Synthesis of Linear Geranylphenols and Their Effect on Mycelial Growth of Plant Pathogen Botrytis cinerea


Abstract
: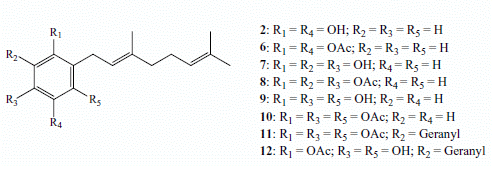
1. Introduction


2. Results and Discussion
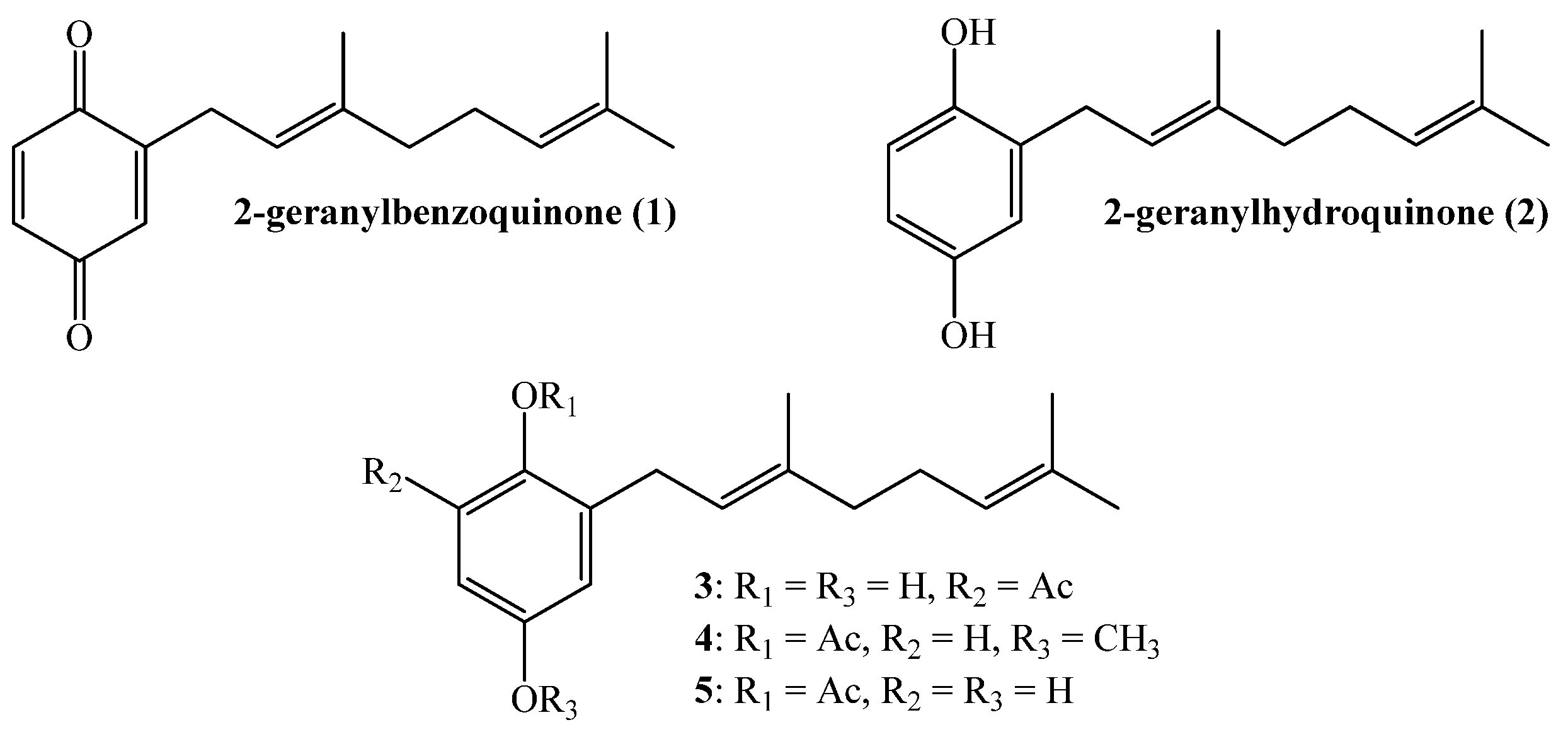
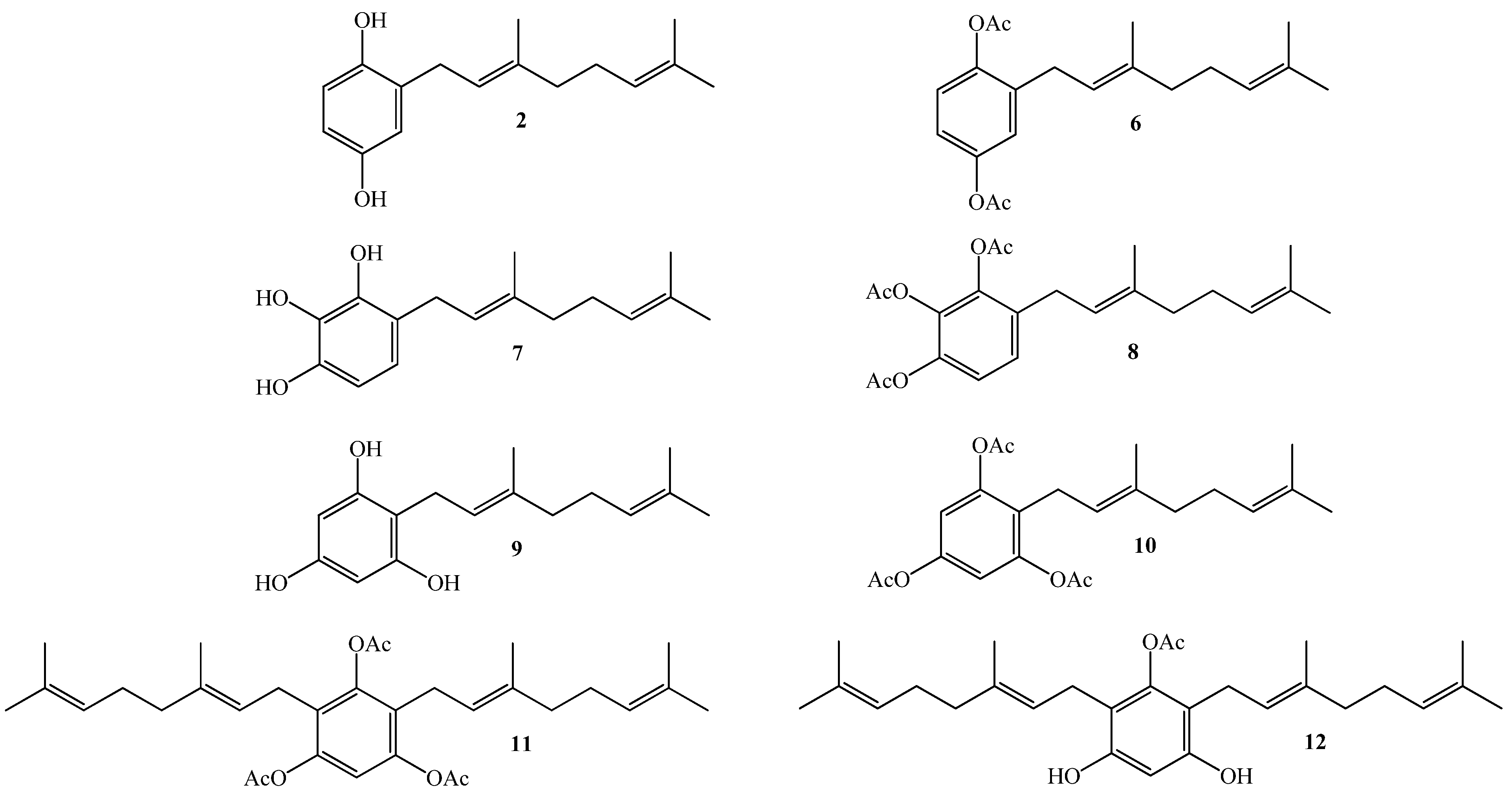
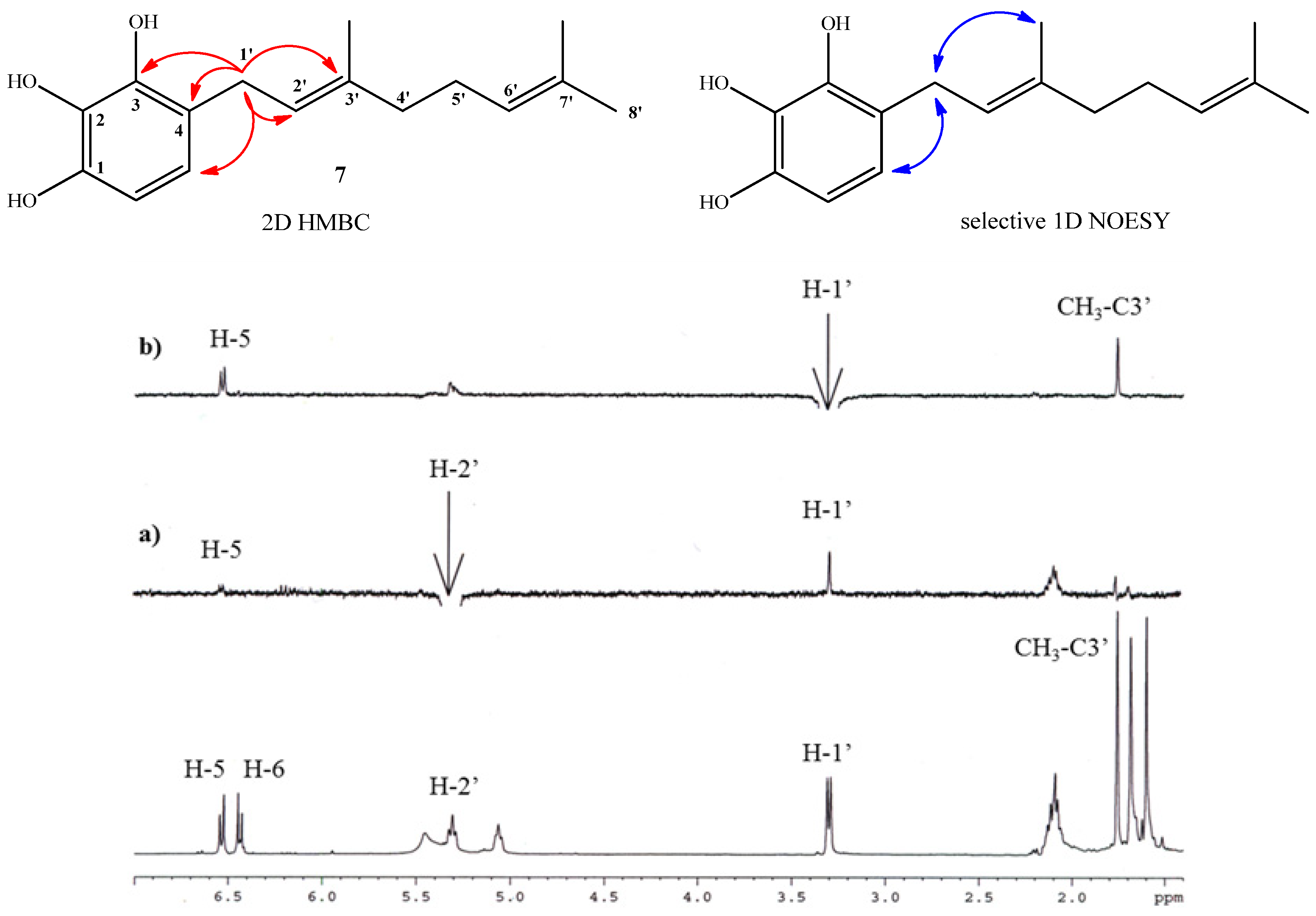
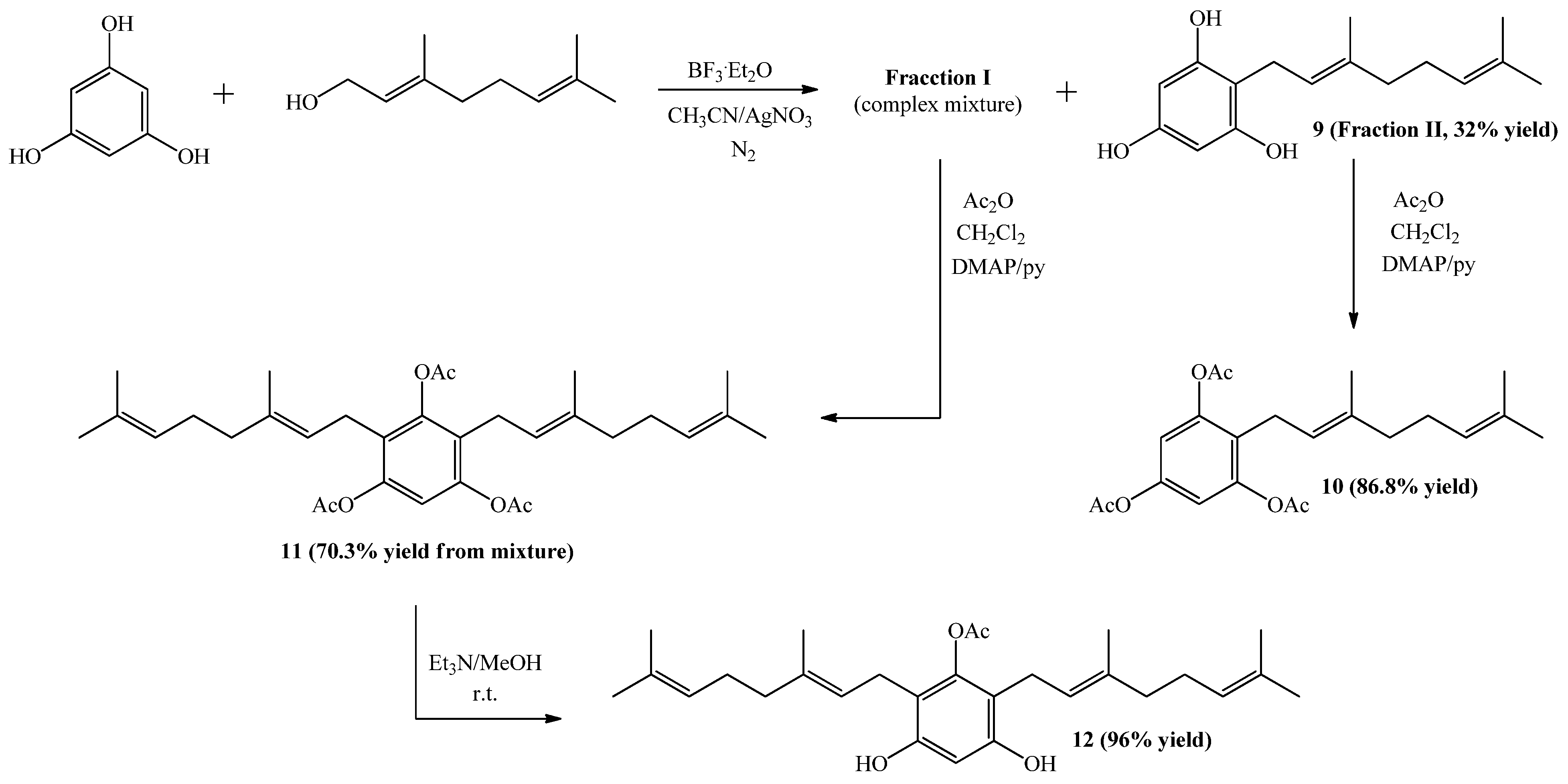
2.1. Chemistry




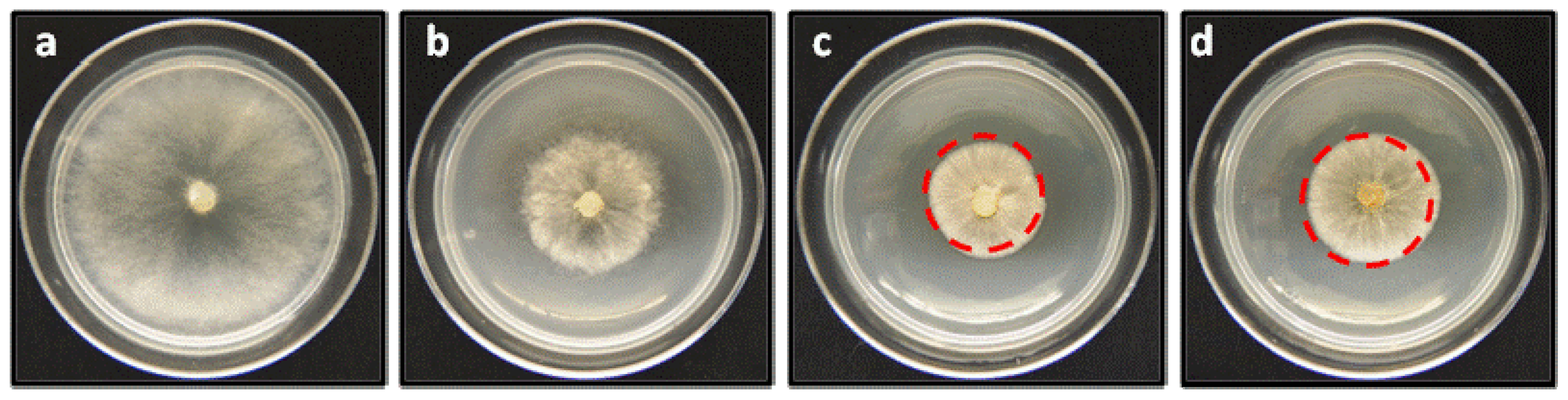
2.2. Anti-phytopathogenic Activity against Botrytis cinerea


{kind=link}
{kind=link}
{kind=link}
{kind=link}
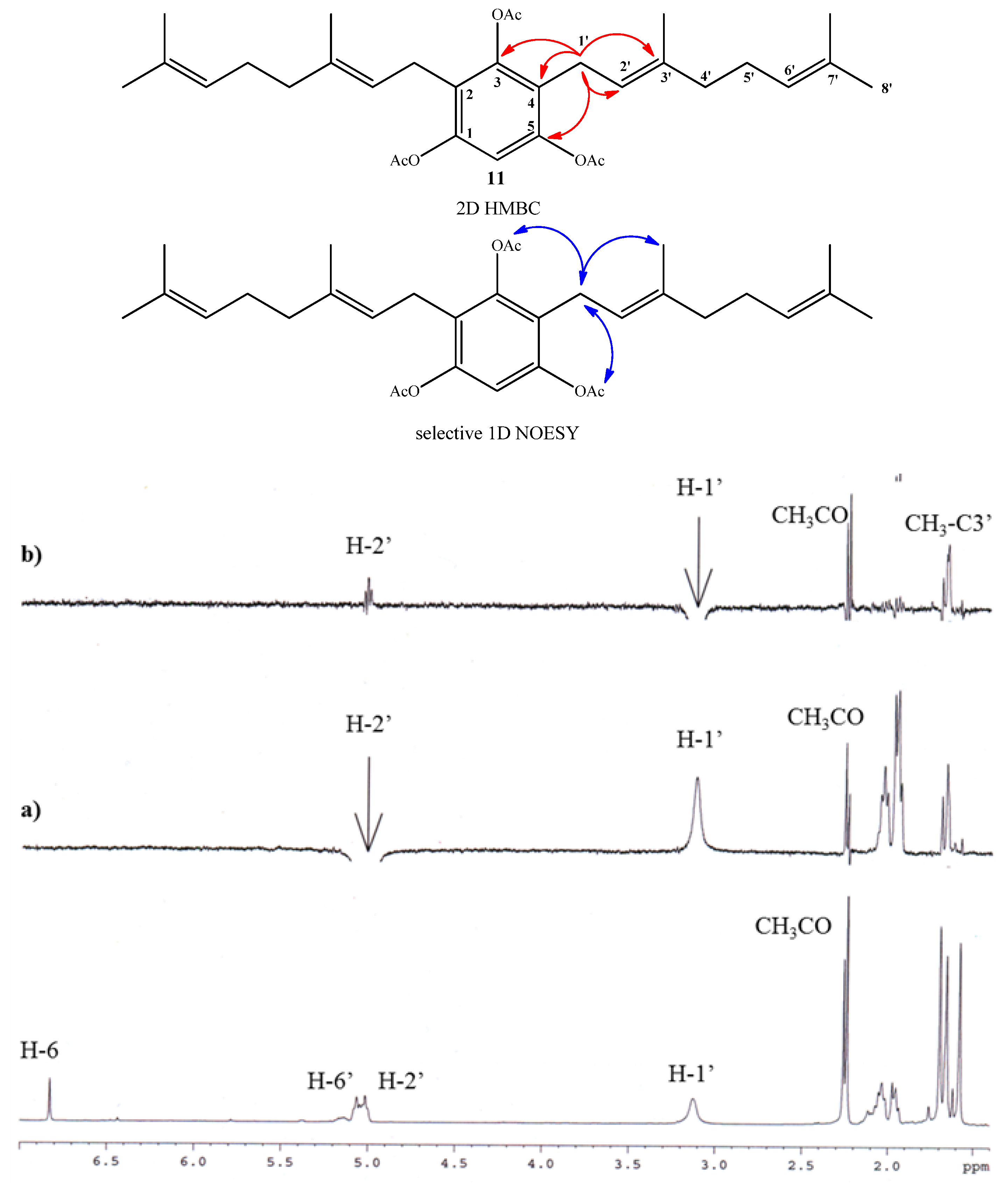{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Percentage of inhibition on in vitro mycelial growth of Botrytis cinerea (%) a | ||
|---|---|---|---|
| 50 mg/L | 150 mg/L | 250 mg/L | |
| 2 | 58 ± 2.15 | 82 ± 3.04 | 86 ± 1.35 |
| 6 | 40 ± 7.20 | 67 ± 1.67 | 76 ± 1.54 |
| 7 | 45 ± 1.36 | 63 ± 0.65 | 72 ± 1.78 |
| 8 | 28 ± 2.44 | 56 ± 5.70 | 64 ± 1.76 |
| 9 | 38 ± 0.21 | 66 ± 7.95 | 81 ± 2.57 |
| 10 | 33 ± 5.48 | 56 ± 1.80 | 72 ± 0.87 |
| 11 | 0 ± 0.00 | 16 ± 4.10 | 26 ± 0.00 |
| 12 | 0 ± 0.00 | 30.4 ± 2.30 | 66 ± 3.90 |
| C– | 0 ± 0.00 | 0 ± 0.00 | 0 ± 0.00 |
| Captan | 39 ± 7.33 | 60 ± 8.45 | 80 ± 2.92 |
3. Experimental
3.1. General
3.2. Synthesis of Geranylphenol Derivatives
3.3. Fungal Isolate and Culture Condition
3.4. Effect of the Compounds on the Mycelial Growth of Botrytis Cinerea In Vitro
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Latorre, B.A.; Flores, V.; Sara, A.M.; Roco, A. Dicarboximide-resistant isolates of Botrytis cinerea from table grape in Chile-survey and characterization. Plant Dis. 1994, 78, 990–994. [Google Scholar] [CrossRef]
- Wedge, D.E.; Camper, N.D. Agrochemicals and Pharmaceuticals. In Biologically Active Natural Products; Cutler, H.G., Cutler, S.J., Eds.; CRC Press: Boca Raton, FL, USA, 2000. [Google Scholar]
- Fenical, W. 4th Proceeding of Food Drugs from the Sea; Marine Technological Society: Washington, DC, USA, 1974. [Google Scholar]
- Lin, H.C.; Chang, W.L. Phytochemical and pharmacological study on Salvia miltiorihiza (I) isolation of new tanshinones. Chin. Pharm. 1991, 43, 11–17. [Google Scholar]
- Manners, G.D. The hydroquinone terpenoids of Cordia alliodora. J. Chem. Soc. Perkin Trans. 1 1977, 1977, 405–410. [Google Scholar] [CrossRef]
- Reynolds, G.; Rodriguez, E. Geranylhydroquinone: A contact allergen from trichomes of Phaceliacrenulata. Phytochemistry 1979, 18, 1567–1568. [Google Scholar] [CrossRef]
- Reynolds, G.; Epstein, W.L.; Terry, D.; Rodriguez, E. A potent contact allergen of Phacelia (Hydrophyllaceae). Contact Derm. 1980, 6, 272–274. [Google Scholar] [CrossRef]
- Reynolds, G.; Rodriguez, E. Prenylated phenols that cause contact dermatitis from trichomes of Phaceliaixodes. Planta Med. 1981, 43, 187–193. [Google Scholar] [CrossRef]
- Sato, A.; Shindo, T.; Kasanuki, N.; Hasegawa, K. Antioxidant metabolites from the tunicate Amaroucium multiplicatum. J. Nat. Prod. 1989, 52, 975–981. [Google Scholar] [CrossRef]
- Benslimane, A.F.; Pouchus, Y.F.; Leboterff, J.; Verbist, J.F.; Roussakis, C.; Monniot, F. Cytotoxic and antibacterial substances from the ascidian Aplidium antillense. J. Nat. Prod. 1988, 51, 582–583. [Google Scholar] [CrossRef]
- De Rosa, S.; de Giulio, A.; Iodice, C. Biological effects of prenylated hydroquinones: Structure-activity relationship studies in antimicrobial, brine shrimp, and fish lethality assays. J. Nat. Prod. 1994, 57, 1711–1716. [Google Scholar] [CrossRef]
- Rudali, G.; Menetrier, L. Action de la géranyl-hydroquinone sur différents cancers spontanés et provoqués chez celles souris. Therapie 1967, 22, 895–899. [Google Scholar]
- Rudali, G. Research on the radioprotective action of geranyl-hydroquinone. C. R. Seances Soc. Biol. Ses. Fil. 1966, 160, 1365–1369. [Google Scholar]
- Rodriguez, E. Cytotoxic and Insecticidal Chemicals of Desert Plants. In Plant Resistance to Insects; Hedin, P., Ed.; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1983; Volume 208, Chapter 17. [Google Scholar]
- Sladic, D.; Gasic, M.J. Reactivity and biological activity of the marine sesquiterpene hydroquinone avarol and related compounds from sponges of the order Dictyoceratida. Molecules 2006, 11, 1–33. [Google Scholar] [CrossRef]
- Baeza, E.; Catalan, K.; Pena-Cortes, H.; Espinoza, L.; Villena, J.; Carrasco, H. Synthesis of geranylhydroquinone derivatives with potential cytotoxic activity. Quim. Nova 2012, 35, 523–526. [Google Scholar] [CrossRef]
- Baeza, E.; Catalan, K.; Villena, J.; Carrasco, H.; Cuellar, M.; Espinoza, L. Synthesis and cytotoxic activity of geranylmethoxyhydroquinone derivatives. J. Chil. Chem. Soc. 2012, 57, 1219–1223. [Google Scholar] [CrossRef]
- Taborga, L.; Vergara, A.; Osorio, M.; Carvajal, M.; Madrid, A.; Marilaf, F.; Carrasco, H.; Espinoza, L. Synthesis and NMR structure determination of new linear geranylphenols by direct geranylation of activated phenols. J. Chil. Chem. Soc. 2013, 58, 1790–1796. [Google Scholar] [CrossRef]
- Takenaka, K.; Tanigaki, Y.; Patil, M.L.; Rao, C.V.L.; Takizawa, S.; Suzuki, T.; Sasai, H. Enantioselective 6-endo-trig Wacker-type cyclization of 2-geranylphenols: Application to a facile synthesis of (−)-cordiachromene. Tetrahedron Asymmetry 2010, 21, 767–770. [Google Scholar] [CrossRef]
- Inouye, H.; Tokura, K.; Tohita, S. Uber die Inhaltsstoffe von Pirolaceen. XV Zur Struktur des Pirolatins. Chem. Ber. 1968, 101, 4057–4065. [Google Scholar] [CrossRef]
- Raikar, S.B.; Nuhant, P.; Delpech, B.; Marazano, C. Synthesis of polyprenylated benzoylphloroglucinols by regioselective prenylation of phloroglucinol in an aqueous medium. Europ. J. Org. Chem. 2008, 2008, 1358–1369. [Google Scholar] [CrossRef]
- Fedorov, S.N.; Radchenko, O.S.; Shubina, L.K.; Balaneva, N.N.; Bode, A.M.; Stonik, V.A.; Dong, Z.G. Evaluation of cancer-preventive activity and structure-activity relationships of 3-demethylubiquinone Q2, isolated from the ascidian Aplidium glabrum, and its synthetic analogs. Pharm. Res. 2006, 23, 70–81. [Google Scholar] [CrossRef]
- Hou, Z.; Yang, R.; Zhang, C.; Zhu, L.; Miao, F.; Yang, X.; Zhou, L. 2-(Substituted phenyl)-3,4-dihydroisoquinolin-2-iums as novel antifungal lead compounds: Biological evaluation and structure-activity relationships. Molecules 2013, 18, 10413–10424. [Google Scholar] [CrossRef]
- Danelutte, A.P.; Lago, J.H.G.; Young, M.C.M.; Kato, M.J. Antifungal flavanones and prenylated hydroquinones from Piper crassinervium Kunth. Phytochemistry 2003, 64, 555–559. [Google Scholar] [CrossRef]
- Lago, J.H.G.; Ramos, C.S.; Casanova, D.C.C.; Morandim, A.D.; Bergamo, D.C.B.; Cavalheiro, A.J.; Bolzani, V.D.; Furlan, M.; Guimaraes, E.F.; Young, M.C.M.; et al. Benzoic acid derivatives from piper species and their fungitoxic activity against Cladosporium cladosporioides and C. sphaerospermum. J. Nat. Prod. 2004, 67, 1783–1788. [Google Scholar] [CrossRef]
- Malami, I. Prenylated benzoica acid derivates from piper species as source of anti-infective agents. Int. J. Pharm. Sci. Res. 2012, 3, 1554–1559. [Google Scholar]
- Mendoza, L.; Espinoza, P.; Urzua, A.; Vivanco, M.; Cotoras, M. In vitro antifungal activity of the diterpenoid 7á-hydroxy-8(17)-labden-15-oic acid and its derivatives against Botrytis cinerea. Molecules 2009, 14, 1966–1979. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 2, 6–12 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Espinoza, L.; Taborga, L.; Díaz, K.; Olea, A.F.; Peña-Cortés, H. Synthesis of Linear Geranylphenols and Their Effect on Mycelial Growth of Plant Pathogen Botrytis cinerea. Molecules 2014, 19, 1512-1526. https://doi.org/10.3390/molecules19021512
Espinoza L, Taborga L, Díaz K, Olea AF, Peña-Cortés H. Synthesis of Linear Geranylphenols and Their Effect on Mycelial Growth of Plant Pathogen Botrytis cinerea. Molecules. 2014; 19(2):1512-1526. https://doi.org/10.3390/molecules19021512
Chicago/Turabian StyleEspinoza, Luis, Lautaro Taborga, Katy Díaz, Andrés F. Olea, and Hugo Peña-Cortés. 2014. "Synthesis of Linear Geranylphenols and Their Effect on Mycelial Growth of Plant Pathogen Botrytis cinerea" Molecules 19, no. 2: 1512-1526. https://doi.org/10.3390/molecules19021512