Pyrrolidine-Mediated Direct Preparation of (E)-Monoarylidene Derivatives of Homo- and Heterocyclic Ketones with Various Aldehydes

Abstract
: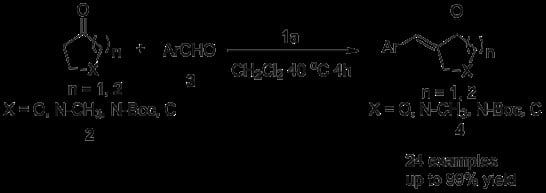
1. Introduction

2. Results and Discussion


{kind=link}
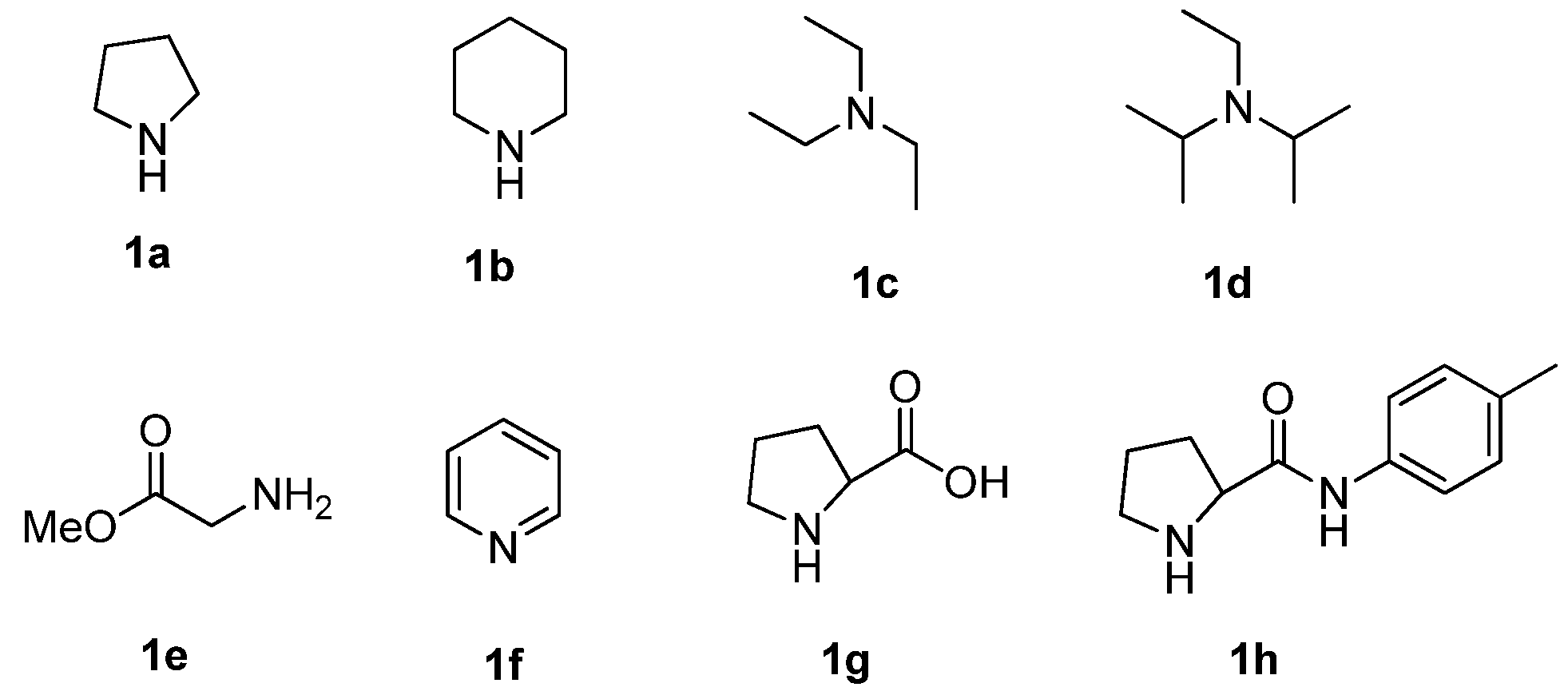{kind=link}
{kind=link}
{kind=link}
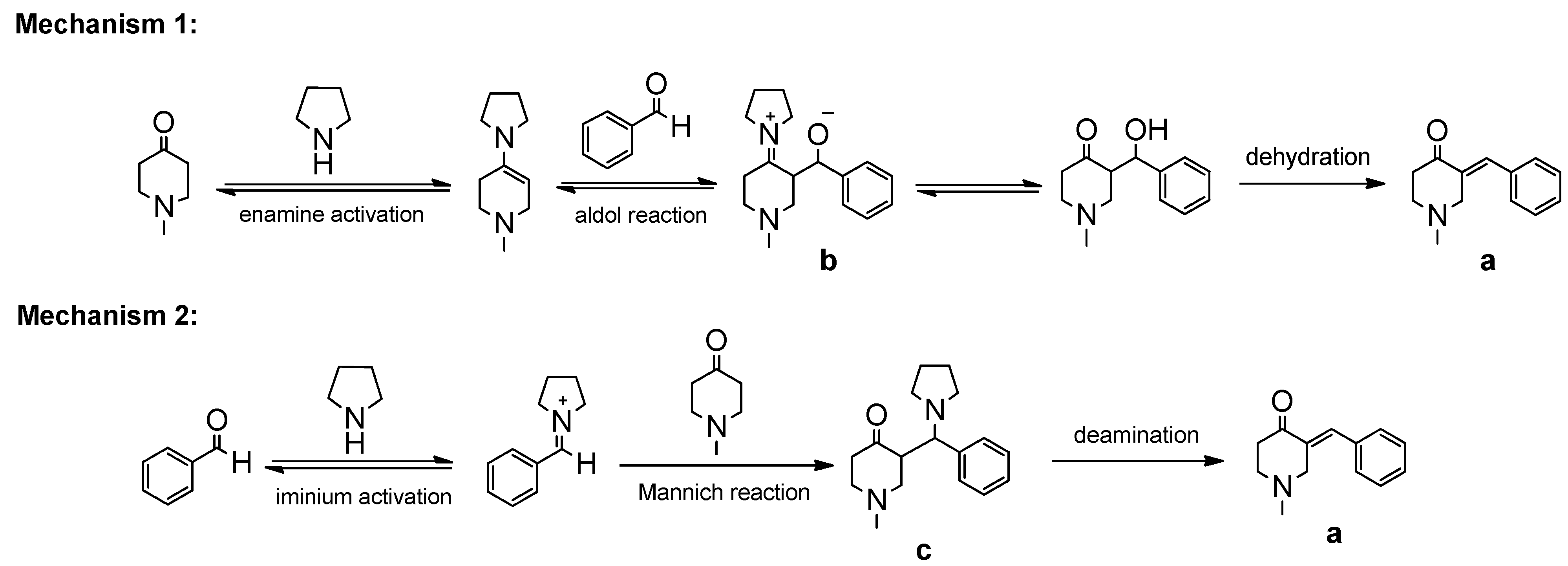{kind=link}
| Entry | Catalyst | Solvent | Temp (°C) | Yield (%) b |
|---|---|---|---|---|
| 1 | 1a | CH2Cl2 | 25 | 46 c |
| 2 | 1a | CH2Cl2 | 25 | 77 |
| 3 | 1b | CH2Cl2 | 25 | 18 d |
| 4 | 1c | CH2Cl2 | 25 | N.R e |
| 5 | 1d | CH2Cl2 | 25 | N.R |
| 6 | 1e | CH2Cl2 | 25 | 22 |
| 7 | 1f | CH2Cl2 | 25 | N.R |
| 8 | 1g | CH2Cl2 | 25 | N.D f |
| 9 | 1h | CH2Cl2 | 25 | trace |
| 10 | 1a | CHCl3 | 25 | 42 |
| 11 | 1a | Et2O | 25 | 41 |
| 12 | 1a | toluene | 25 | 18 |
| 13 | 1a | dioxane | 25 | trace |
| 14 | 1a | CH3OH | 25 | 68 |
| 15 | 1a | EtOH | 25 | 73 |
| 16 | 1a | EtOH/H2O | 25 | 38 f |
| 17 | 1a | CH2Cl2 | 0 | 59 |
| 18 | 1a | CH2Cl2 | 40 | 94 |
| Entry | 2 | 3 | 4 | Yield (%) b |
|---|---|---|---|---|
| 1 | X = N-CH3, n = 2 | R1 = Ph | 4a | 94 |
| 2 | X = N-CH3, n = 2 | R1 = 4-NO2-Ph | 4b | 90 |
| 3 | X = N-CH3, n = 2 | R1 = 4-CN-Ph | 4c | 99 |
| 4 | X = N-CH3, n = 2 | R1 = 4-F-Ph | 4d | 75 |
| 5 | X = N-CH3, n = 2 | R1 = 4-Br-Ph | 4e | 73 |
| 6 | X = N-CH3, n = 2 | R1 = 3,4-diCl-Ph | 4f | 80 |
| 7 | X = N-CH3, n = 2 | R1 = 4-CH3-Ph | 4g | 54 |
| 8 | X = N-CH3, n = 2 | R1 = 4-CH3O-Ph | 4h | 50 |
| 9 | X = N-CH3, n = 2 | R1 = 3-Cl-Ph | 4i | 81 |
| 10 | X = N-CH3, n = 2 | R1 = 3-Br-Ph | 4j | 86 |
| 11 | X = N-CH3, n = 2 | R1 = 3-CH3O-Ph | 4k | 65 |
| 12 | X = N-CH3, n = 2 | R1 = 2-F-Ph | 4l | 84 |
| 13 | X = N-CH3, n = 2 | R1 = 2-Br-Ph | 4m | 94 |
| 14 | X = N-CH3, n = 2 | R1 = 2-naphthyl | 4n | 60 |
| 15 | X = N-CH3,n = 2 | R1 = 1-naphthyl | 4o | 93 |
| 16 | X = N-CH3, n = 2 | R1 = 2-pyridinyl | 4p | 78 |
| 17 | X = N-CH3, n = 2 | R1 = 4-pyridinyl | 4q | 59 |
| 18 | X = N-CH3, n = 2 | R1 = 2-thienyl | 4r | 46 |
| 19 | X = N-CH3, n = 2 | R1 = CH3CH2CH2 | 4s | 53 |
| 20 | X = N-Boc, n = 2 | R1 = Ph | 4t | 92 |
| 21 | X = O, n = 2 | R1 = Ph | 4u | 53 |
| 22 | X = O, n = 2 | R1 = 4-NO2-Ph | 4v | 64 |
| 23 | X = C, n = 2 | R1 = 4-NO2-Ph | 4w | 84c |
| 24 | X = C, n = 1 | R1 = 4-NO2-Ph | 4x | 95 |


3. Experimental
3.1. General
3.2. General Experimental Procedure
3.3. Spectral Data
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nowakowska, Z. A review of anti-infective and anti-inflammatory chalcones. Eur. J. Med. Chem. 2007, 42, 125–137. [Google Scholar] [CrossRef]
- Dimmock, J.R.; Elias, D.W.; Beazel, M.A.; Kandepu, N.M. Bioactivities of chalcones. Curr. Med. Chem. 1999, 6, 1125–1149. [Google Scholar]
- Lin, L.M.; Zhou, Y.; Flavin, M.T.; Zhou, L.M.; Nie, W.G.; Chen, F.C. Chalcones and flavonoids as anti-Tuberculosis agents. Bioorg. Med. Chem. 2002, 10, 2795–2798. [Google Scholar] [CrossRef]
- Wang, S.; Yu, G.; Lu, J.; Xiao, K.; Hu, Y.; Hu, H.W. A regioselective tandem reaction between chalcones and 2-acetamidoacetamide promoted by Cs2CO3 for the preparation of 3-unsubstituted 2-pyridones. Synthesis 2003, 487–490. [Google Scholar]
- Wei, X.D.; Fang, J.; Hu, Y.F.; Hu, H.W. A convenient preparation of 3,5-diarylisooxazoles. Synthesis 1992, 1205–1206. [Google Scholar]
- Zheng, H.Z.; Yi, L.T.; Li, X.W.; Guo, F.Z.; Qi, L.Z.; Chu, C.T. Synthesis of optically active α,β-unsaturated triazolyl alcohols via asymmetric NaBH4 reduction of the corresponding ketones. Synth. Commun. 2004, 1359–1365. [Google Scholar]
- Sugawara, Y.; Yamada, W.; Yoshida, S.; Ikeno, T.; Yamada, T. Carbon dioxide-mediated catalytic rearrangement of propargyl alcohols into α,β-unsaturated ketones. J. Am. Chem. Soc. 2007, 129, 12902–12903. [Google Scholar] [CrossRef]
- Petrov, O.; Ivanova, Y.; Gerova, M. SOCl2/EtOH: Catalytic system for synthesis of chalcones. Catal. Commun. 2008, 9, 315–316. [Google Scholar] [CrossRef]
- Sano, S.; Yokoyama, K.; Shiro, M.; Nagao, Y. A facile method for the stereoselective Horner-Wadsworth-Emmons reaction of aryl alkyl ketones. Chem. Pharm. Bull. 2002, 50, 706–709. [Google Scholar]
- Shibuya, M.; Ito, S.; Takahashi, M.; Iwabuchi, Y. Oxidative rearrangement of cyclic tertiary allylic alcohols with IBX in DMSO. Org. Lett. 2004, 6, 4303–4306. [Google Scholar] [CrossRef]
- Jurado-González, M.; Sullivan, A.C.; Wilson, J.R. Selective oxidations of allylic alcohols using vanadyl and cobalt(II) alkyl phosphonate modified silicas. Tetrahedron Lett. 2004, 45, 4465–4468. [Google Scholar] [CrossRef]
- Clive, D.; Stephen, P. Synthesis of the bicyclic dienone core of the antitumor agent ottelione B. Chem. Commun. 2002, 1940–1941. [Google Scholar] [CrossRef]
- Taber, D.F.; Herr, R.J.; Pack, S.K.; Geremia, J.M. A convenient method for the preparation of (Z)-α,β-unsaturated carbonyl compounds. J. Org. Chem. 1996, 61, 2908–2910. [Google Scholar]
- Xi, Z.; Fan, H.T.; Mito, S.; Takahashi, T.J. CO insertion reaction of zirconacyclopentadienes. Organomet. Chem. 2003, 682, 108–112. [Google Scholar]
- Deshong, P.; Sidler, D.R.; Slough, G.A. Synthesis of enones and butenolides using organomanganese pentacarbonyl complexes. Tetrahedron Lett. 1987, 28, 2233–2236. [Google Scholar] [CrossRef]
- Mogilaiah, K.; Reddy, N.V. LiCl Catalyzed Claisen-Schmidt condensation in solvent-free conditions using microwaves. Synth. Commun. 2003, 33, 73–78. [Google Scholar] [CrossRef]
- Hatsuda, M.; Kuroda, T.; Seki, M. An improved synthesis of (E)-cinnamic acid derivatives via the Claisen–Schmidt condensation. Synth. Commun. 2003, 33, 427–434. [Google Scholar] [CrossRef]
- Choudary, B.M.; Kantam, M.L.; Ranganath, K.V.S.; Mahendar, K.; Sreedher, B. Bifunctional nanocrystalline MgO for chiral epoxy ketones via Claisen-Schmidt condensation-asymmetric epoxidation reactions. J. Am. Chem. Soc. 2004, 126, 3396–3397. [Google Scholar]
- Sabitha, G.; Reddy, G.S.K.K.; Reddy, K.B.; Yadav, J.S. Iodotrimethylsilane-mediated cross aldol condensation: a facile synthesis of α,α'-bis(substituted benzylidene)cycloalkanones. Synthesis 2004, 263–266. [Google Scholar] [CrossRef]
- Toda, F.; Tanaka, K.; Hamai, K. Aldol condensations in the absence of solvent: acceleration of the reaction and enhancement of the stereoselectivity. J. Chem. Soc. Perkin Trans. 1 1990, 28, 3207–3209. [Google Scholar]
- Noyce, D.S.; Pryor, W.A. Carbonyl Reactions. I. Kinetics and mechanism of the acid-catalyzed aldol condensation of benzaldehyde and acetophenone. J. Am. Chem. Soc. 1955, 77, 1397–1401. [Google Scholar] [CrossRef]
- Zheng, M.; Wang, L.; Shao, J.; Zhong, Q. A facile synthesis of α,α'-bis(substituted benzylidene)cycloalkanones catalyzed by bis(p-ethoxyphenyl)telluroxide(bmpto) under micro-wave irradiation. Synth. Commun. 1997, 27, 351–354. [Google Scholar] [CrossRef]
- Iranpoor, N.; Kazemi, F. RuCl3 catalyses aldol condensations of aldehydes and ketones. Tetrahedron 1998, 54, 9475–9480. [Google Scholar] [CrossRef]
- Raston, C.L.; Scott, J.L. Chemoselective, solvent-free aldol condensation reaction. Green Chem. 2000, 2, 49–52. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Banno, K.; Narasaka, K. New cross-aldol reactions. Reactions of silyl enol ethers with carbonyl compounds activated by titanium tetrachloride. J. Am. Chem. Soc. 1974, 96, 7503–7509. [Google Scholar] [CrossRef]
- Denmark, S.E.; Stavenger, R.A. Asymmetric catalysis of aldol reactions with chiral Lewis bases. Acc. Chem. Res. 2000, 33, 432–440. [Google Scholar] [CrossRef]
- Yanagisawa, A.; Goudu, R.; Arai, T. Selective synthesis of α,β-unsaturated ketones by dibutyltin dimethoxide-catalyzed condensation of aldehydes with alkenyl trichloroacetates. Org. Lett. 2004, 6, 4281–4283. [Google Scholar] [CrossRef]
- Trost, B.M. A biological catalysis for synthetic efficiency. Pure Appl. Chem. 1992, 64, 315–322. [Google Scholar] [CrossRef]
- Kreher, U.P.; Rosamilia, A.E.; Raston, C.L.; Scott, J.L.; Strauss, C.R. Direct preparation of monoarylidene derivatives of aldehydes and enolizable ketones with DIMCARB. Org. Lett. 2003, 5, 3107–3110. [Google Scholar]
- Wang, W.; Mei, Y.; Li, H.; Wang, J. A novel pyrrolidine imide catalyzed direct formation of α,β-unsaturated ketones from unmodified ketones and aldehydes. Org. Lett. 2005, 7, 601–604. [Google Scholar] [CrossRef]
- Mojtahedi, M.M.; Abaee, M.S.; Khakbaz, M.; Alishiri, T.; Samianifard, M.; Mesbah, A.W.; Harms, K. An efficient procedure for the synthesis of α,β-unsaturated ketones and its application to heterocyclic systems. Synthesis 2011, 23, 3821–3826. [Google Scholar]
- Kumar, R.R.; Perumal, S.; Senthilkumar, P.; Yogeeswari, P.; Sriram, D. A facile synthesis and antimycobacterial evaluation of novel spiro-pyrido-pyrrolizines and pyrrolidines. Eur. J. Med. Chem. 2009, 44, 3821–3826. [Google Scholar] [CrossRef]
- Mojtahedi, M.M.; Abaee, M.S.; Samianifard, M.; Shamloo, A.; Padyab, M.; Mesbah, A.W.; Harms, K. Ultrasound mediation for efficient synthesis of monoarylidene derivatives of homo- and heterocyclic ketones. Ultrason. Sonochem. 2013, 20, 924–930. [Google Scholar] [CrossRef]
- Erkkila, A.; Majander, I.; Pihko, P.M. Iminium catalysis. Chem. Rev. 2007, 107, 5416–5470. [Google Scholar] [CrossRef]
- Moyano, A.; Rios, R. Asymmetric organocatalytic cyclization and cycloaddition reactions. Chem. Rev. 2011, 111, 4703–4832. [Google Scholar] [CrossRef]
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric enamine catalysis. Chem. Rev. 2007, 107, 5471–5569. [Google Scholar] [CrossRef]
- Yang, J.W.; Chandler, C.; Stadler, M.; Kampe, D.; List, B. Proline-catalysed Mannich reactions of acetaldehyde. Nature 2008, 452, 453–455. [Google Scholar] [CrossRef]
- Schmid, M.B.; Zeitler, K.; Gschwind, R.M. NMR investigations on the proline-catalyzed aldehyde self-condensation: mannich mechanism, dienamine detection, and erosion of the aldol addition selectivity. J. Org. Chem. 2011, 76, 3005–3015. [Google Scholar] [CrossRef]
- Sample Availability:Samples of the compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gu, X.; Wang, X.; Wang, F.; Sun, H.; Liu, J.; Xie, Y.; Xiang, M. Pyrrolidine-Mediated Direct Preparation of (E)-Monoarylidene Derivatives of Homo- and Heterocyclic Ketones with Various Aldehydes. Molecules 2014, 19, 1976-1989. https://doi.org/10.3390/molecules19021976
Gu X, Wang X, Wang F, Sun H, Liu J, Xie Y, Xiang M. Pyrrolidine-Mediated Direct Preparation of (E)-Monoarylidene Derivatives of Homo- and Heterocyclic Ketones with Various Aldehydes. Molecules. 2014; 19(2):1976-1989. https://doi.org/10.3390/molecules19021976
Chicago/Turabian StyleGu, Xin, Xiaoyan Wang, Fengtian Wang, Hongbao Sun, Jie Liu, Yongmei Xie, and Mingli Xiang. 2014. "Pyrrolidine-Mediated Direct Preparation of (E)-Monoarylidene Derivatives of Homo- and Heterocyclic Ketones with Various Aldehydes" Molecules 19, no. 2: 1976-1989. https://doi.org/10.3390/molecules19021976