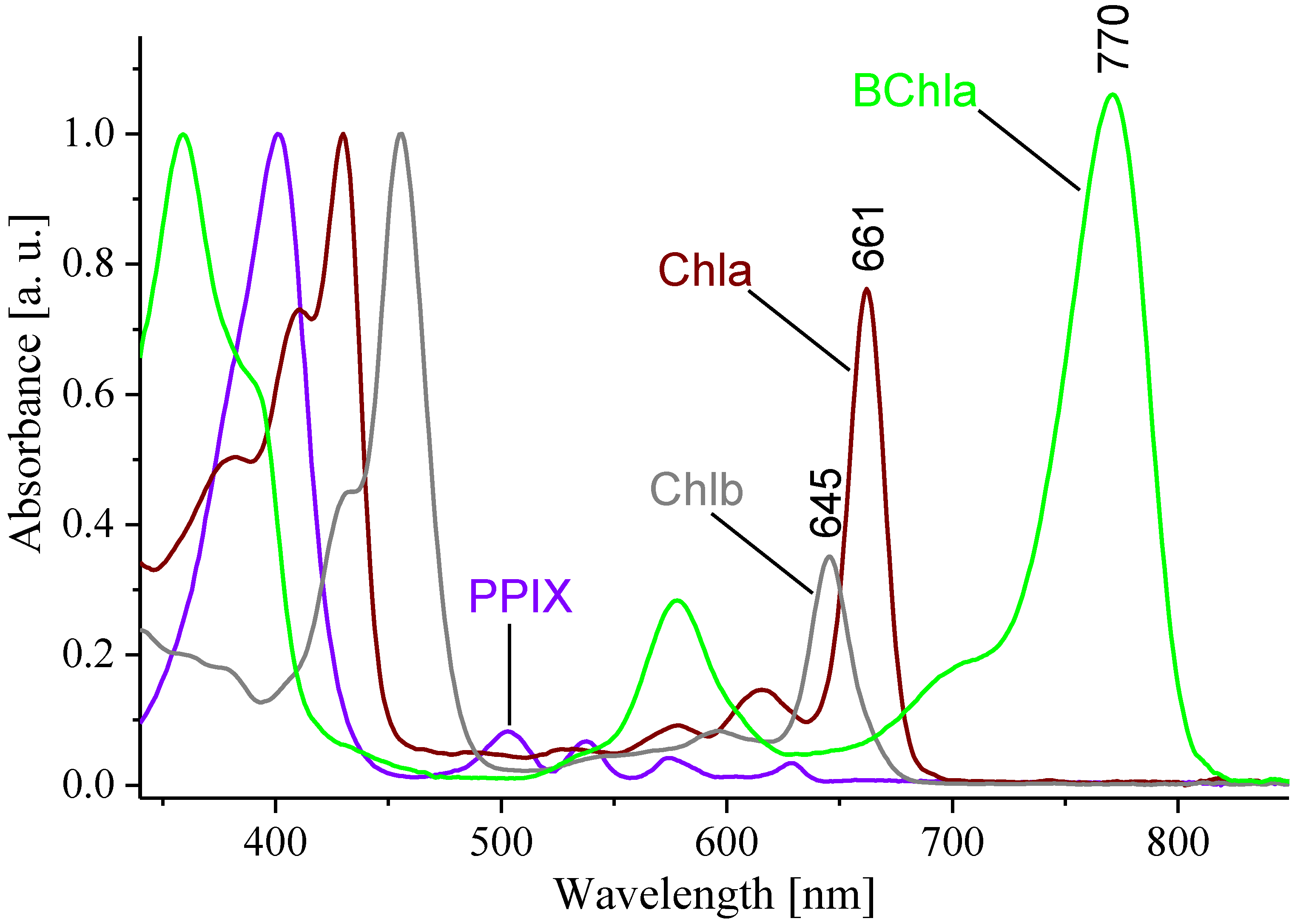{kind=link}
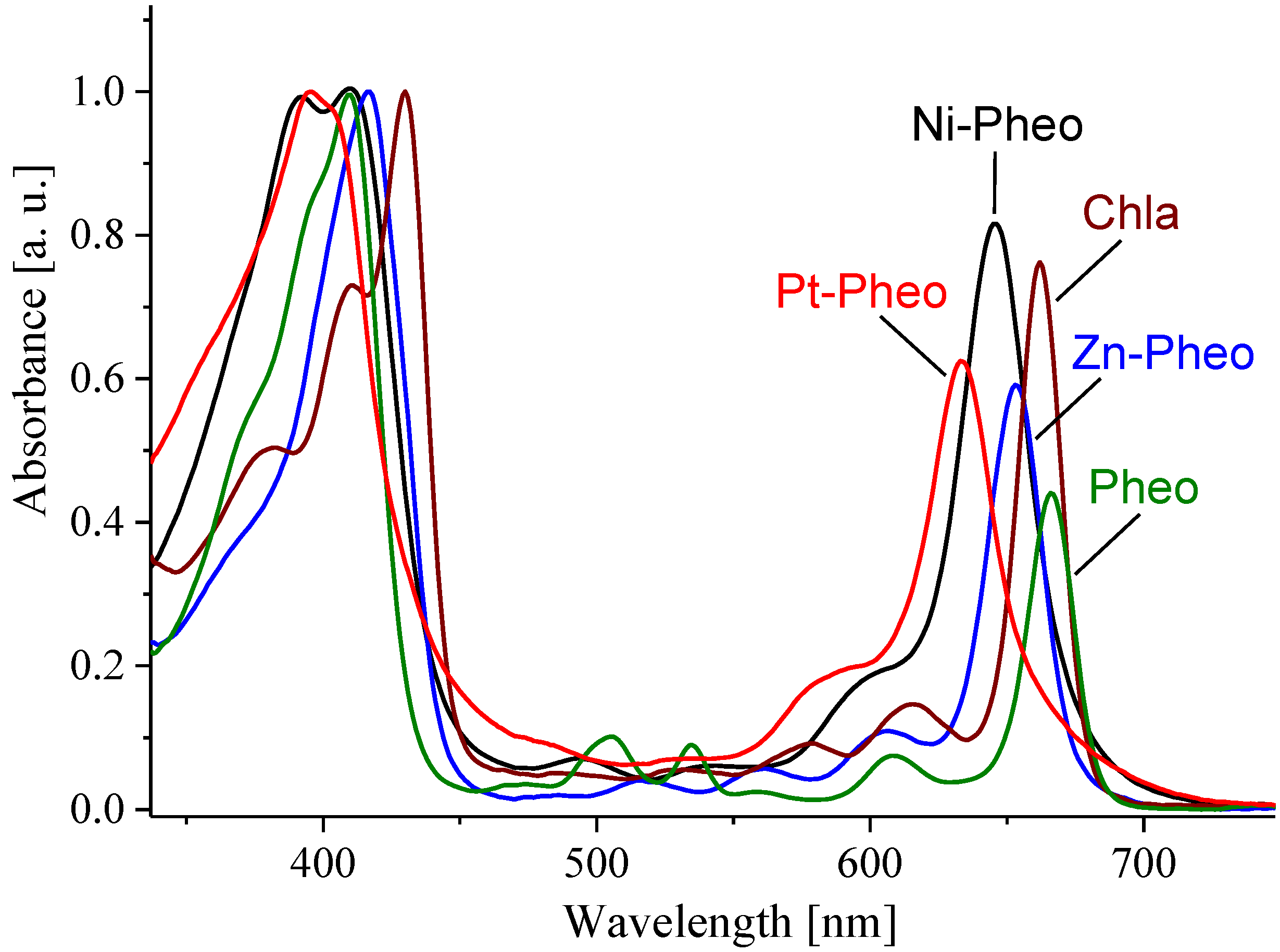{kind=link}
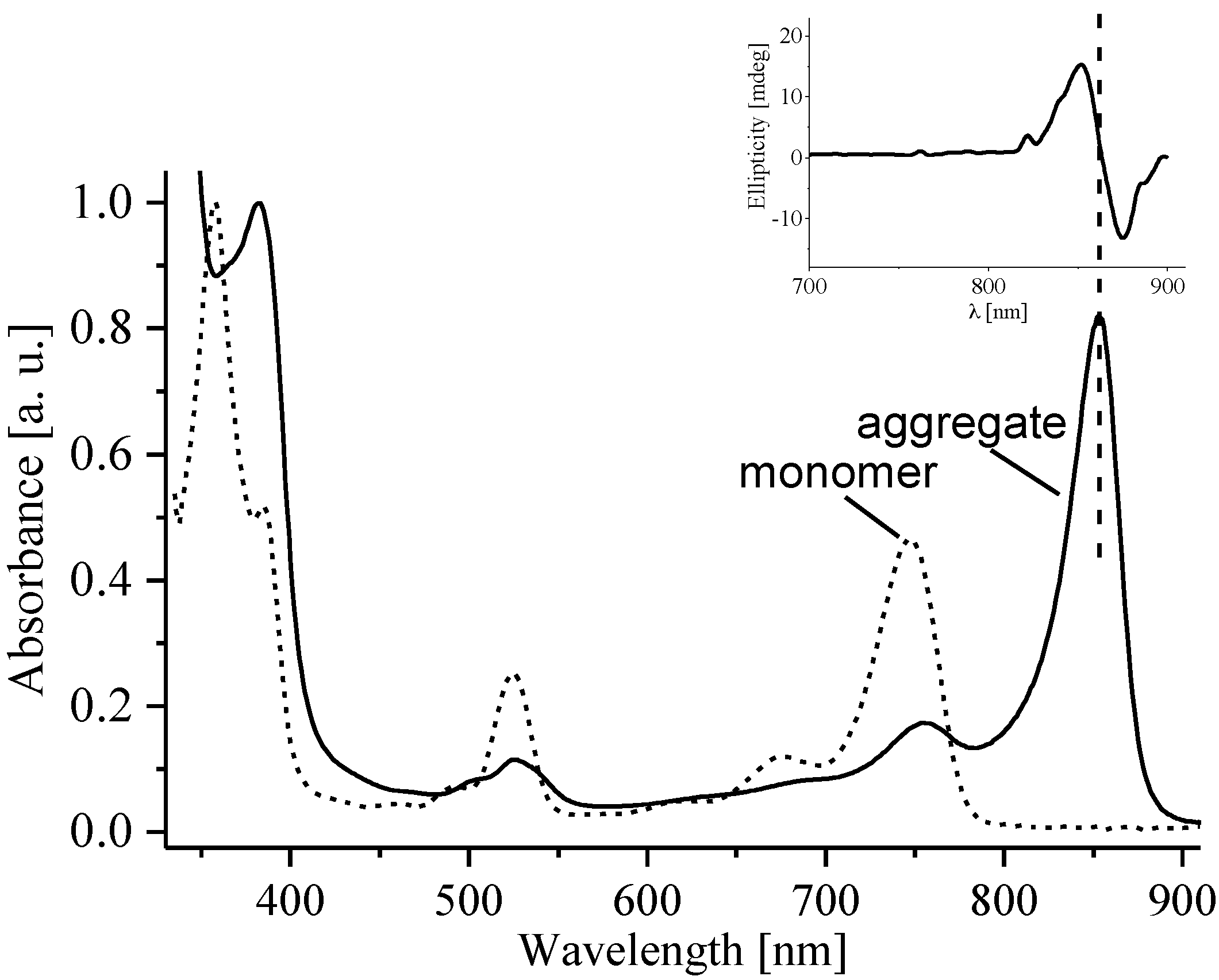{kind=link}
{kind=link}
| Pigment | S1 energy [cm−1] (± 2%) | τfl [ns] (± 2%) | Φfl (± 5%) |
|---|---|---|---|
| PPIX | 15833 | 11.2 | 0.03 |
| Chla | 15025 | 5.7 | 0.27 |
| Chlb | 15401 | 3.7 | 0.20 |
| BChla | 12758 | 2.9 | 0.23 |
| Pheo | 14924 | 6.6 | 0.27 |
| BPheo | 13234 | 2.4 | 0.09 |
| Zn-Pheo | 15216 | 3.3 | 0.13 |
| Ni-Pheo | 15218 a | nd | nd |
| Pt-Pheo | 15205 a | nd | nd |
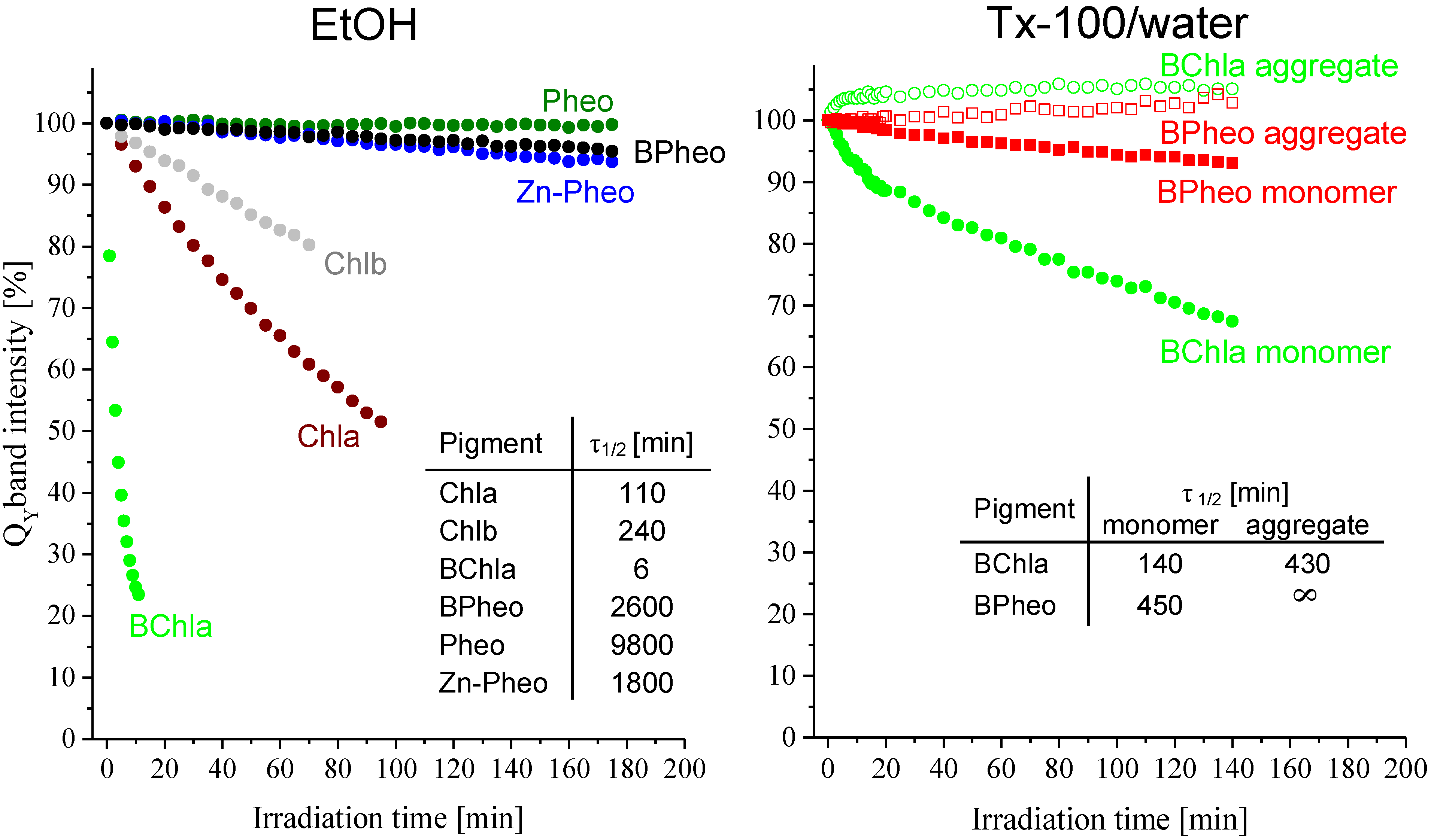
2.1.2. Pigment Photostability

2.2. Discussion
2.2.1. Model System
2.2.2. Symmetry of the Macrocyclic π-Electron System
2.2.3. Type of Central Metal Ion
2.2.4.Peripheral Substituents
2.2.5. Isocyclic Ring
2.2.6. Pigment Photostability
3. Experimental Section
3.1. Materials
3.2. Methods
3.2.1. Isolation and Purification of Pigments
3.2.2. Pigment Aggregation
3.2.3. Photostability Study
3.2.4. Spectroscopy
4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhang, S.F.; Yang, X.D.; Numata, Y.H.; Han, L.Y. Highly efficient dye-sensitized solar cells: Progress and future challenges. Energ. Environ. Sci. 2013, 6, 1443–1464. [Google Scholar] [CrossRef]
- Nocera, D.G. The artificial leaf. Acc. Chem. Res. 2012, 45, 767–776. [Google Scholar] [CrossRef]
- Kobuke, Y. Artificial light-harvesting systems by use of metal coordination. Eur. J. Inorg. Chem. 2006, 2333–2351. [Google Scholar]
- Young, K.J.; Martini, L.A.; Milot, R.L.; Snoeberger, R.C.; Batista, V.S.; Schmuttenmaer, C.A.; Crabtree, R.H.; Brudvig, G.W. Light-driven water oxidation for solar fuels. Coord. Chem. Rev. 2012, 256, 2503–2520. [Google Scholar] [CrossRef]
- Li, Y.; Cai, Z.-L.; Chen, M. Spectroscopic properties of chlorophyll f. J. Phys. Chem. B 2013, 117, 11309–11317. [Google Scholar] [CrossRef]
- Grimm, B.; Porra, R.J.; Rüdiger, W.; Scheer, H. Chlorophylls and Bacteriochlorophylls; Springer: Dordrecht, The Netherlands, 2006; Volume 25, pp. 1–94. [Google Scholar]
- Scheer, H. Chlorophylls; CRC Press: Boca Raton, FL, USA, 1991. [Google Scholar]
- Willows, R.D.; Li, Y.; Scheer, H.; Chen, M. Structure of chlorophyllf. Org. Lett. 2013, 15, 1588–1590. [Google Scholar] [CrossRef]
- Orf, G.S.; Tank, M.; Vogl, K.; Niedzwiedzki, D.M.; Bryant, D.A.; Blankenship, R.E. Spectroscopic insights into the decreased efficiency of chlorosomes containing bacteriochlorophyll f. BBA-Bioenergetics 2013, 1827, 493–501. [Google Scholar] [CrossRef]
- Norris, J.R.; Raghavan, M. Strategies for mimicking the primary events of bacterial photosynthesis—structure, function, and mechanism. In Photochemical Conversion and Storage of Solar Energy; Kluwer Academic Publ.: Dordrecht, The Netherlands, 1991; pp. 141–150. [Google Scholar]
- Henderson, B.W.; Sumlin, A.B.; Owczarczak, B.L.; Dougherty, T.J. Bacteriochlorophyll-a as photosensitizer for photodynamic treatment of transplantable murine tumors. J. Photochem. Photobiol. B 1991, 10, 303–313. [Google Scholar] [CrossRef]
- Fiedor, L.; Gorman, A.A.; Hamblett, I.; Rosenbach-Belkin, V.; Salomon, Y.; Scherz, A.; Tregub, I. A pulsed laser and pulse radiolysis study of amphiphilic chlorophyll derivatives with pdt activity toward malignant melanoma. Photochem. Photobiol. 1993, 58, 506–511. [Google Scholar] [CrossRef]
- Rosenbach-Belkin, V.; Chen, L.; Fiedor, L.; Tregub, I.; Pavlotsky, F.; Brumfeld, V.; Salomon, Y.; Scherz, A. Serine conjugates of chlorophyll and bacteriochlorophyll: Photocytotoxicity in vitro and tissue distribution in mice bearing melanoma tumors. Photochem. Photobiol. 1996, 64, 174–181. [Google Scholar] [CrossRef]
- Blankenship, R.E. Photosynthetic pigments: Structure and spectroscopy. In Molecular Mechanisms of Photosynthesis; Blankenship, R.E., Ed.; Blackwell Science Ltd.: Oxford, UK, 2008; pp. 42–60. [Google Scholar]
- Tamiaki, H.; Kunieda, M. Photochemistry of chlorophylls and their synthetic analogs. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific Publishing Company: Singapore, 2011; Volume 11, pp. 223–290. [Google Scholar]
- Chen, M.; Scheer, H. Extending the limits of natural photosynthesis and implications for technical light harvesting. J. Porphyrins Phthalocyanines 2013, 17, 1–15. [Google Scholar] [CrossRef]
- Brancaleon, L.; Magennis, S.W.; Samuel, I.D.W.; Namdas, E.; Lesar, A.; Moseley, H. Characterization of the photoproducts of protoporphyrin ix bound to human serum albumin and immunoglobuling. Biophys. Chem. 2004, 109, 351–360. [Google Scholar] [CrossRef]
- Weber, G.; Teale, F.W.J. Determination of the absolute quantum yield of fluorescent solutions. Trans. Faraday Soc. 1957, 53, 646–655. [Google Scholar] [CrossRef]
- Fiedor, L.; Stąsiek, M.; Myśliwa-Kurdziel, B.; Strzałka, K. Phytol as one of the determinants of chlorophyll interactions in solution. Photosynth. Res. 2003, 78, 47–57. [Google Scholar] [CrossRef]
- Losev, A.P.; Sagun, E.I.; Kochubeev, G.A.; Nichiporovich, I.N. Fluorescence quantum yields, lifetimes, and critical distances for energy transfer for chlorophyll and its pheophytin in solutions. J. Appl. Spectrosc. 1986, 45, 798–803. [Google Scholar] [CrossRef]
- Fiedor, L.; Kania, A.; Myśliwa-Kurdziel, B.; Stochel, G. Understanding chlorophylls: Central magnesium and phytyl as structural determinants. Biochim. Biophys. Acta 2008, 1777, 1491–1500. [Google Scholar] [CrossRef]
- Drzewiecka-Matuszek, A.; Skalna, A.; Karocki, A.; Stochel, G.; Fiedor, L. Effects of heavy central metal on the ground and excited states of chlorophyll. J. Biol. Inorg. Chem. 2005, 10, 453–462. [Google Scholar] [CrossRef]
- Pilch, M.; Dudkowiak, A.; Jurzyk, B.; Łukasiewicz, J.; Susz, A.; Stochel, G.; Fiedor, L. Molecular symmetry determines the mechanism of a very efficient ultrafast excitation-to-heat conversion in ni-substituted chlorophylls. Biochim. Biophys. Acta 2013, 1827, 30–37. [Google Scholar] [CrossRef]
- Fery-Forgues, S.; Lavabre, D. Are fluorescence quantum yields so tricky to measure? A demonstration using familiar stationery products. J. Chem. Educ. 1999, 76, 1260–1264. [Google Scholar] [CrossRef]
- Morris, J.V.; Mahaney, M.A.; Huber, J.R. Fluorescence quantum yield determinations. 9,10-diphenylanthracene as a reference standard in different solvents. J. Phys. Chem. 1976, 80, 969–974. [Google Scholar] [CrossRef]
- Niedzwiedzki, D.M.; Blankenship, R.E. Singlet and triplet excited state properties of natural chlorophylls and bacteriochlorophylls. Photosynth. Res. 2010, 106, 227–238. [Google Scholar] [CrossRef]
- Rosenbach-Belkin, V.; Fisher, J.R.E.; Scherz, A. Effect of nonexcitonic interactions among the paired molecules on the qy transition of bacteriochlorophyll dimers—applications to the primary electron-donors p-860 and p-960 in bacterial reaction centers. J. Am. Chem. Soc. 1991, 113, 676–678. [Google Scholar] [CrossRef]
- Gouterman, M. Optical spectra and electronic structure of porphyrins and related rings. In The Porphyrins; Dolphin, D., Ed.; Academic Press, Inc: New York, NY, USA, 1978; Volume III, pp. 1–165. [Google Scholar]
- Vernon, L.P.; Seely, G.R. The Chlorophylls; Academic Press: New York, NY, USA, 1966. [Google Scholar]
- Gouterman, M.; Wagniere, G.H.; Snyder, L.C. Spectra of porphyrins. Part ii. Four orbital model. J. Mol. Spectr. 1972, 11, 108–127. [Google Scholar] [CrossRef]
- De Paula, J.C.; Robblee, J.H.; Pasternack, R.F. Aggregation of chlorophyll a probed by resonance light scattering spectroscopy. Biophys. J. 1995, 68, 335–341. [Google Scholar] [CrossRef]
- Beddard, G.S.; Porter, G. Concentration quenching in chlorophyll. Nature 1976, 260, 366–367. [Google Scholar] [CrossRef]
- Hartwich, G.; Fiedor, L.; Simonin, I.; Cmiel, E.; Schäfer, W.; Noy, D.; Scherz, A.; Scheer, H. Metal-substituted bacteriochlorophylls. 1. Preparation and influence of metal and coordination on spectra. J. Am. Chem. Soc. 1998, 120, 3675–3683. [Google Scholar] [CrossRef]
- Küpper, H.; Dedic, R.; Svoboda, A.; Hala, J.; Kroneck, P.M.H. Kinetics and efficiency of excitation transfer from chlorophylls, their heavy metal-substituted derivatives, and pheophytins to singlet oxygen. Biochim. Biophys. Acta 2002, 1572, 107–113. [Google Scholar] [CrossRef]
- Teuchner, K.; Stiel, H.; Leupold, D.; Scherz, A.; Noy, D.; Simonin, I.; Hartwich, G.; Scheer, H. Fluorescence and the excited state absorption in modified pigments of bacterial photosynthesis. A comparative study of metal-substituted bacteriochlorophylls a. J. Lumin. 1997, 612–614. [Google Scholar]
- Callahan, P.M.; Cotton, T.M. Assignment of bacteriochlorophyll a ligation state from absorption and resonance raman spectra. J. Am. Chem. Soc. 1987, 109, 7001–7007. [Google Scholar] [CrossRef]
- Noy, D.; Fiedor, L.; Hartwich, G.; Scheer, H.; Scherz, A. Metal-substituted bacteriochlorophylls. 2. Changes in redox potentials and electronic transition energies are dominated by intramolecular electrostatic interactions. J. Am. Chem. Soc. 1998, 120, 3684–3693. [Google Scholar] [CrossRef]
- Karweik, D.H.; Winograd, N. Nitrogen charge-distributions in free-base porphyrins, metalloporphyrins, and their reduced analogs observed by x-ray photoelectron-spectroscopy. Inorg. Chem. 1976, 15, 2336–2342. [Google Scholar] [CrossRef]
- Kania, A.; Pilch, M.; Rutkowska-Żbik, D.; Susz, A.; Heriyanto; Stochel, G.; Fiedor, L. High-pressure and theoretical studies reveal significant differences in the electronic structure and bonding of magnesium, zinc, and nickel ions in metalloporphyrinoids. Inorg. Chem. 2014, 53, 8473–8484. [Google Scholar] [CrossRef]
- Kania, A.; Fiedor, L. Steric control of bacteriochlorophyll ligation. J. Am. Chem. Soc. 2006, 128, 454–458. [Google Scholar] [CrossRef]
- Sauer, K.; Smith, J.R.L.; Schultz, A.J. The dimerization of chlorophyll a, chlorophyll b, and bacteriochlorophyll in solution. J. Am. Chem. Soc. 1966, 88, 2681–2688. [Google Scholar] [CrossRef]
- Fajer, J. Chlorophyll chemistry before and after crystals of photosynthetic reaction centers. Photosynth. Res. 2004, 80, 165–172. [Google Scholar] [CrossRef]
- Fiedor, J.; Fiedor, L.; Kammhuber, N.; Scherz, A.; Scheer, H. Photodynamics of the bacteriochlorophyll-carotenoid system. 1. Influence of central metal, solvent and β-carotene on photobleaching of bacteriochlorophyll derivatives. Photochem. Photobiol. 2002, 76, 145–152. [Google Scholar]
- Watanabe, T.; Kobayashi, M. Electrochemistry of chlorophylls. In Chlorophylls; Scheer, H., Ed.; CRC Press: Boca Raton, PL, USA, 1991; pp. 287–315. [Google Scholar]
- Barzda, V.; Peterman, E.J.G.; van Grondelle, R.; van Amerongen, H. The influence of aggregation on triplet formation in light-harvesting chlorophyll a/b pigment-protein complex ii of green plants. Biochemistry 1998, 37, 546–551. [Google Scholar] [CrossRef]
- Brody, S.S.; Brody, M. Fluorescence properties of aggregated chlorophyll in vivo and in vitro. Trans. Faraday Soc. 1962, 58, 416–428. [Google Scholar] [CrossRef]
- Krieger-Liszkay, A. Singlet oxygen production in photosynthesis. J. Exp. Bot. 2005, 56, 337–346. [Google Scholar] [CrossRef]
- Iriyama, K.; Ogura, N.; Takamiya, A. A simple method for extraction and partial purification of chlorophyll from plant material, using dioxane. J. Biochem. 1974, 76, 901–904. [Google Scholar]
- Omata, T.; Murata, N. Preparation of chlorophyll a, chlorophyll b and bacteriochlorophyll a by column chromatography with deae-sepharose CL-6B and sepharose CL-6B. Plant Cell Physiol. 1983, 24, 1093–1100. [Google Scholar]
- Jones, I.D.; White, R.C.; Gibbs, E.; Denard, C.D. Absorption spectra of copper and zinc complexes of pheophytins and pheophorbides. J. Agric. Food Chem. 1968, 16, 80–83. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karcz, D.; Boroń, B.; Matwijczuk, A.; Furso, J.; Staroń, J.; Ratuszna, A.; Fiedor, L. Lessons from Chlorophylls: Modifications of Porphyrinoids Towards Optimized Solar Energy Conversion. Molecules 2014, 19, 15938-15954. https://doi.org/10.3390/molecules191015938
Karcz D, Boroń B, Matwijczuk A, Furso J, Staroń J, Ratuszna A, Fiedor L. Lessons from Chlorophylls: Modifications of Porphyrinoids Towards Optimized Solar Energy Conversion. Molecules. 2014; 19(10):15938-15954. https://doi.org/10.3390/molecules191015938
Chicago/Turabian StyleKarcz, Dariusz, Bożena Boroń, Arkadiusz Matwijczuk, Justyna Furso, Jakub Staroń, Alicja Ratuszna, and Leszek Fiedor. 2014. "Lessons from Chlorophylls: Modifications of Porphyrinoids Towards Optimized Solar Energy Conversion" Molecules 19, no. 10: 15938-15954. https://doi.org/10.3390/molecules191015938