Synthesis and Biological Evaluation of Liguzinediol Mono- and Dual Ester Prodrugs as Promising Inotropic Agents

Abstract
:
1. Introduction

2. Results and Discussion
2.1. Synthesis


{kind=link}
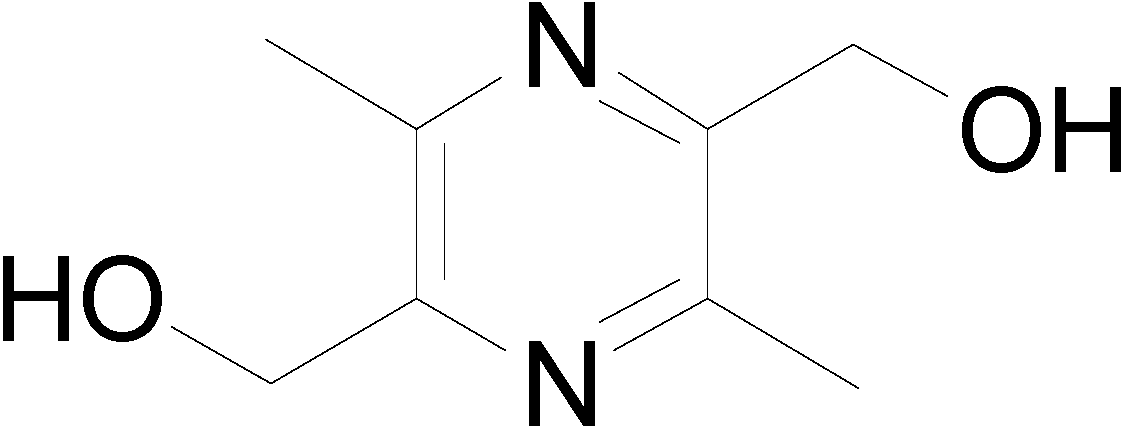{kind=link}
{kind=link}
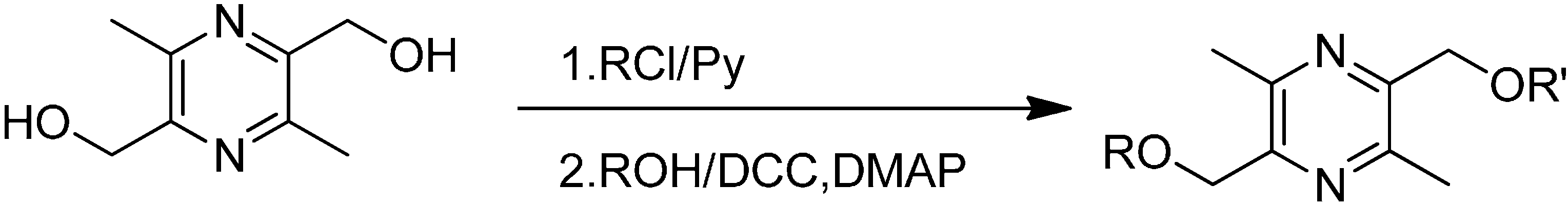{kind=link}
 | |||||
|---|---|---|---|---|---|
| Compound | R | R' | Yield (%) | M.p. (°C) | MW (g/mol) |
| M5 | H | Pivaloyl | 35 | 66–67 | 252.15 |
| M10 | H | n-Decanoyl | 33 | 31–32 | 322.23 |
| M12 | H | n-Dodecanoyl | 37 | 40–41 | 350.26 |
| M16 | H | n-Hexadecanoyl | 30 | 60–62 | 406.32 |
| M18 | H | n-Octadecanoyl | 35 | 65–66 | 434.35 |
| D5 | Pivaloyl | Pivaloyl | 46 | 80–82 | 336.20 |
| D10 | n-Decanoyl | n-Decanoyl | 45 | 46–47 | 476.36 |
| D12 | n-Dodecanoyl | n-Dodecanoyl | 47 | 55–56 | 532.42 |
| D16 | n-Hexadecanoyl | n-Hexadecanoyl | 48 | 74–75 | 644.55 |
| D18 | n-Octadecanoyl | n-Octadecanoyl | 47 | 75–76 | 700.61 |
2.2. Physiochemical Properties
| Compound | Capacity Factor | Log Pa (C log Pb) | Solubility (µg/mL) |
|---|---|---|---|
| Liguzinediol | 0.100 ± 0.004 | −1.12 ± 0.11 (−1.49) | 2.24 × 105 ± 1.4 × 104 |
| M5 | 0.210 ± 0.020 | 1.33 ± 0.10 (0.60) | 8.0 × 103 ± 3.5 × 102 |
| M10 | 0.400 ± 0.015 | 2.48 ± 0.04 (3.60) | 8.89 ± 0.21 |
| M12 | 0.514 ± 0.012 | 3.12 ± 0.15 (4.66) | 2.23 ± 0.11 |
| M16 | 0.834 ± 0.012 | 3.25 ± 0.13 (6.77) | n.d c. |
| M18 | 1.055 ± 0.061 | 3.38 ± 0.20 (7.83) | n.d c. |
| D5 | 0.364 ± 0.039 | 2.50 ± 0.18 (2.70) | 29.90 ± 1.36 |
| D10 | 1.068 ± 0.057 | 2.87 ± 0.16 (8.69) | n.d c. |
| D12 | 1.640 ± 0.032 | 3.77 ± 0.21 (10.80) | n.d c. |
| D16 | 3.911 ± 0.042 | n.d c. | n.d c. |
| D18 | 6.134 ± 0.084 | n.d c. | n.d c. |
2.3. Chemical Stabilities
| Compound | % Unchanged at 24 h (pH 7.4) a | % Max of LZDO Released (80% Human Plasma) b | % Max of LZDO Released (Human Liver Microsomes) b | t1/2 (h) (80%Human Plasma) d | t1/2 (h) (Human Liver Microsomes) d |
|---|---|---|---|---|---|
| M5 | 98.9 ± 0.3 | 79.2 ± 2.2 | 37.5 ± 2.3 | 2.6 ± 0.1 | 5.1 ± 0.2 |
| M10 | 98.9 ± 0.3 | 85.4 ± 3.1 | 108.1 ± 3.2 | 2.3 ± 0.2 | 2.1 ± 0.6 |
| M12 | 99.7 ± 0.2 | 64.2 ± 2.3 | 105.1 ± 3.4 | 2.6 ± 0.5 | 2.2 ± 0.1 |
| M16 | 99.4 ± 0.4 | 44.5 ± 2.2 | 46.3 ± 2.3 | 3.3 ± 0.6 | 3.3 ± 0.7 |
| M18 | 98.2 ± 0.6 | 15.6 ± 1.6 | 15.9 ± 1.2 | 9.6 ± 1.1 | 9.5 ± 0.9 |
| D5 | 96.1 ± 0.2 | 28.6 ± 2.4 | 31.3 ± 1.4 | 3.4 ± 0.4 | 3.2 ± 0.3 |
| D10 | 98.7 ± 0.4 | 23.4 ± 1.2 | 30.9 ± 1.8 | 4.2 ± 0.4 | 4.1± 0.5 |
| D12 | 99.8 ± 0.5 | 8.7 ± 1.7 | 8.6 ± 1.1 | 7.9 ± 1.0 | 7.9 ± 0.9 |
| D16 | stable c | n.d. e | n.d. e | n.d. e | n.d. e |
| D18 | stable c | n.d. e | n.d. e | n.d. e | n.d. e |
2.4. In Vitro Metabolism
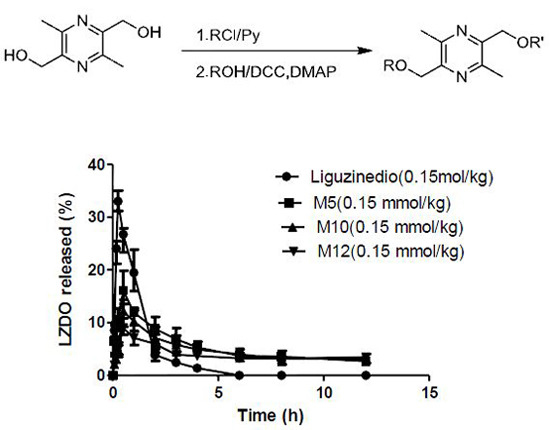
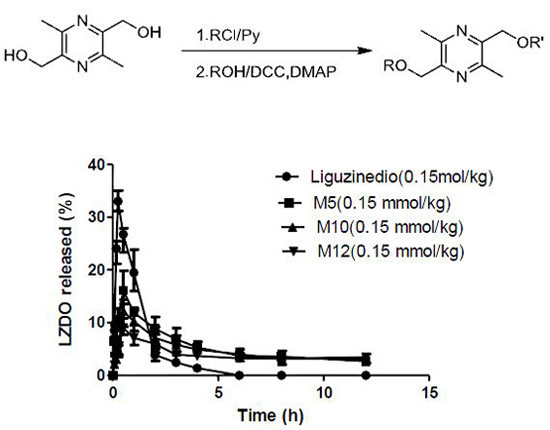
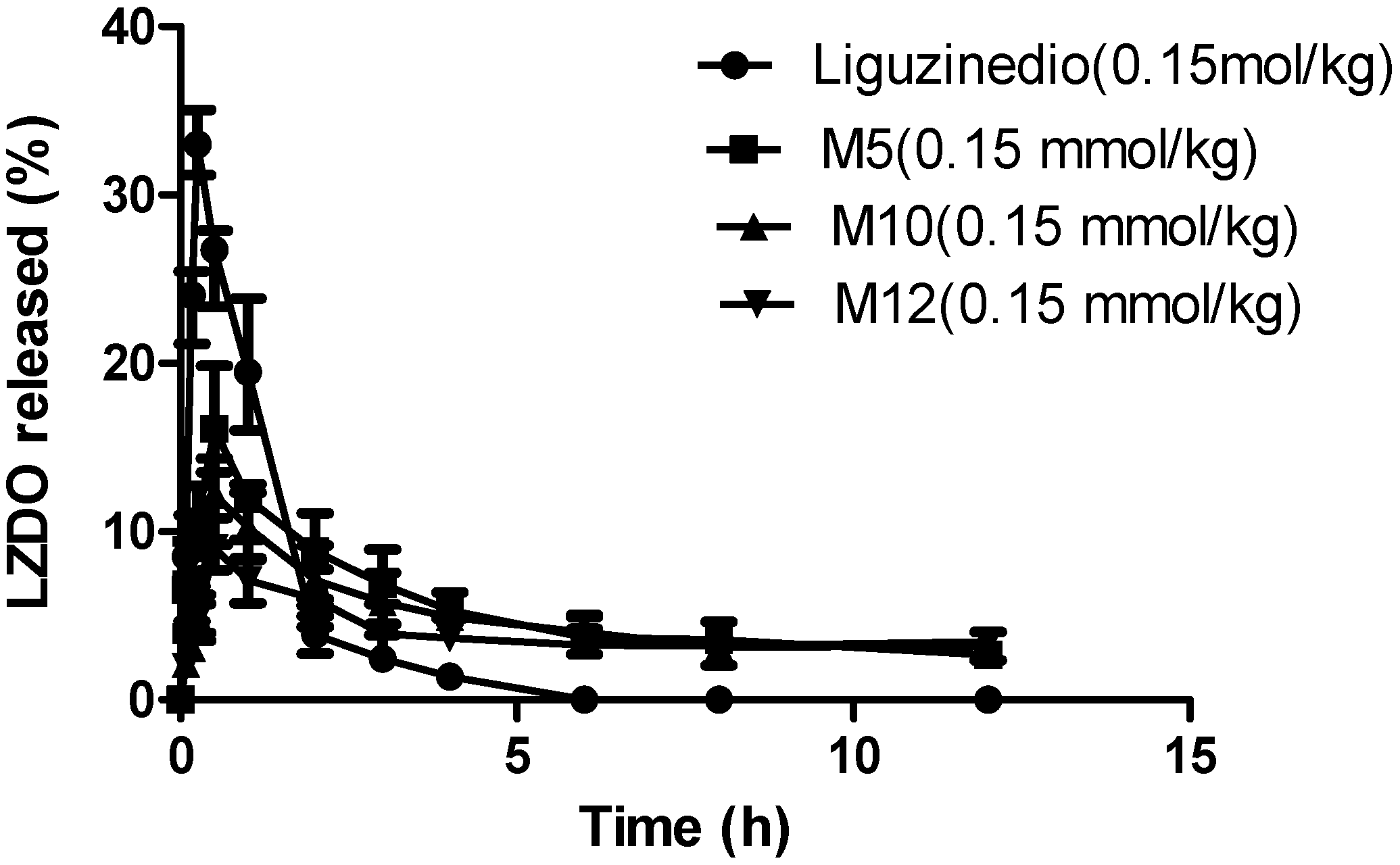
2.5. In Vivo Pharmacokinetics
| Compound | Liguzinediol | M5 | M10 | M12 |
|---|---|---|---|---|
| Dose | 25.2 mg/kg (0.15 mmol/kg) | 37.8 mg/kg (0.15 mmol/kg) | 48.3 mg/kg (0.15 mmol/kg) | 52.5 mg/kg (0.15 mmol/kg) |
| Tmax (h) | 0.25 | 0.50 | 0.50 | 0.50 |
| Cmax (μg·mL−1) | 33.00 ± 1.59 | 16.56 ± 2.54 | 12.07 ± 1.69 | 9.07 ± 1.11 |
| t1/2 (h) | 1.06 ± 0.47 | 7.81 ± 5.43 | 9.99 ± 8.64 | 19.96 ± 9.96 |
| CL/F (L·h−1·kg−1) | 0.59 ± 0.04 | 0.45 ± 0.22 | 0.57 ± 0.24 | 0.43 ± 0.19 |
| AUC0–12h (μg·h·mL−1) | 40.43 ± 2.26 | 64.58 ± 9.03 | 57.78 ± 3.31 | 49.06 ± 3.24 |

3. Experimental Section
3.1. Materials and Solutions
3.2. Synthesis
3.2.1. Preparation of 3,6-Dimethyl-5-hydroxymethyl-2-pivaloyloxymethylpyrazine (M5) and 3,6-Dimethyl-2,5-dipivaloyloxymethylpyrazine (D5)
3.2.2. General Preparation Procedure for Linear Chain Esters
3.3. Physicochemical Properties
3.3.1. Determination of Lipophilic Index
3.3.2. Partition Coefficient Determination
3.3.3. Solubility Studies
3.4. Hydrolysis in Phosphate Buffer (pH 7.4)
3.5. Hydrolysis in Human Serum
3.6. Metabolism Studies in Human Liver Microsomes
3.7. In Vivo Pharmacokinetic Studies
3.7.1. Rat Experiment
3.7.2. Sample Treatment
3.7.3. Pharmacokinetics and Statistical Analysis
3.8. Analytical Procedures
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cowie, M.R.; Mosterd, A.; Wood, D.A.; Deckers, J.W.; Poole-Wilson, P.A.; Sutton, G.C.; Grobbeef, D.E. The epidemiology of heart failure. Eur. Heart J. 1997, 18, 208–225. [Google Scholar] [CrossRef] [PubMed]
- Toller, W.G.; Stranz, C. Levosimendan, a new inotropic and vasodilator agent. Anesthesiology 2006, 104, 556–569. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.N.; Goldstein, S.O.; Greenberg, B.H.; Lorell, B.H.; Bourge, R.C.; Jaski, B.E.; Gottlieb, S.O.; Demets, D.L.; White, B.G. A dose-dependent increase in mortality with vesnarinone among patients with severe heart failure. New. Engl. J. Med. 1998, 339, 1810–1816. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Gattis, W.A.; Uretsky, B.F.; Adams, K.F.; McNulty, S.E.; Grossman, S.H.; McKenna, W.J.; Zannad, F.; Swedberg, K.; Gheorghiade, M.; et al. Continuous intravenous dobutamine is associated with an increased risk of death in patients with advanced heart failure: Insights from the Flolan International Randomized Survival Trial (FIRST). Am. Heart J. 1999, 138, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Cuffe, M.S.; Califf, R.M.; Adams, K.F., Jr.; Benza, R.; Bourge, R.; Colucci, W.S.; Massie, B.M.; O’Connor, C.M.; Pina, I.; Quigg, R.; et al. Short-term intravenous milrinone for acute exacerbation of chronic heart failure: A randomized controlled trial. JAMA 2002, 287, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Gjesdal, K.; Feyzi, J.; Olsson, S.B. Digitalis: A dangerous drug in atrial fibrillation? An analysis of the SPORTIF III and V data. Heart 2008, 94, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xu, Y.; Li, W.; Wu, H.; Luo, Z.K.; Li, X.H.; Huang, F.F.; Young, C.; Liu, Z.; Zhou, S.Y. The novel compound LZDO exerts positive inotropic effects in isolated rat heart via sarcoplasmic reticulum Ca2+ ATPase-dependent mechanism. Life Sci. 2012, 91, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Larissa, L.; Elie, R.C.; Lahouaria, H.; Anne-Marie, L.; Roger, J.H. Sarcoplasmic reticulum Ca2+ ATPase as a therapeutic target for heart failure. Expert Opin. Biol. Ther. 2010, 10, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, S.Y.; Li, W.; Wen, H.M.; Bian, H.M.; Chen, L. Liguzinediol induced positive inotropic effect in normal isolated rat hearts. Chin. J. New Drugs Clin. Remedies 2009, 28, 293–296. [Google Scholar]
- Liu, Z.; Bian, H.M.; Chen, L.; Li, W.; Wen, H.M. Effect of liguzinediol on cardiac hemodynamics in healty rats. Chin. Pharm. J. 2009, 44, 1155–1158. [Google Scholar]
- Zhang, D.N.; Guo, Y.; Li, Z.Q.; Li, W.; Bian, H.M. Effect of liguzinediol on ventricular remodeling in press-overloaded rats. Chin. Pharmacol. Bull. 2012, 28, 1699–1704. [Google Scholar]
- Guo, Y.; Zhou, J.; Bian, H.M.; Zhu, Q.; Li, W.; Wu, Y. The effect of liguzinediol on hemodynamics in rats with acute heart failure induced by pentobarbital. Chin. J. Exp. Tradit. Med. Formulae 2012, 18, 170–174. [Google Scholar]
- Xu, Y.; Luo, Z.K; Liu, Q.M.; Huang, F.F.; Li, X.H.; Liu, L.; Li, W.; Chen, L. Positive inotropic mechanism of liguzinediol and heart safety evaluation. Chin. J. Pharmacol. Toxicol. 2012, 26, 151–156. [Google Scholar]
- Wen, K.; Liu, Z.; Bian, H.M.; Li, W. Acute toxicity of liguzinediol. Chin. J. New Drugs Clin. Remedies 2011, 30, 234–235. [Google Scholar]
- Li, W.; Chen, L.; Bian, H.M.; Wen, H.M.; Liu, Z. Application of 2,5-dihydroxymethyl-3,6-dimethyl-pyrazine and its derivatives in pharmacy. U.S. Patent 8158630 B2, 17 April 2012. [Google Scholar]
- Li, W.; Chen, L.; Bian, H.M.; Wen, H.M.; Liu, Z. Application of 2,5-dihydroxymethyl-3,6-dimethyl-pyrazine and its derivatives in pharmacy. CN Patent ZL200810157140.4, 17 November 2010. [Google Scholar]
- Zhang, L.; Li, W.; Wen, H.M.; Shan, C.X.; Bian, H.M. An LC-MS/MS method for determining liguzinediol in rat plasma and studying its pharmacokinetics. Chin. J. New Drugs 2013, 22, 1024–1046. [Google Scholar]
- Shan, C.X.; Li, W.; Wen, H.M.; Wang, X.Z.; Zhu, Y.H.; Cui, X.B. Application of 2,5-dihydroxymethyl-3,6-dimethyl pyrazine and its derivatives in pharmacy. J. Pharm. Biomed. Anal. 2012, 62, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.X.; Li, W.; Chen, L.; Wen, H.M.; Zhu, X.W. Synthesis and positive inotroic effect of methyl-substituted derivatives of liguzinediol. Chin. J. New Drugs Clin. Remedies 2014, 33, 357–363. [Google Scholar]
- Chen, L.; Li, W.; Chen, L.; Liu, Z. Synthesis and positive inotropic effects of the secondary alcohol derivatives of liguzinediol. Chin. Pharm. J. 2013, 48, 1118–1122. [Google Scholar]
- Ettmayer, P.; Amidon, G.L.; Clement, B.; Testa, B. Lessons learned from marketed and investigational prodrugs. J. Med. Chem. 2004, 47, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Bonina, F.; Puglia, C.; Rimoli, M.G.; Avallone, L.; Abignente, E.; Boatto, G.; Nieddu, M.; Meli, R.; Amorena, M.; Caprariis, P.D. Synthesis and in vitro chemical and enzymatic stability of glycosyl 3'-azido-3'-deoxythymidine derivatives as potential anti-HIV agents. Eur. J. Pharm. Sci. 2002, 16, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Järvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, W.; Wen, H.M.; Bian, H.M.; Zhang, J.; Chen, L.; Chen, L.; Yang, K.D. Synthesis, biological evaluation, and pharmacokinetic study of novel LZDO prodrugs. Molecules 2013, 18, 4561–4572. [Google Scholar] [CrossRef]
- Simões, M.F.; Valente, E.; Gómez, J.R.; Anes, E.; Constantino, L. Lipophilic pyrazinoic acid amide and ester prodrugs. Eur. J. Pharm. Sci. 2009, 37, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Høyemd, S.; Bruheim, S.; Mælandsmoa, G.; Standal, M.; Olberg, D.E.; Brudeli, B.; Åsberg, A.; Klaveness, J.; Rongvedd, P. Didanosine ester prodrugs: Synthesis, albumin binding properties and pharmacokinetic studies in rats. Eur. J. Med. Chem. 2009, 44, 3874–3879. [Google Scholar] [CrossRef] [PubMed]
- Vacondio, F.; Silva, C.; Lodola, A.; Carmi, C.; Rivara, S.; Duranti, A.; Tontini, A.; Sanchini, S.; Clapper, J.R.; Piomelli, D.; et al. Biphenyl-3-yl alkylcarbamates as fatty acid amide hydrolase (FAAH) inhibitors: Steric effects of N-alkyl chain on rat plasma and liver stability. Eur. J. Med. Chem. 2011, 46, 4466–4473. [Google Scholar] [CrossRef] [PubMed]
- Rai, B.J.; Liu, Z.D.; Liu, D.Y.; Lu, S.L.; Hider, R.C. Synthesis, physiochemical properties and biological evaluation of ester prodrugs of 3-hydroxypyridin-4-ones: Design of orally active chelators with clinical potential. Eur. J. Med. Chem. 1999, 34, 475–485. [Google Scholar] [CrossRef]
- Neises, B.; Steglich, W. Simple method for the esterification of carboxylic acids. Angew. Chem. Int. Ed. Engl. 1978, 17, 522–524. [Google Scholar] [CrossRef]
- Rautio, J.; Nevalainen, T.; Taipale, H.; Vepsalainen, J.; Gynther, J.; Pedersen, T.; Järvinen, T. Synthesis and In vitro Evaluation of aminoacyloxyalkyl esters of 2–(6-methoxy-2-naphthyl) propionic acid as novel naproxen prodrugs for dermal drug delivery. Pharm. Res. 1999, 16, 1172–1178. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.V.; Maddalena, D.J.; Agatonovic-Kustrin, S. Bioavailability Prediction Based on Molecular Structure for a Diverse Series of Drugs. Pharm. Res. 2004, 21, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.D.; Sung, J.H.; Lee, D.W.; Kim, J.S.; Jeon, E.M.; Kim, D.D.; Kim, D.W.; Kim, J.O.; Piao, M.G.; Li, D.X.; et al. Evaluation of physicochemical properties, skin permeation and accumulation profiles of salicylic acid amide prodrugs as sunscreen agent. Int. J. Pharm. 2011, 419, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Dias, C.S.; Anand, B.S.; Mitra, A.K. Effect of mono- and di-acylation on the ocular disposition of ganciclovir: Physicochemical properties, ocular bioreversion, and antiviral activity of short chain ester prodrugs. J. Pharm. Sci. 2001, 91, 660–668. [Google Scholar] [CrossRef]
- Shao, Z.; Park, G.B.; Krishnamoorthy, R.; Mitra, A.K. The physiochemical properties, plasma enzymatic hydrolysis, and nasal absorption of acyclovir and its 2'-ester prodrugs. Pharm. Res. 1994, 11, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Lazzarato, L.; Donnola, M.; Rolando, B.; Chegaev, K.; Marini, E.; Cena, C.; Stilo, A.D.; Fruttero, R.; Biondi, S.; Ongini, E.; et al. (Nitrooxyacyloxy)methyl Esters of Aspirin as Novel Nitric Oxide Releasing Aspirins. J. Med. Chem. 2009, 52, 5058–5068. [Google Scholar] [CrossRef] [PubMed]
- National Research Council. Guide for the Care and Use of Laboratory Animals, 1st ed.National Academy Press: Washington, DC, USA, 1996; pp. 21–25.
- Yan, Y.D.; Kim, H.K.; Seo, K.H.; Lee, W.S.; Lee, G.S.; Woo, J.S.; Yong, C.S.; Choi, H.G. The Physicochemical Properties, in vitro Metabolism and Pharmacokinetics of a Novel Ester Prodrug of EXP3174. Mol. Pharm. 2010, 7, 2132–2140. [Google Scholar] [CrossRef] [PubMed]
- Anand, B.S.; Katragadda, S.; Mitra, A.K. Pharmacokinetics of novel dipeptide ester prodrugs of acyclovir after oral administration: Intestinal absorption and liver metabolism. J. Pharmacol. Exp. Ther. 2004, 311, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Sample Availability: Samples of all the compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Li, W.; Wen, H.-M.; Zhu, H.-H.; Wang, T.-L.; Cheng, D.; Yang, K.-D.; Chen, Y.-Q. Synthesis and Biological Evaluation of Liguzinediol Mono- and Dual Ester Prodrugs as Promising Inotropic Agents. Molecules 2014, 19, 18057-18072. https://doi.org/10.3390/molecules191118057
Zhang J, Li W, Wen H-M, Zhu H-H, Wang T-L, Cheng D, Yang K-D, Chen Y-Q. Synthesis and Biological Evaluation of Liguzinediol Mono- and Dual Ester Prodrugs as Promising Inotropic Agents. Molecules. 2014; 19(11):18057-18072. https://doi.org/10.3390/molecules191118057
Chicago/Turabian StyleZhang, Jing, Wei Li, Hong-Mei Wen, Hao-Hao Zhu, Tian-Lin Wang, Dong Cheng, Kun-Di Yang, and Yu-Qing Chen. 2014. "Synthesis and Biological Evaluation of Liguzinediol Mono- and Dual Ester Prodrugs as Promising Inotropic Agents" Molecules 19, no. 11: 18057-18072. https://doi.org/10.3390/molecules191118057