Theoretical Study on the Allosteric Regulation of an Oligomeric Protease from Pyrococcus horikoshii by Cl− Ion

Abstract
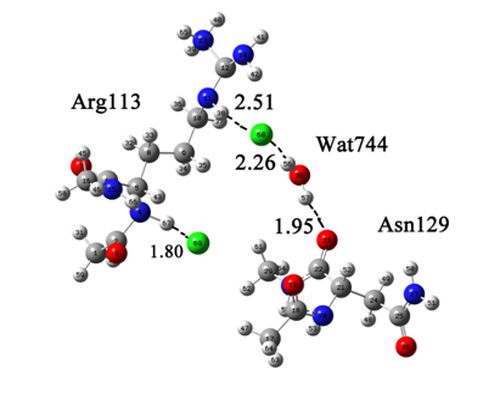
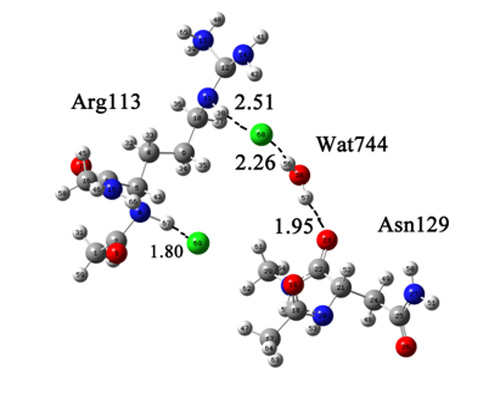
:
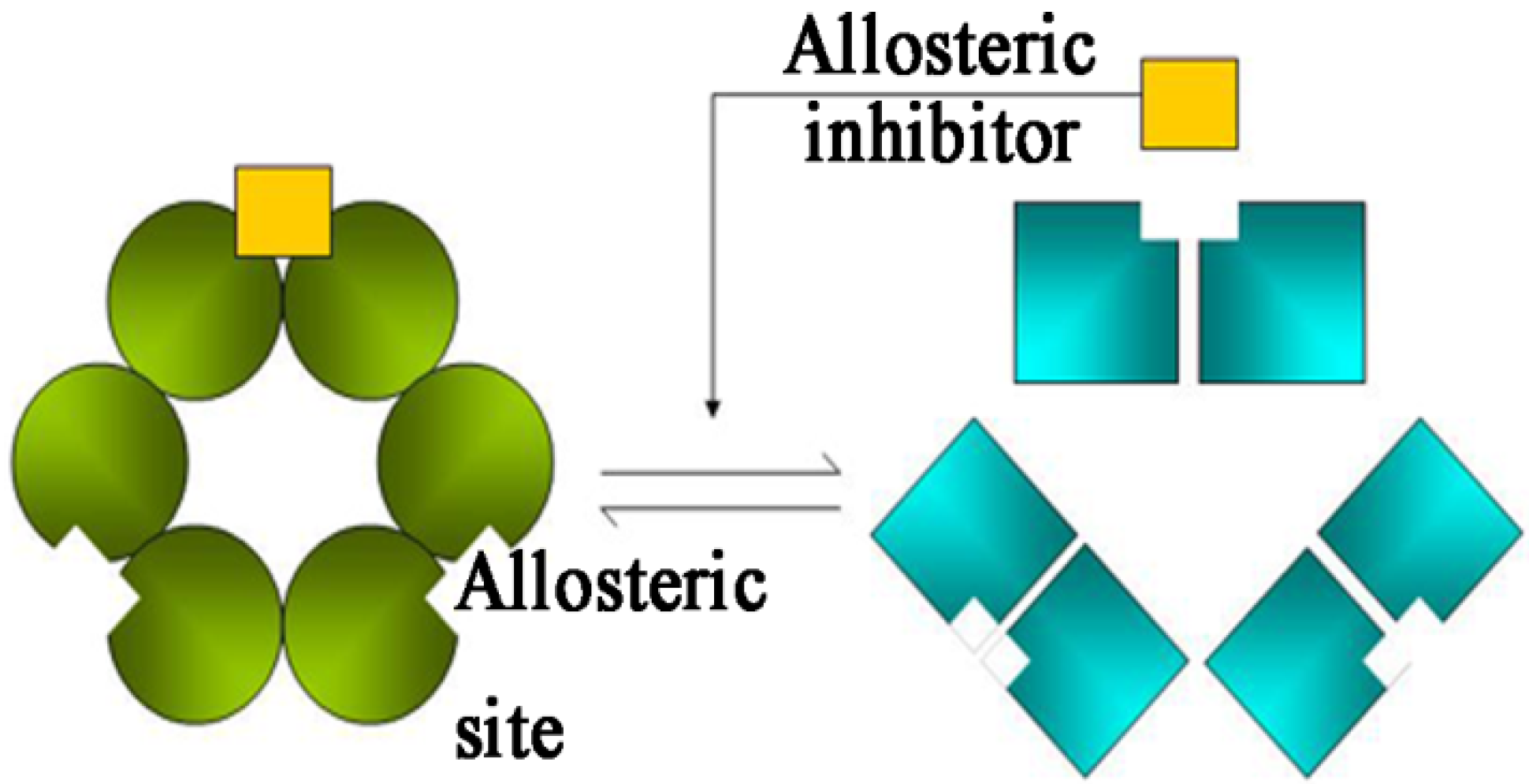
1. Introduction

2. Results and Discussion
2.1. Quantum Mechanical Calculation to Determine the Cl− Binding Mode


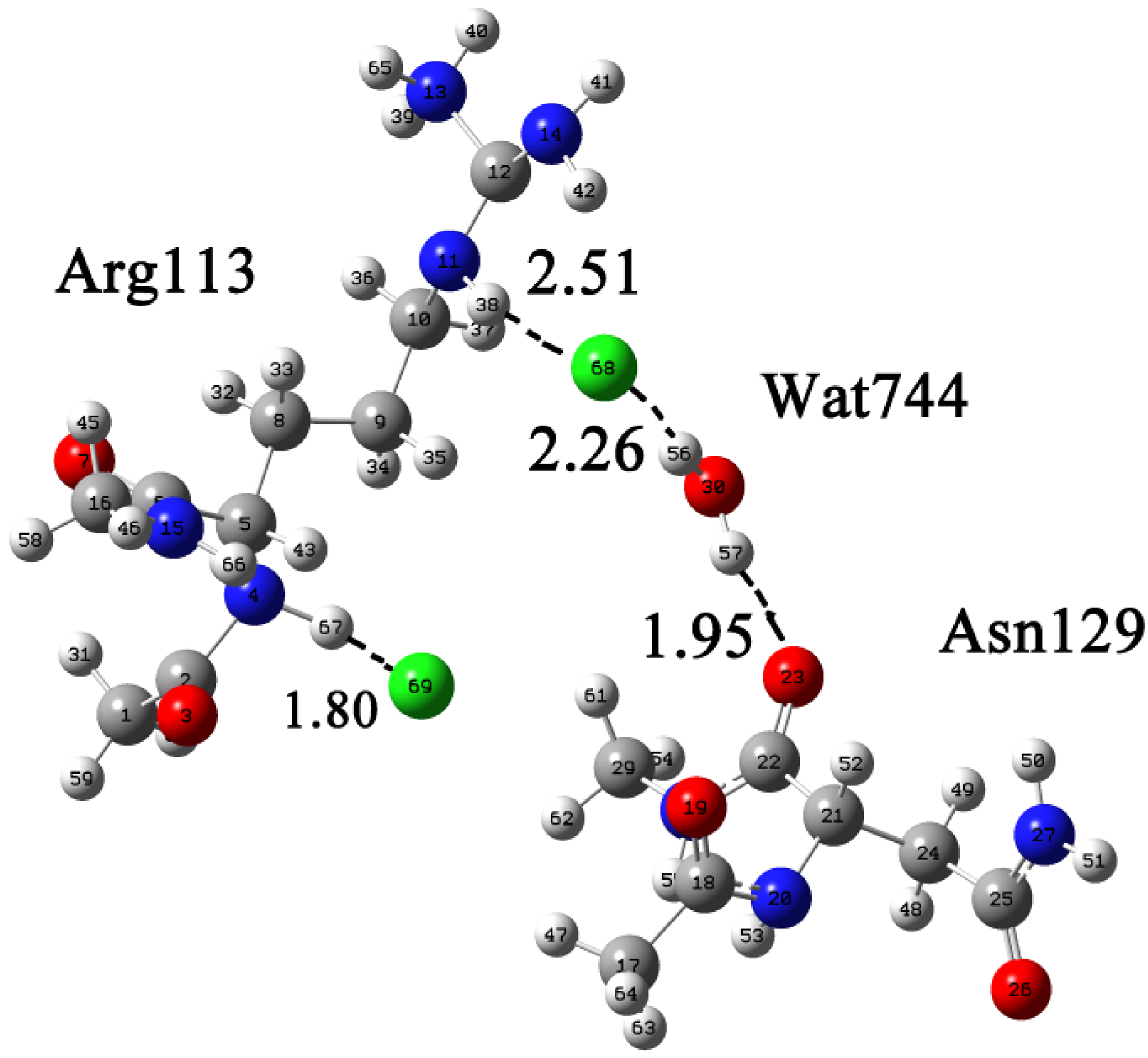
2.2. Protein-Substrate Complex Preparation

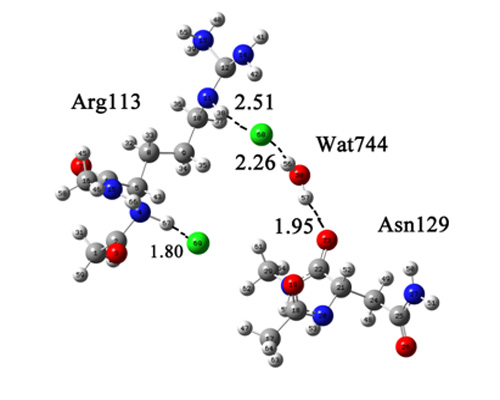{kind=link}
{kind=link}
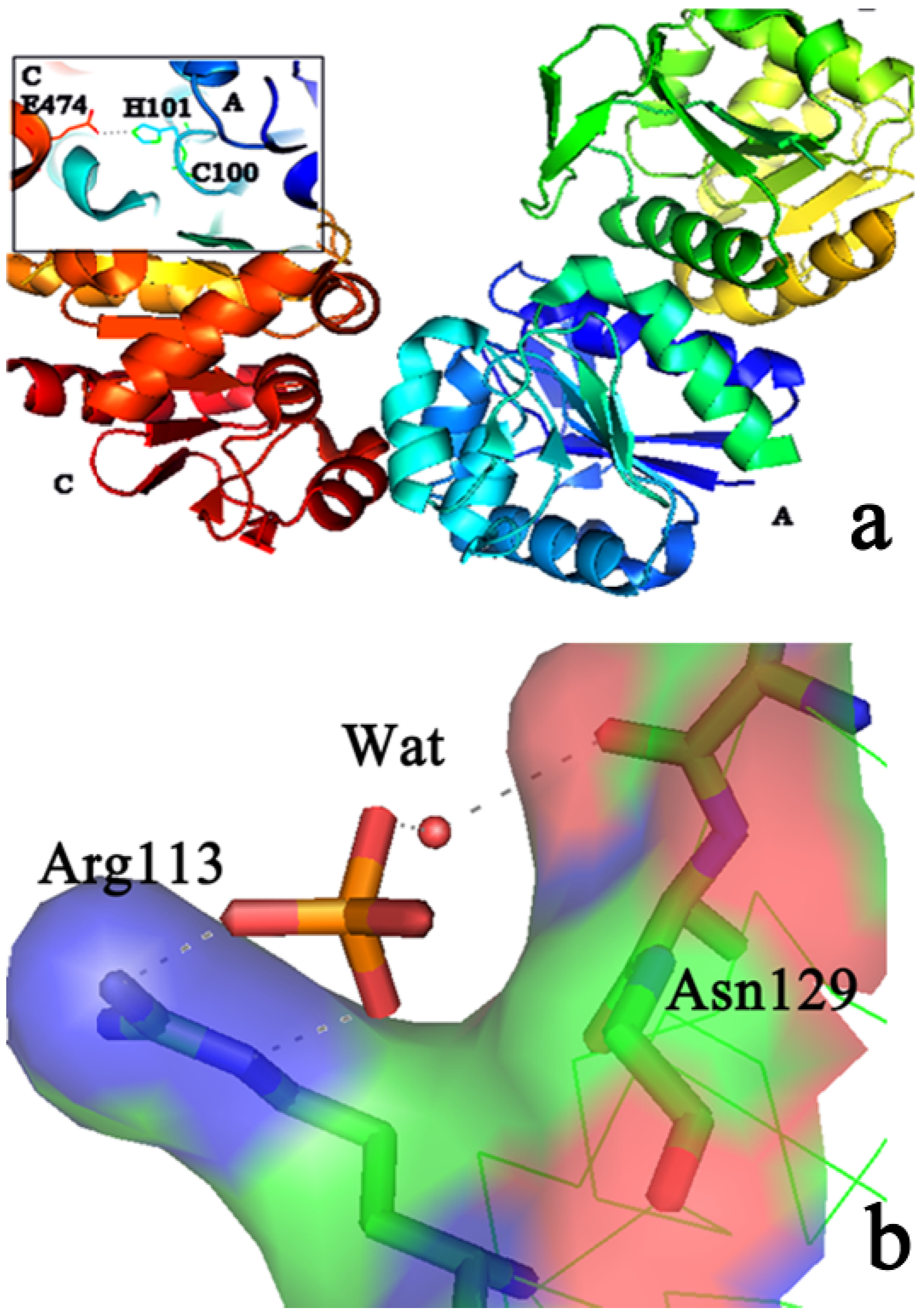{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
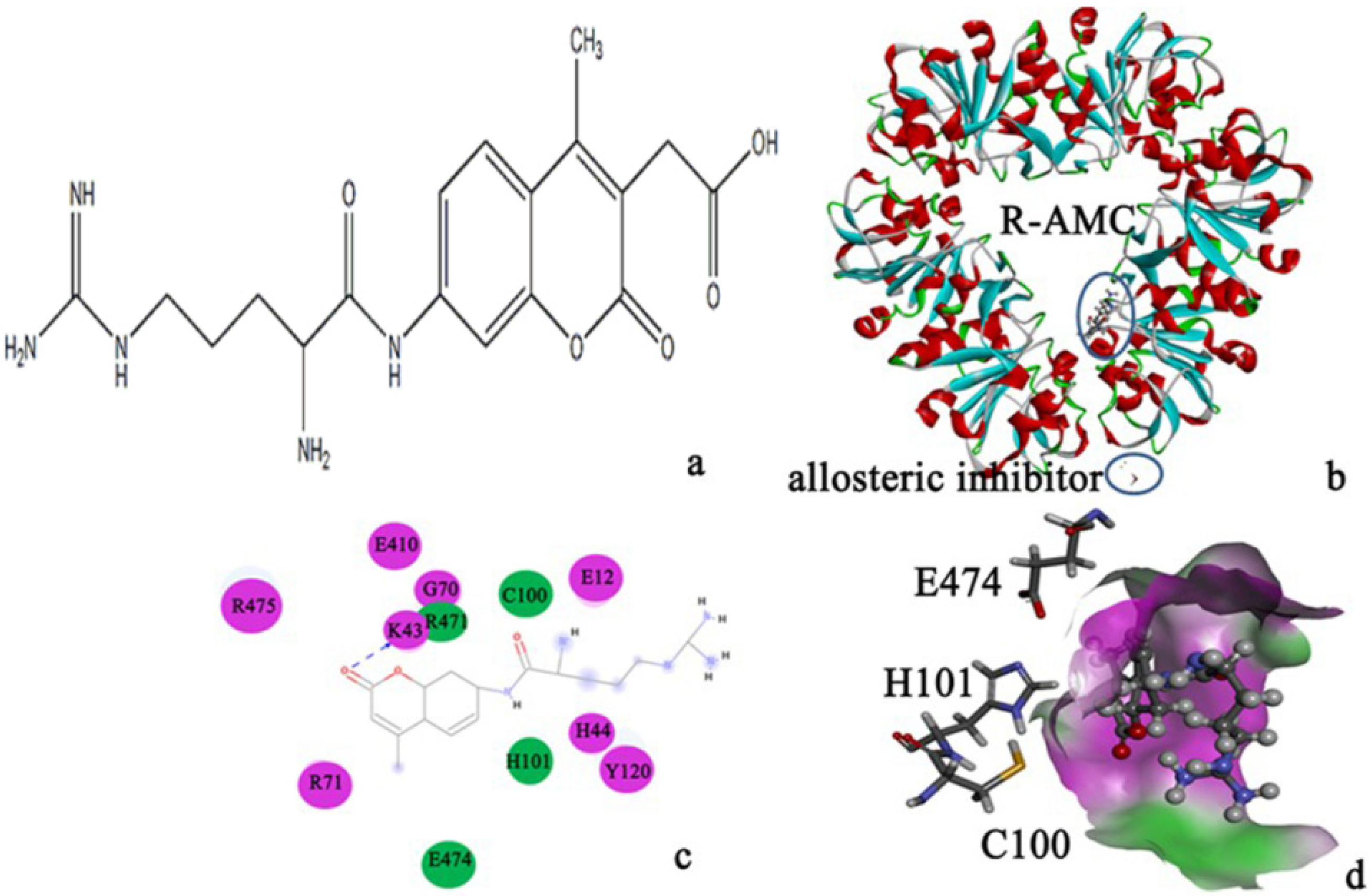
| Substrate/PH1704 | AutoDock vina | AutoDock 4.2 | Dock 6.6. |
|---|---|---|---|
| R-AMC | −7.17 | −6.65 | −6.55 |

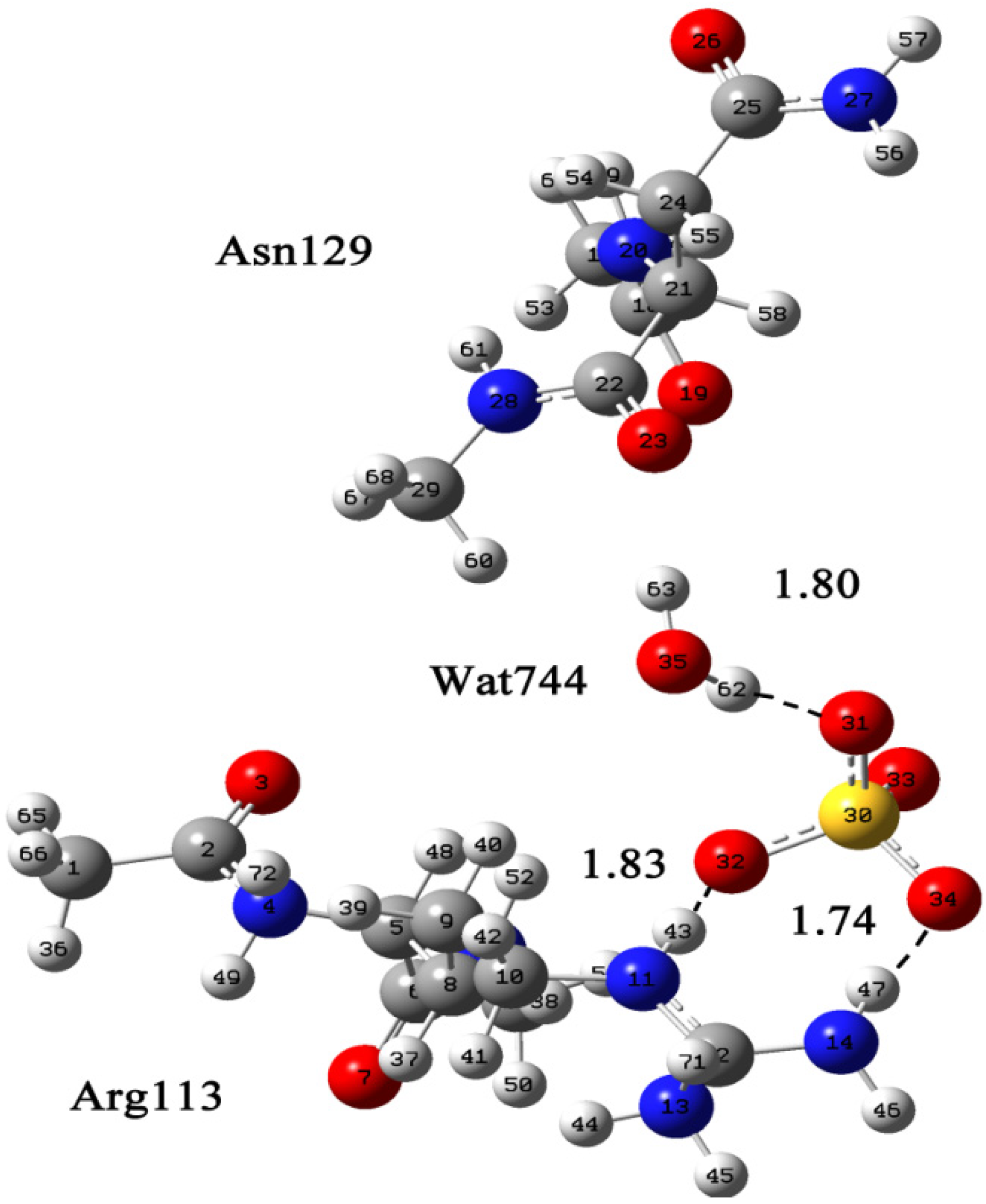
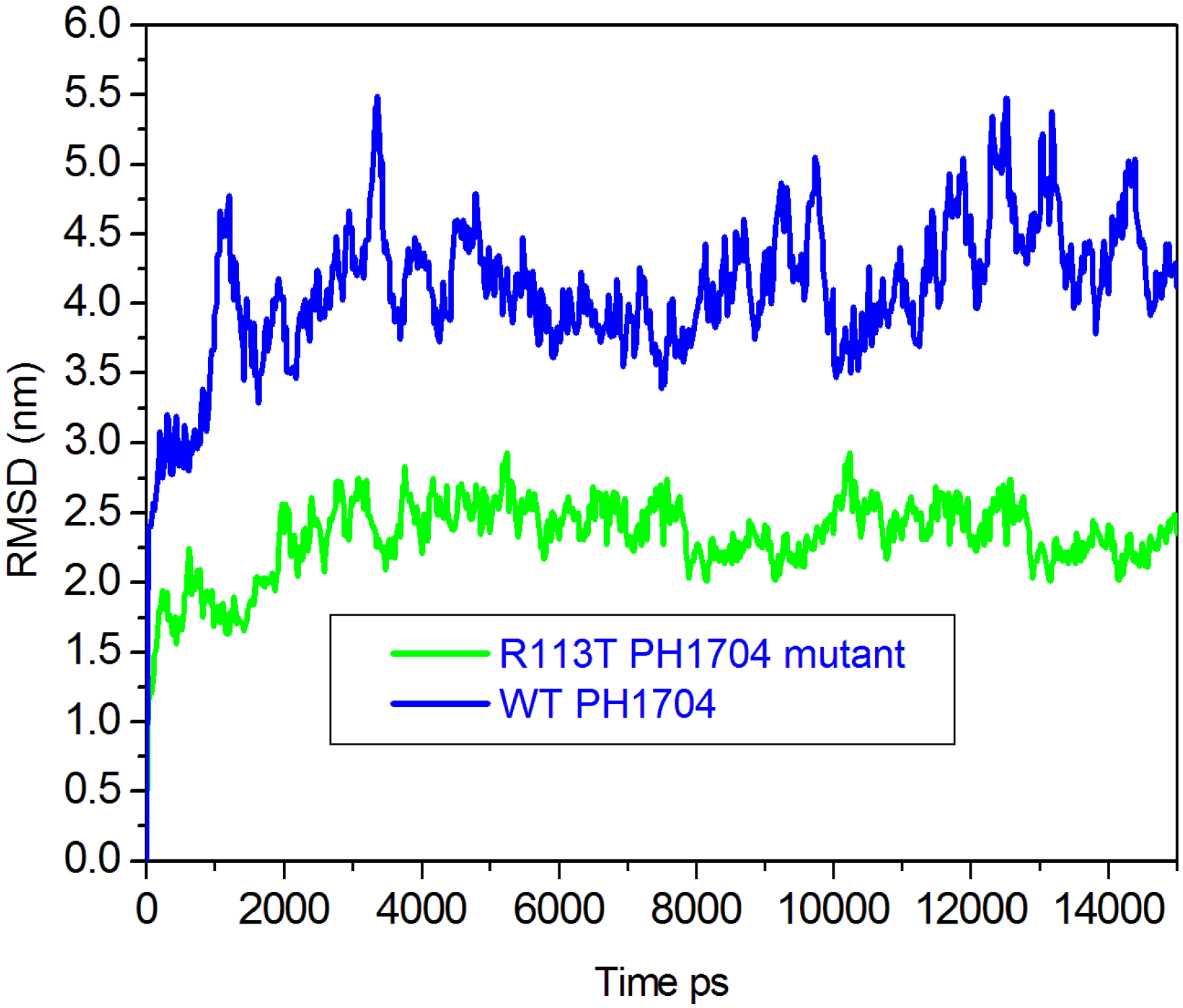
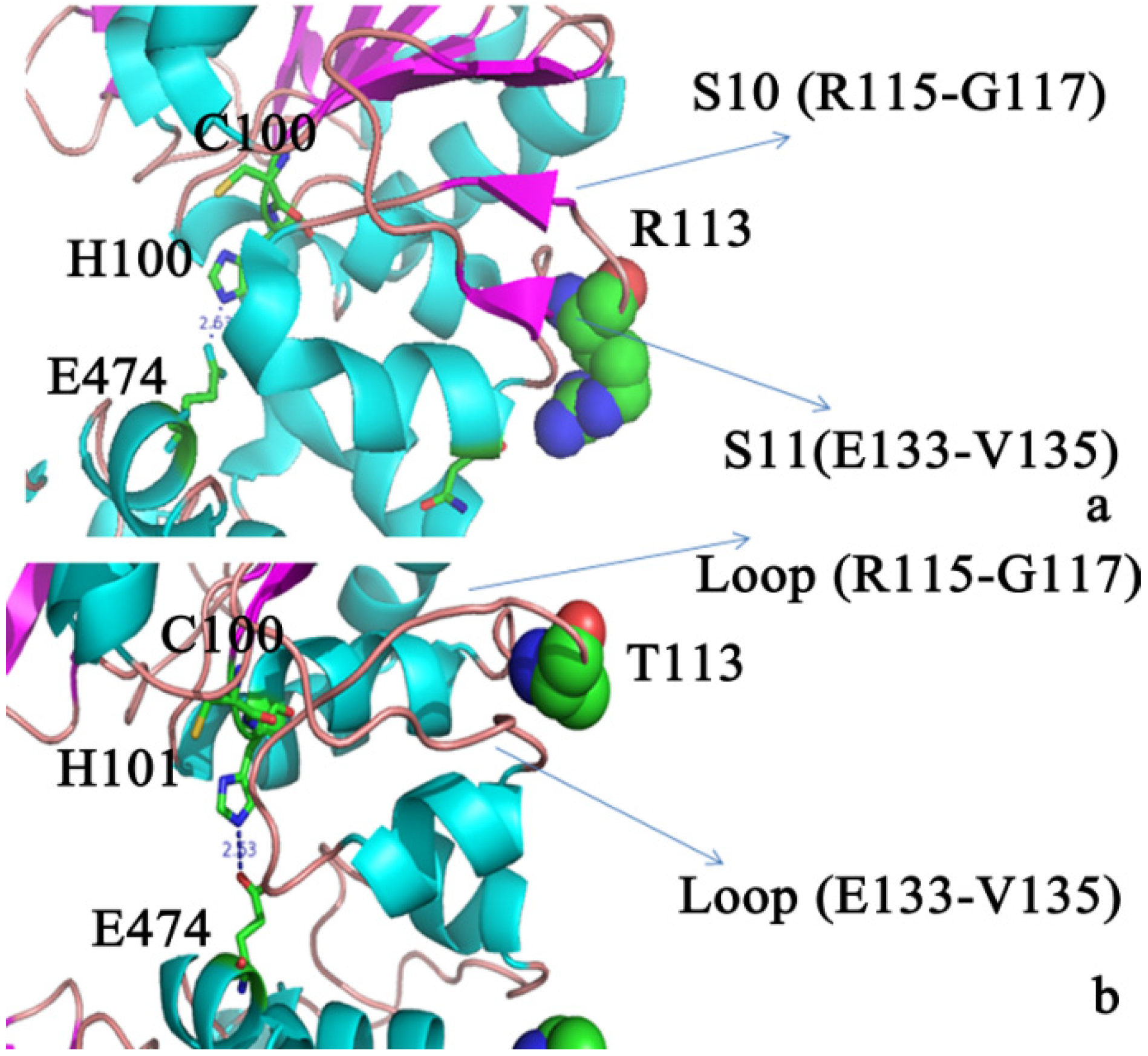
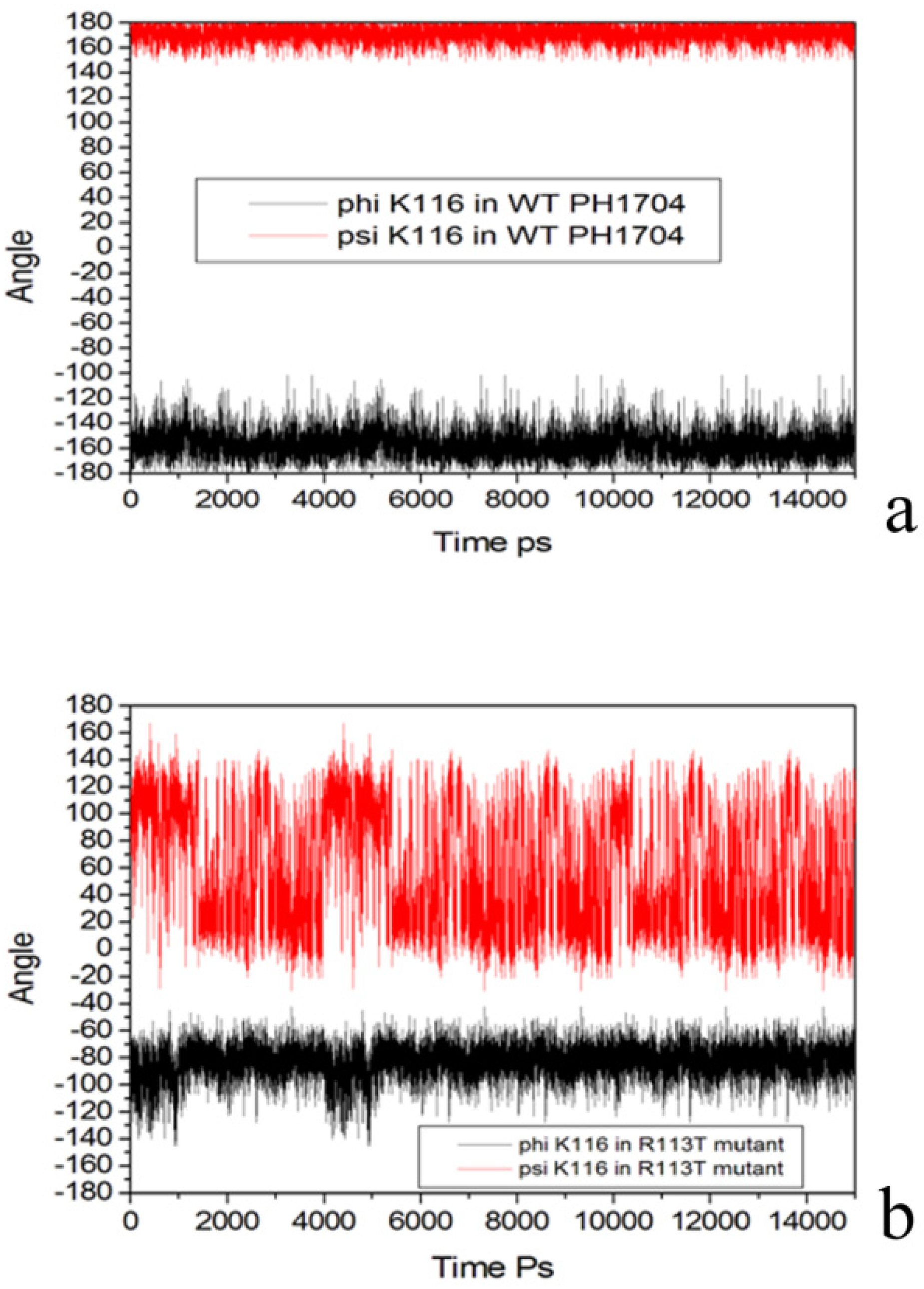
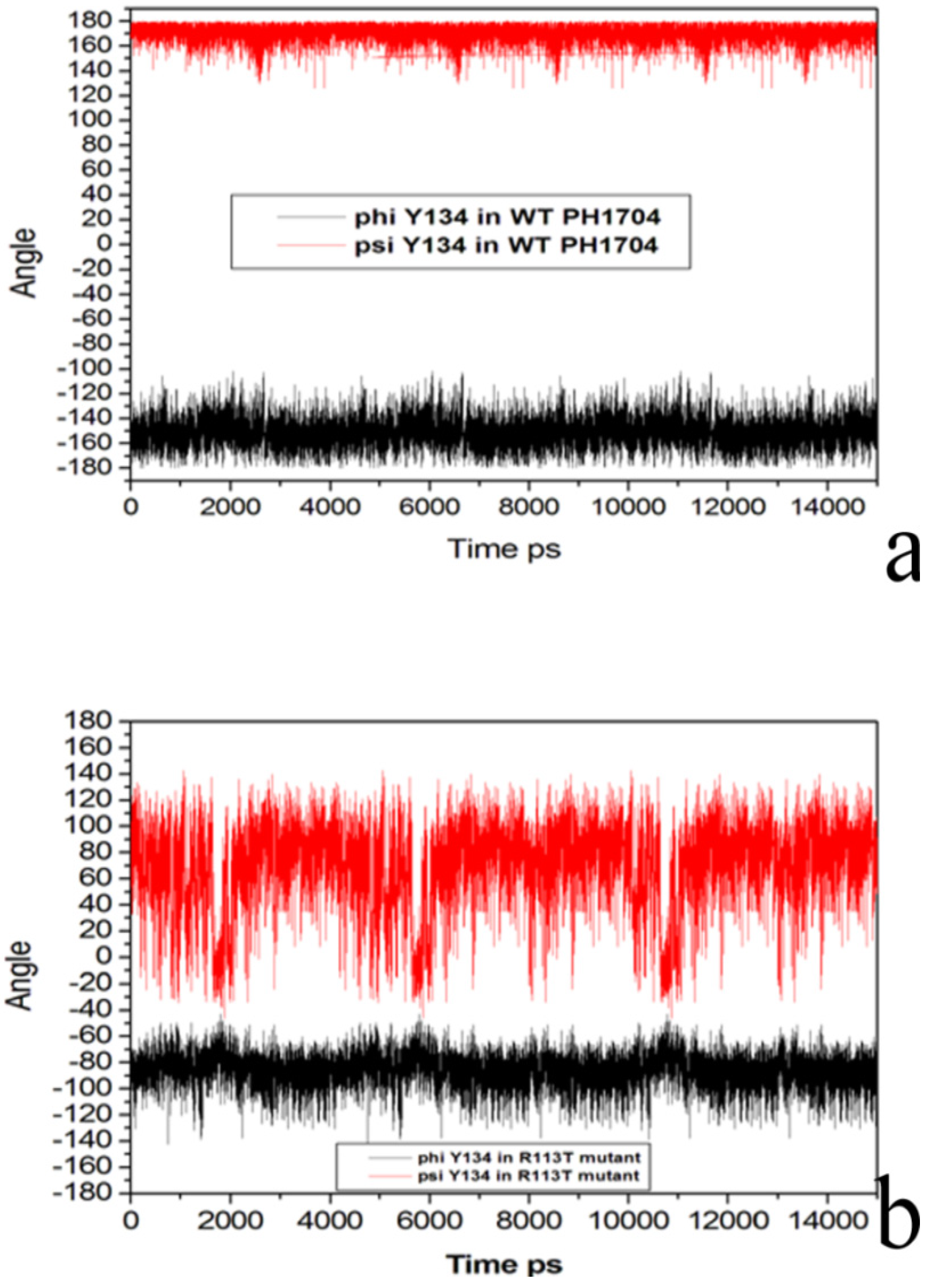
2.3. Molecular Dynamics Simulations to Study Allosteric Regulation by Cl−





| A subunit | C subunit | Distance (Å) |
|---|---|---|
| S108 OH | D525 COO− | 2.64 |
| H101 NH | E474 COO− | 1.66 |
| D126 COO− | R477 NH+ | 2.08 |
| Residue | Amino acid group | Residue | Amino acid group | Occupancy (%) | |
|---|---|---|---|---|---|
| R113T | WT | ||||
| A S108 | OH | C D525 | COO− | 97.35 | 49.43 |
| A H101 | NH | C E474 | COO− | 90.25 | 42.71 |
| A D126 | COO- | C R477 | NH+ | 72.43 | 32.12 |

3. Experimental
3.1. Quantum Mechanical Calculation Method
3.2. Protein-Substrate Complex Preparation
3.3. Molecular Dynamics Simulations
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Du, X.; Choi, I.G.; Kim, R.; Wang, W.; Jancarik, J.; Yokota, H.; Kim, S.H. Crystal structure of an intracellular protease from Pyrococcus horikoshii at 2-Å resolution. Proc. Natl. Acad. Sci. USA 2000, 97, 14079–14084. [Google Scholar]
- Zhan, D.L.; Han, W.W.; Feng, Y. Experimental and computational studies indicate the mutation of Glu12 to increase the thermostability of oligomeric protease from Pyrococcus horikoshii. J. Mol. Model. 2011, 17, 1241–1249. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Morton, F.R.; Barrett, A.J. MEROPS the peptidase database. Nucleic Acids Res. 2006, 34, D270–D272. [Google Scholar] [CrossRef]
- Halio, S.B.; Bauer, M.W.; Mukund, S.; Adams, M.; Kelly, R.M. Purification and characterization of two functional forms of intracellular protease PfpI from the hyperthermophilic archaeon Pyrococcus furiosus. App. Environ. Micromol. 1997, 1, 289–295. [Google Scholar]
- Halio, S.B.; Blumentals, I.I.; Short, S.A.; Merrill, B.M.; Kelly, R.M. Sequence, expression in Escherichia coli, and analysis of the gene encoding a novel intracellular protease (PfpI) from the hyperthermophilic archaeon Pyrococcus furiosus. J. Bacteriol. 1996, 178, 2605–2612. [Google Scholar]
- Barbe, V.; Cruveiller, S.; Kunst, F.; Lenoble, P.; Meurice, G.; Sekowska, A.; Vallenet, D.; Wang, T.; Moszer, I.; Médigue, C.; et al. From a consortium sequence to a unified sequence: The Bacillus subtilis 168 reference genome a decade later. Microbiology 2009, 155, 1758–1775. [Google Scholar] [CrossRef]
- Wei, Y.; Ringe, D.; Wilson, M.A.; Ondrechen, M.J. Identification of functional subclasses in the DJ-1 superfamily proteins. PLoS Comput. Biol. 2007, 3, e120–e126. [Google Scholar] [CrossRef]
- Jung, H.J.; Kim, S.; Kim, Y.J.; Kim, M.K.; Kang, S.G.; Lee, J.H.; Kim, W.; Cha, S.S. Dissection of the dimerization modes in the DJ-1 superfamily. Mol. Cells 2012, 33, 163–171. [Google Scholar] [CrossRef]
- Fioravanti, E.; Asunción Durá, M.; Lascoux, D.; Micossi, E.; Franzetti, B.; Sweeney, S.M. Structure of the Stress Response Protein DR1199 from Deinococcus radiodurans: A Member of the DJ-1 Superfamily. Biochemistry 2008, 47, 11581–11589. [Google Scholar] [CrossRef]
- Chain, P.S.; Comerci, D.J.; Tolmasky, M.E.; Larimer, F.W.; Malfatti, S.A.; Vergez, L.M.; Aguero, F.; Land, M.L.; Ugalde, R.A.; Garcia, E. Whole-genome analyses of speciation events in pathogenic Brucellae. Infect. Immun. 2005, 73, 8353–8261. [Google Scholar] [CrossRef]
- Häcker, H.G.; Sisay, M.T.; Gütschow, M. Allostericmodulation of caspases. Phar. Ther. 2011, 132, 180–195. [Google Scholar] [CrossRef]
- Velázquez-Delgado, E.M.; Hardy, J.A. Zinc-mediatedallostericinhibition of caspase-6. J. Biol. Chem. 2012, 287, 36000–36011. [Google Scholar] [CrossRef]
- Abdel-Aziz, M.H.; Sidhu, P.S.; Liang, A.; Kim, J.Y.; Mosier, P.D.; Zhou, Q.; Farrell, D.H.; Desai, U.R. Designingallostericregulators of thrombin. Monosulfated benzofuran dimers selectively interact with Arg173 of exosite 2 to induce inhibition. J. Med. Chem. 2012, 55, 6888–6897. [Google Scholar] [CrossRef]
- Koschubs, T.; Dengl, S.; Dürr, H.; Kaluza, K.; Georges, G.; Hartl, C.; Jennewein, S.; Lanzendörfer, M.; Auer, J.; Stern, A.; et al. Allostericantibody inhibition of human hepsin protease. Biochem. J. 2012, 442, 483–494. [Google Scholar] [CrossRef]
- Grimsley, J.K.; Calamini, B.; Wild, J.R.; Mesecar, A.D. Structural and mutational studies of organophosphorus hydrolase reveal a cryptic and functional allosteric-binding site. Arch. Biochem. Biophys. 2005, 442, 169–179. [Google Scholar] [CrossRef]
- Wiesmann, C.; Barr, K.J.; Kung, J.; Zhu, J.; Erlanson, D.A.; Shen, W.; Fahr, B.J.; Zhong, M.; Taylor, L.; Randal, M.; et al. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 2004, 11, 730–737. [Google Scholar]
- Hardy, J.A.; Lam, J.; Nguyen, J.R.; O’Brien, T.; Wells, J.A. Discovery of an allosteric site in the caspases. Proc. Natl. Acad. Sci. USA 2004, 101, 12461–12466. [Google Scholar]
- Zhan, D.L.; Gao, N.; Han, W.W.; Feng, Y. Quantum chemistry calculation of thermophilic protease PH1704 allosteric center and mutant dynamics. Chem. J. Chin. Univ. 2013. [Google Scholar] [CrossRef]
- Zhan, D.L.; Gao, N.; Han, W.W.; Feng, Y. Molecular docking and dynamics simulation improving thermophilic protease activity of PhpI. Chem. J. Chin. Univ. 2013, 3, 628–633. [Google Scholar]
- Motif Scan. Available online: http://myhits.isb-sib.ch/cgi-bin/motif_scan (accessed on 30 June 2013).
- Sheng, L.; Lu, Y.C.; Peng, B.Z.; Ding, J.P. Crystal structure of human phosphoribosylpyrophosphate synthetase 1 reveals a novel allosteric site. Biochem. J. 2007, 401, 39–47. [Google Scholar] [CrossRef]
- Peterson, A.W.; Cockrell, G.M.; Kantrowitz, E.R. A second allosteric site in Escherichia coliaspartatetranscarbamoylase. Biochemistry 2012, 51, 4776–4778. [Google Scholar] [CrossRef]
- Protein Data Bank. Available online: http://www.rcsb.org/pdb/home/home.do (accessed on 30 June 2013).
- Micka, M.; Ivan, J. Acid–base controllable recognition properties of a highly versatile calix[6] crypturea. Chem. Eur. J. 2010, 16, 2159–2169. [Google Scholar] [CrossRef]
- Zhan, D.L.; Zhou, Z.H.; Guan, S.S.; Han, W.W. The Effect of conformational variability of phosphotriesterase upon N-acyl-L-homoserine lactone and paraoxon binding: Insights from molecular dynamics studies. Molecules 2013, 18, 15501–15518. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulation. Nucl. Aci. Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Monod, J.; Wyman, J.; Changeux, J.P. On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 1965, 12, 88–118. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. Condens. Matter. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Cervantes-Navarro, F.; Glossman-Mitnik, D. DFT study of the effect of substituents on the absorption and emission spectra of Indigo. Chem. Cent. J. 2012, 6, 70–75. [Google Scholar] [CrossRef]
- Han, W.W.; Wang, Y.; Zhou, Y.H.; Yao, Y.; Li, Z.S.; Feng, Y. Understanding structural/functional properties of amidase from Rhodococcus erythropolis by computational approaches. J. Mol. Model. 2009, 15, 481–487. [Google Scholar] [CrossRef]
- Decherchi, S.; Colmenares, J.; Catalano, C.E.; Spagnuolo, M.; Alexov, E. Between algorithm and model: Different Molecular Surface definitions for the Poisson-Bolztman based electrostatic characterization of biomolecules in solution. Commun. Comput. Phys. 2013, 13, 61–89. [Google Scholar]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nuc. Aci. Res. 2007, 35, W522–W525. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Norgan, A.P.; Coffman, P.K.; Kocher, J.P. Multilevel parallelization of autodock 4.2. J. Cheminform. 2011, 28, 3–12. [Google Scholar]
- Brozell, S.R.; Mukherjee, S.; Balius, T.E.; Roe, D.R.; Case, D.A.; Rizzo, R.C. Evaluation of DOCK 6 as a pose generation and database enrichment tool. J. Comput. Aided. Mol. Des. 2012, 26, 749–773. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Crowley, M.; Walker, R.C.; Zhang, W.; et al. AMBER 10; University of California: San Francisco, CA, USA, 2008. [Google Scholar]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madurs, J.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Kräutler, V.; Wilfred, F.G.; Philippe, H.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Com. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·Log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhan, D.; Sun, J.; Feng, Y.; Han, W. Theoretical Study on the Allosteric Regulation of an Oligomeric Protease from Pyrococcus horikoshii by Cl− Ion. Molecules 2014, 19, 1828-1842. https://doi.org/10.3390/molecules19021828
Zhan D, Sun J, Feng Y, Han W. Theoretical Study on the Allosteric Regulation of an Oligomeric Protease from Pyrococcus horikoshii by Cl− Ion. Molecules. 2014; 19(2):1828-1842. https://doi.org/10.3390/molecules19021828
Chicago/Turabian StyleZhan, Dongling, Jiao Sun, Yan Feng, and Weiwei Han. 2014. "Theoretical Study on the Allosteric Regulation of an Oligomeric Protease from Pyrococcus horikoshii by Cl− Ion" Molecules 19, no. 2: 1828-1842. https://doi.org/10.3390/molecules19021828