3.2. Chemistry
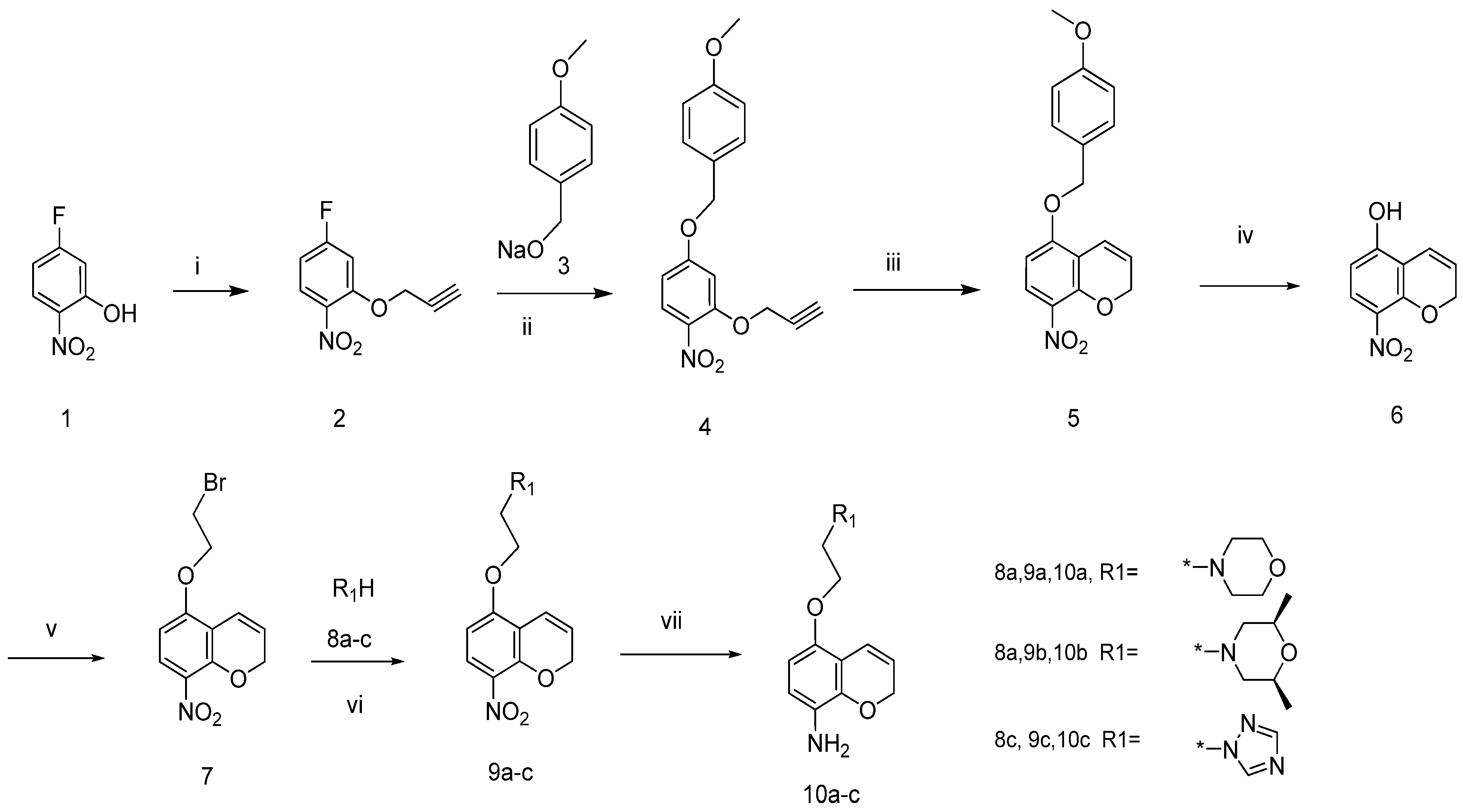
4-Fluoro-1-nitro-2-(prop-2-ynyloxy) benzene (2). To a stirred solution of 5-fluoro-2-nitrophenol (30 g, 191 mmol) in DMF (300 mL) anhydrous K2CO3 (52.83 g, 382 mmol) and 3-bromopropyne (27.27 g, 229 mmol) were successively added The mixture was stirred at room temperature for 5 h and then poured into ice water (1,500 mL). A buff precipitate separated out upon standing overnight. The solid was collected by filtration and air-dried to give compound 2 as a buff solid (33.91 g, yield: 85%), mp: 50–52 °C, 1H-NMR (CDCl3, 400 MHz), δ: 7.98 (m, 1H), 6.98 (m, 1H), 6.80 (m, 1H), 4.86 (s, 2H), 2.66 (s, 1H). FAB-MS (m/z): 196 [M+H]+.
4-(4-Methoxybenzyloxy)-1-nitro-2-(prop-2-ynyloxy)benzene (4). To a stirred solution of (4-methoxyphenyl)methanol (22.63 g, 163.8 mmol) in anhydrous DMF (200 mL), 60% sodium hydride (8.74 g, 218.4 mmol) was added in portions. The mixture was stirred at ambient temperature until no further gas was released, then cooled to −35 °C. To this solution, compound 2 (21.84 g, 109.2 mmol) was added in one batch. The reaction mixture was stirred for a further 6 h at −35 °C under nitrogen, and then was poured into ice water (1,000 mL). The precipitate was collected, washed with water and air-dried to give compound 4 as a buff solid (27.82 g, yield: 93%), mp: 53–54 °C, 1H-NMR (CDCl3, 400 MHz), δ: 8.03 (d, J = 9.24 Hz, 1H), 7.36 (2H, d, J = 8.44 Hz), 6.94 (2H, d, J = 8.44 Hz), 6.79 (1H, d, J = 2.52 Hz), 6.66 (dd, J = 9.24 Hz, 2.52 Hz, 1H), 5.08 (2H, s), 4.82 (2H, d, J = 2.24 Hz), 384 (3H, s), 2.59 (1H, t, J = 2.52 Hz).
5-(4-Methoxybenzyloxy)-8-nitro-2H-chromene (5). Compound 4 (22 g, 71.8 mmol) was dissolved in N,N-diethylaniline (330 mL). The reaction mixture was heated to 195 °C and kept at this temperature for 1 h. After cooling to room temperature, the solvent was distilled off under reduced pressure. The residue was purified by column chromatography on silica gel with ethyl acetate/petroleum ether (3:2) as eluent to give compound 5 as a yellow solid (6.47 g, yield: 29%). 1H-NMR (CDCl3, 400 MHz), δ: 7.85 (1H, d, J = 9.52 Hz), 7.32 (2H, d, J = 5.64 Hz), 6.94 (2H, d, J = 5.64 Hz), 6.79 (1H, dt, J = 10.36 Hz, 1.68 Hz), 6.55 (1H, d, J = 9.52 Hz), 5.83 (1H, dt, J = 10.36 Hz, 3.64 Hz), 5.08 (2H, s), 4.94 (2H, dd, J = 3.64 Hz), 3.81(3H, s). ESI-MS (+Q, m/z): 314 [M+H]+, 336 [M+Na]+.
8-Nitro-2H-chromen-5-ol (6). To a solution of compound 5 (18.2 g, 58 mmol) in dichloromethane (200 mL), trifluoroethylacetic acid (10.0 mL) was added dropwise with stirring at −10 °C. The reaction mixture was stirred at this temperature for 8 h, then quenched by addition of ice water (5 mL). The aqueous solution was then adjusted to pH = 10 with 1 N sodium hydroxide and the two phases were separated. The water phase was extracted twice with dichloromethane. The organic phases were combined and washed with water and brine, dried with Na2SO4 and the solvent removed under vacuum to yield the crude product, which was purified by column chromatography (dichloromethane/methanol 100:2) to give compound 6 as a yellow solid (7.5 g, yield: 67%). 1H-NMR (CDCl3, 400 MHz), δ: 11.11 (1H, s) 7.73 (d, 1H, J = 9.24 Hz), 6.68 (dt, 1H, J = 10.08 Hz, 1.96 Hz), 6.53 (1H, d, J = 9.24 Hz), 5.92 (1H, dt, J = 10.08 Hz, 3.64 Hz), 4.87 (2H, dd, J = 3.64 Hz, 1.96 Hz).
5-(2-Bromoethoxy)-8-nitro-2H-chromene (7). To a stirred solution of compound 6 (5.97 g, 31 mmol) in acetonitrile (150 mL) potassium carbonate (5.13 g, 37 mmol) and 1,2-dibromoethane (23.23 g, 124 mmol) were continuously added. The resulting mixture was heated to reflux for 2.5 h and then concentrated. The residue was partitioned between water (50 mL) and ethyl acetate (50 mL). The organic layer was separated, and the aqueous phase extracted with several additional portions of ethyl acetate. The combined organic phase was washed with brine, dried (MgSO4) and concentrated to dryness. The residue was separated by column chromatography on silica gel with ethyl acetate/petroleum ether (1/1) as eluent to give compound 7 as a yellow solid (4.69 g, yield: 34%). 1H-NMR (CDCl3, 400 MHz), δ: 7.84 (d, 1H, J = 9.2 Hz), 6.82 (dd, 1H, J = 10.8 Hz, 1.7 Hz), 6.41 (1H, d, J = 9.52 Hz), 6.80 (1H, dt, J = 10.36 Hz, 1.68 Hz), 6.44 (1H, d, J = 9.52 Hz), 5.88 (1H, dt, J = 10.08 Hz, 3.68 Hz). ESI-MS (+Q, m/z): 300 [M+H]+, 302 [M+H]+, 322 [M+Na]+, 324 [M+Na]+.
4-(2-(8-Nitro-2H-chromen-5-yloxy)ethyl)morpholine (9a). To a stirred solution of compound 7 (2 g, 6.66 mmol) in DMF (60 mL) potassium carbonate (1.4 g, 10.1 mmol) and morpholine (908 mg, 10.42 mmol) were continuously added. The resulting mixture was heated to 80 °C for 2 h. and then poured into cold water and extracted with ethyl acetate. The organic phase was washed with brine, dried over Na2SO4, and concentrated to dryness. The residue was separated by column chromatography on silica gel with ethyl acetate/petroleum ether (2/1) as eluent to give compound 9a as a white solid (1.06 g, yield: 52%). mp: 92–94 °C, 1H-NMR (CDCl3, 400), δ: 7.86 (d, J = 9.2Hz, 1H), 6.73 (dt, J = 10.0, 2.0 Hz, 1H), 6.47 (d, J = 9.2 Hz, 1H), 5.86 (dt, J = 10, 3.6 Hz, 1H), 4.95 (m, 2H), 4.22 (m, 2H), 3.75 (m, 4H), 2.86 (m, 2H), 2.61 (m, 4H). FAB-MS(m/z): 307 [M+H]+.
cis-2,6-Dimethyl-4-(2-(8-nitro-2H-chromen-5-yloxy)ethyl) morpholine (9b). Compound 9b was prepared following the method described for compound 9a, except cis-2,6-dimetylmopholine was used instead of morpholine as a reactant. Treating compound 7 (2 g, 6.63 mmol) in this manner to afford the title compound resulted in 1.80 g (81% yield) of compound 9b as a white solid. 1H-NMR (CDCl3, 400 MHz), δ: 7.85 (1H, d, J = 9.24 Hz), 6.74 (1H, dt, J = 10.12 Hz, 1.96 Hz), 6.47 (1H, d, J = 9.24 Hz), 6.74 (1H, dt, J = 10.12 Hz, 3.64 Hz), 4.94 (2H, dd, J = 3.64 Hz, 1.96 Hz), 4.19 (2H, m), 3.68 (2H, m), 2.81 (4H, m), 1.92 (2H, m), 1.17 (3H, s), 1.16 (3H, s). ESI-MS (+Q, m/z): 335 [M+H]+, 142.
1-(2-(8-Nitro-2H-chromen-5-yloxy)ethyl)-1H-1,2,4-triazole (9c). Compound 9c was prepared following the method described for compound 9a, except 1H-1,2,4-triazole was used instead of morpholine as a reactant. Treating compound 7 (2 g, 6.66 mmol) by this method resulted in 1.11 g (58% yield) of compound 9c as a white solid. 1H-NMR (CDCl3, 400 MHz), δ: 8.21 (1H, s), 8.00 (1H, s), 7.81 (1H, d, J = 9.24 Hz), 6.56 (1H, dt, J = 10.08 Hz, 1.96 Hz), 6.42 (1H, d, J = 9.52 Hz), 5.87 (1H, dt, J = 10.08 Hz, 3.64 Hz), 4.94 (2H, dd, J = 3.64 Hz, 1.96 Hz), 4.64 (2H, m), 4.54 (2H, m). ESI-MS (+Q, m/z): 289 [M+H]+, 311 [M+Na]+.
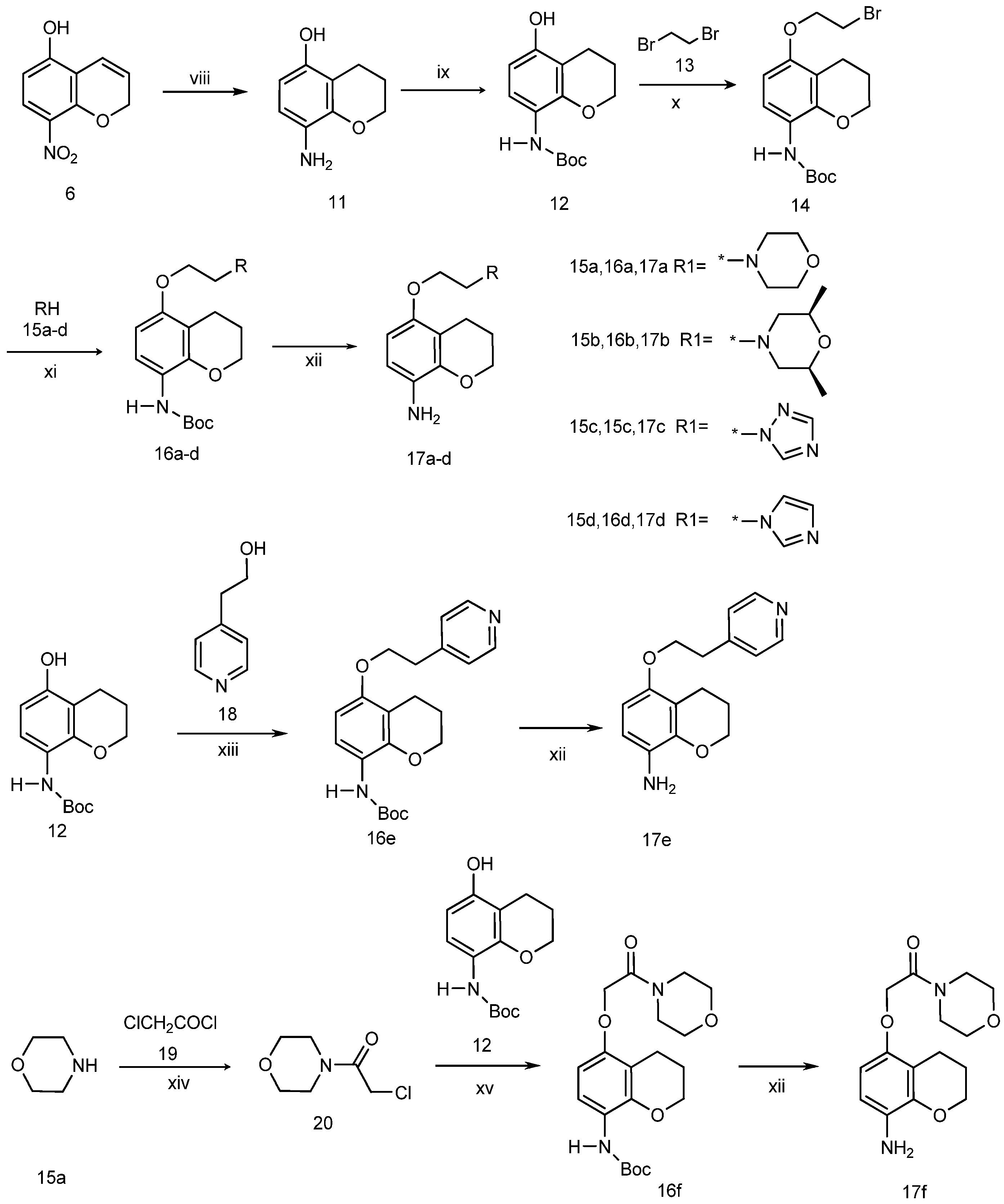
5-Hydroxy-8-aminochromane (11). To a solution of 5-hydroxy-8-nitro-2H-chromene (7, 5.0 g, 25.9 mmol) in anhydrous ethanol (100 mL) two drops of hydrochloric acid and palladium/carbon (2.0 g, 10%) were added. The mixture was heated to 60 °C, then hydrazine hydrate (10.0 mL, 85%) was added dropwise. After refluxing for 6 h, the mixture was cooled and filtered. The filtrate was dried and concentrated to give 2.12 g (50% yield) of compound 11 as an oily product which was sufficiently pure for use in the next step without further purification. 1H-NMR (CDCl3, 400 MHz), δ: 6.44 (d, 1H, J = 8.0 Hz), 6.19 (d, 1H, J = 8.4 Hz), 4.18 (t, 2H, J = 5.2 Hz), 3.54 (s, 2H), 2.65 (t, 2H, J = 6.8 Hz), 2.00 (p, 2H).
N-Boc-5-hydroxy-8-aminochromane (12). To a stirred solution of crude 11 (2.12 g, 12.8 mmol) and triethylamine (3.0 mL) in dichloromethane (100 mL), a solution of (Boc)2O (4.0 g, 18.3 mmol) in dichloromethane (20 mL) was added at 0 °C. The mixture was stirred at room temperature overnight, poured into water, extracted with dichloromethane, dried, and concentrated to dryness. The residue was separated by column chromatography on silica gel with ethyl acetate/petroleum ether (2/1) as eluent to give 2.69 g (79% yield) of compound 12 as a white solid. 1H-NMR (CDCl3, 400 MHz), δ: 7.61 (d, 1H), 6.76 (s, 1H), 6.30 (d, 1H, J = 12 Hz), 5.30 (s, 1H), 4.17 (t, 2H, J = 6 Hz), 2.65 (t, 2H, J = 8 Hz), 1.98 (p, 2H), 1.52 (s, 9H).
N-Boc-5-(2-bromoethoxy)-8-aminochromane (14). A mixture of compound 12 (2.20 g, 8.3 mmol), anhydrous potassium carbonate (1.38 g, 10 mmol), and 1,2-dibromopropane (6.23 g, 33 mmol) in acetonitrile (50 mL) was heated under reflux for 72 h. The resulting mixture was cooled to room temperature and water was added to the mixture, which was then extracted three times with ethyl acetate. The combined ethyl acetate layers were washed with water, brine, and then dried over Na2SO4, filtered and concentrated to dryness. The residue was separated by column chromatography on silica gel with ethyl acetate/petroleum ether (1/1) as eluent to give 0.83 g (27% yield) of compound 14 as a white solid. 1H-NMR (CDCl3, 400 MHz), δ: 7.78 (d, 1H), 6.85 (s, 1H), 6.35 (d, 1H, J = 8 Hz), 4.27 (t, 2H, J = 6 Hz), 4.20 (t, 2H, J = 8 Hz), 3.65 (t, 2H, J = 6 Hz), 2.73 (t, 2H, J = 6 Hz), 2.00 (p, 2H), 1.53 (s, 9H).
N-Boc-5-(2-morpholinoethoxy)-8-aminochromane (16a). A mixture of compound 14 (1.86 g, 5 mmol), anhydrous potassium carbonate (0.83 g, 6.01 mmol) and morpholine (6 mmol) in DMF (20 mL) was heated to 80 °C for 2 h. The resulting mixture was cooled to room temperature and poured onto cold water, and extracted three times with ethyl acetate. The combined ethyl acetate layers were washed with water, brine, and then dried over Na2SO4, filtered and concentrated to dryness. The residue was separated by column chromatography on silica gel with ethyl acetate/petroleum ether (2/1) as eluent to give 1.56 g (82.4% yield) of compound 16a as a white solid. 1H-NMR (CDCl3, 400 MHz), δ: 7.77 (d, 1H), 6.81 (s, 1H), 6.35 (d, 1H, J = 9.2 Hz), 4.17 (t, 2H, J = 5.6 Hz), 4.08 (t, 2H), 3.74 (t, 2H, J = 4.4 Hz), 2.80 (t, 2H), 2.60–2.65 (m, 6H), 1.97 (p, 2H), 1.50 (s, 9H).
N-Boc-5-(2-cis-2,6-dimethyl)-morpholinoethoxy)-8-aminochromane (16b). Compound 16b was prepared following the method described for compound 16a, except cis-1,6-dimetylmophiline was used instead of morpholine as a reactant. Treating compound 14 (2 g, 5.37 mmol) in this manner produced 1.71 g (90% yield) of compound 16b as a white solid. 1H-NMR (CDCl3, 400 MHz), δ: 7.76 (d, 1H), 6.81 (s, 1H), 6.35 (d, 1H, J = 8.8 Hz), 4.17 (t, 2H, J = 4.8 Hz), 4.07 (t, 2H), 3.70 (m, 2H), 2.77–2.84 (m, 4H), 2.65 (t, 2H, J = 6.8 Hz), 1.91–1.98 (m, 4H), 1.50 (s, 9H), 1.17 (d, 3H), 1.15 (d, 3H).
N-Boc-5-(2-(1H-1,2,4-triazol-1-yl)-ethoxy)-8-aminochromane (16c). Compound 16c was prepared following the method described for compound 16a, except 1,2,4-triazole was used instead of morpholine as a reactant. Treating compound 14 (2 g, 5.37 mmol) in this manner produced 1.28 g (66% yield) of compound 16c as a white solid. 1H-NMR (CDCl3, 400 MHz), δ: 8.23 (s, 1H), 7.97 (s, 1H), 7.77 (d, 1H), 6.81 (s, 1H), 6.31 (d, 1H, J = 8.8 Hz), 4.57 (t, 2H, J = 5.2 Hz), 4.29 (t, 2H, J = 5.2 Hz), 4.15 (t, 2H, J = 5.2 Hz), 2.51 (t, 2H, J = 6.4 Hz), 1.94 (p, 2H), 1.50 (s, 9H).
N-Boc-5-(2-(1H-imidazol-1-yl)-ethoxy)-8-aminochromane (16d). Compound 16d was prepared following the method described for compound 16a, except imidazole was used instead of morpholine as a reactant. Treating compound 14 (2 g, 5.37 mmol) in this manner produced 1.29 g (67% yield) of compound 16d as a white solid. 1H-NMR (CDCl3, 400 MHz), δ: 7.76 (d, 1H), 7.66 (s, 1H), 7.08 (s, 1H), 7.02 (s, 1H), 6.82 (s, 1H), 6.30 (d, 1H, J = 8.8 Hz), 4.34 (t, 2H), 4.17 (t, 2H), 2.58 (t, 2H, J = 6.6 Hz), 1.96 (p, 2H), 1.50 (s, 9H).
N-Boc-5-(2-(pyridin-4-yl)ethoxy)-8-aminochromane (16e). A solution of diethyl azodicarboxylate (DEAD, 1.05g, 5.0 mmol) in THF (25 mL) was slowly added to a solution of triphenylphosphine (1.57 g, 5.0 mmol), compound 12 (1.59 g, 6 mmol), and 2-(pyridin-4-yl)ethanol (616 mg, 5.0 mmol) in CH2Cl2 (25 mL), and the resulting cloudy mixture was stirred at room temperature for 3 h. After filtration of the mixture, the filtrate was concentrated in vacuo. Flash chromatography of the crude product (ethyl acetate/petroleum ether(1/1) afforded 1.76 g (95% yield) of compound 16e as a colourless oil that gradually crystallized upon standing at room temperature. 1H-NMR (CDCl3, 400 MHz), δ: 8.45 (d, 2H, J = 6.0 Hz), 7.80 (d, 1H), 6.87 (d, 2H, J = 5.2 Hz), 6.86 (s, 1H), 6.39 (d, 2H, J = 8.8 Hz), 4.36 (t, 2H), 4.30 (t, 2H), 4.17 (t, 2H), 2.62 (t, 2H, J = 6.4 Hz), 1.94 (p, 2H), 1.51 (s, 9H).
3.2.1. N-Boc-5-(2-morpholino-2-oxoethoxy)-8-aminochromane (16f)
Step 1: preparation of 4-(2-chloroacetyl)morpholine (20). To a stirred solution of morpholine (17.4 g, 200 mmol) and triethylamine (24.24 g, 220 mmol) in CH2Cl2 (200 mL), acetyl chloride (24.00 g, 210 mmol) in CH2Cl2 (volume) was added dropwise. The resulting reaction mixture was stirred at this temperature for a further 4 h, then poured into water, and the aqueous layer extracted twice with methylene chloride. The organic chloride phase was combined, washed with dilute hydrochloric acid and water, and dried over Na2SO4, and evaporated to dryness to give compound 20 (22.66 g, 69.2% yield) as a pale yellow oil.
Step 2: preparation of N-boc-5-(2-morpholino-2-oxoethoxy)-8-aminochromane (16f). A solution of compound 12 (1.86 g, 5.0 mmol), anhydrous potassium carbonate (0.83 g, 6.0 mmol) and 4-(2-chloroacetyl)-morpholine (20, 982 mg, 6.0 mmol) in DMF (20 mL) was heated to 80 °C for 2 h. The resulting mixture was cooled to room temperature, poured onto cold water and extracted three times with ethyl acetate. The combined ethyl acetate layers were washed with water, brine, and then dried over Na2SO4, filtrated and concentrated to dryness. The residue was separated by column chromatography on silica gel with ethyl acetate/petroleum ether (1/1) as eluent to give 1.35 g (69% yield) of compound 16f as a white solid. 1H-NMR (CDCl3, 400 MHz), δ: 7.77 (d, 1H), 6.89 (s, 1H), 6.35 (d, 1H, J = 8.8 Hz), 4.63 (s, 2H), 4.19 (t, 2H), 3.60–3.68 (m, 8H), 2.68 (t, 2H, J = 6.0 Hz), 1.99 (p, 2H), 1.50 (s, 9H).
Step 3: preparation of 5-(morpholinoethoxy)-8-aminochromane (17a). To a solution of compound 16a (1.48 g, 4.0 mmol) in CH2Cl2 (40 mL), precooled trifluoroacetic acid (4.0 mL) was added at 0–4 °C, and the reaction was stirred at this temperature for 5 h. After evaporation, water was added to the residue, and the pH of the mixture was adjusted to 10 by addition of 1 M aqueous NaOH solution. The aqueous layer was extracted with ethyl acetate, washed with water, dried over anhydrous sodium sulfate, filtered, and concentrated to give 930 mg (86% yield) of compound 17a as a grey solid. This product is unstable and was therefore used without delay for the next step.
5-(2-(2,6-Dimethyl)-morpholinoethoxy)-8-aminochromane (17b). Compound 17b was prepared following the method described for compound 17a employing 16b (1.64 g, 4.0 mmol) as a reactant, producing compound 17b as a pale white solid (906 mg, 74% yield). This product is unstable and was therefore used without delay for the next step.
5-(2-(1,2,4-Triazole)-ethoxy)-8-aminochromane (17c). Compound 17c was prepared following the method described for the compound 17a employing 16c (1.23 g, 4.0 mmol) as reactant, to produce compound 17c as a white solid (729 mg, 70% yield). This product is unstable and was therefore used without delay for the next step.
5-(2-Imidazolylethoxy)-8-aminochromane (17d). Compound 17d was prepared following the method described for the compound 17a employing 16d (1.23 g, 4.0 mmol) as reactant, to produce compound 17d as a white solid (778 mg, 75% yield). This product is unstable and was therefore used without delay for the next step.
5-(2-(Pyridin-4-yl)ethoxy)-8-aminechromane (17e). Compound 17e was prepared following the method described for compound 17a employing 16e (1.48 g, 4 mmol) as reactant, to produce compound 17e as a white solid (789 mg, 73% yield). This product is unstable and was therefore used without delay for the next step.
5-(2-Morpholino-2-oxoethoxy)-8-aminochroman (17f). Compound 17f was prepared following the method described for compound 17a employing 16f (1.17 g, 4 mmol) as reactant, to afford the compound 17f as a pale white solid (795 mg, 68% yield). This product is unstable and was therefore used without delay for the next step.
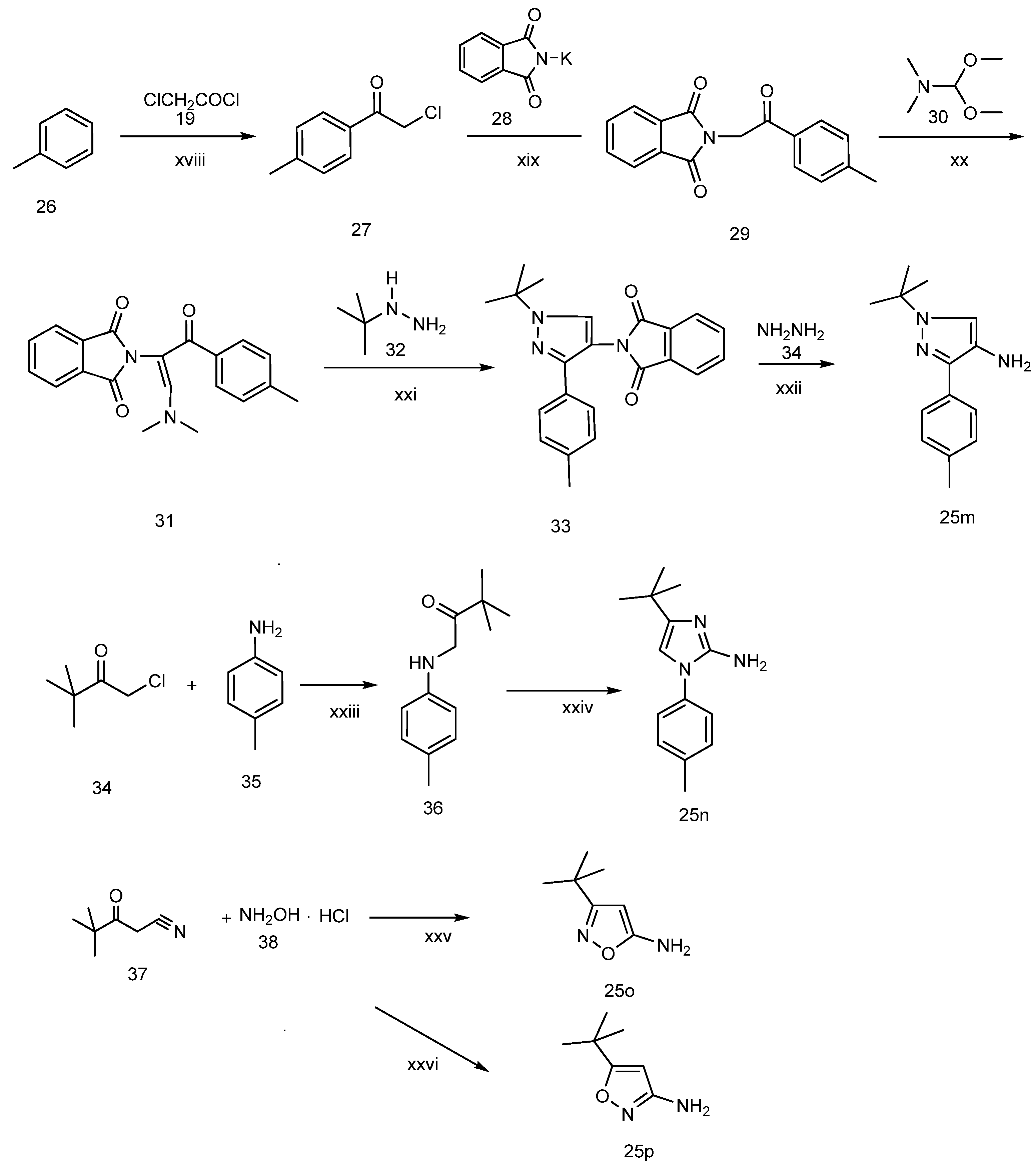
3-tert-Butyl-1-p-tolyl-1H-pyrazol-5-amine (25a). A solution of 4-tolyllhydrazine hydrochloride (5.20 g, 33 mmol) and pentylacyl acetonitrile (3.75 g, 30 mmol) in 0.4 M ethanolic solution of HCl (100 mL) was heated under reflux during 48 h. After cooling to room temperature, 1M NaOH was added to the mixture until the pH reached 10–11. The mixture was partitioned between water and ethyl acetate. The water phase was extracted twice with dichloromethane. The organic phases were combined and washed with water and brine, then dried with Na2SO4. Evaporation of the solvent in vacuo afforded the crude product, which was subjected to recrystallization from ethyl acetate and petroleum ether to produce compound 25a as a white solid (5.88 g, 86% yield). 1H-NMR (CDCl3, 400 MHz), δ: 7.40 (d, 2H, J = 8.4 Hz), 7.25 (d, 2H, J = 3.08 Hz), 5.50 (s, 1H), 3.72 (brs, 2H, NH2), 2.37 (s, 3H), 1.32 (s, 9H).
3-tert-Butyl-1-(4-fluorophenyl)-1H-pyrazol-5-amine (25b). The title compound was prepared according to the method used for 25a except (4-fluorophenyl)hydrazine hydrochloride was used instead of 4-tolylhydrazine hydrochloride. Yield: 79%. 1H-NMR (CDCl3, 400 MHz), δ: 7.59 (d, 2H), 7.10 (d, 2H), 5.58 (s, 1 H), 3.62 (brs, 2H, NH2), 1.32 (s, 9H).
3-tert-Butyl-1-(4-methoxyphenyl)-1H-pyrazol-5-amine (25c). The title compound was prepared according to the method used for 25a, except (4-methoxyphenyl)hydrazine hydrochloride was used instead of 4-tolylhydrazine hydrochloride. Yield: 87%. 1H-NMR (CDCl3, 400 MHz): 7.41 (d, 2H), 6.97 (d, 2H), 5.43 is, 1H), 3.83 (s, 3H), 1.35 (s, 9H).
3-tert-Butyl-1-(4-trifluoromethylphenyl)-1H-pyrazol-5-amine (25d). The title compound was prepared according to the method used for 25a, except (4-trifluoromethylphenyl)hydrazine hydrochloride was used instead of 4-tolyllhydrazine hydrochloride. Yield: 79%. 1H-NMR (CDCl3, 400 MHz), δ: 7.77 (d, 2H, J = 8.4 Hz), 7.69 (d, 2H, J = 8.4 Hz), 5.56 (s, 1H), 3.76 (s, 2H), 1.31 (s, 9H).
3-tert-Butyl-1-(4-nitrophenyl)-1H-pyrazol-5-amine (25e). The title compound was prepared according to the method used for 25a, except 4-nitrohydrazine hydrochloride was used instead of 4-tolylhydrazine hydrochloride. Yield: 83%. 1H-NMR (DMSO-d6, 400 MHz), δ: 8.28 (d, J = 6.9 Hz, 2H), 7.93 (d, J = 6.9 Hz, 2H), 5.55 (s, 2H), 5.46 (s, 1H), 1.20 (s, 9H).
3-tert-Butyl-1-(4-isopropylphenyl)-1H-pyrazol-5-amine (25f). The title compound was prepared according to the method used for 25a, except (4-isopropylphenyl) hydrazine hydrochloride was used instead of 4-tolylhydrazine hydrochloride. Yield: 81%. 1H-NMR (CDCl3, 400 MHz), δ: 7.44 (d, 2H, J = 8.4 Hz), 7.29 (d, 2H, J = 8.4 Hz), 5.51 (s, 1H), 3.72 (s, 2H), 2.93 (m, 1H), 1.31 (s, 9H), 1.25 (d, 3H), 1.24 (d, 3H).
3-tert-Butyl-1-m-tolyl-1H-pyrazol-5-amine (25g). The title compound was prepared according to the method used for 25a, except m-tolylhydrazine hydrochloride was used instead of p-tolylhydrazine hydrochloride. Yield: 79%. 1H-NMR (CDCl3, 400 MHz), δ: 7.38 (s, 1H), 7.31–7.33 (m, 2H), 7.13 (m, 1H), 5.51 (s, 1H), 3.73 (bs, 2H), 2.39 (s, 3H), 1.31 (s, 9H).
3-tert-Butyl-1-(4-chlorophenyl)-1H-pyrazol-5-amine (25h), 3-tert-butyl-1-(4-bromophenyl)-1H-pyrazol-5-amine (25i), 3-tert-butyl-1-(4-ethylphenyl)-1H-pyrazol-5-amine (25j) and 3-tert-butyl-1-phenyl-1H-pyrazol-5-amine (25n) are all commercially available.
3-tert-Butyl-1-(naphthalen-1-yl)-1H-pyrazol-5-amine (25o). Compound 25o was prepared according to the method used for 1, except (naphthalen-1-yl)hydrazine hydrochloride was used instead of 4-tolylhydrazine hydrochloride. Yield: 82%. 1H-NMR (CDCl3, 400 MHz), δ: 7.89–7.91 (m, 2H), 7.51–7.55 (m, 5H), 5.59 (s, 1H), 3.50 (s, 2H), 1.34 (s, 9H).
3.2.2. 1-tert-Butyl-3-(p-tolyl)-4-amine-1H-pyrazole (25p)
Step 1: preparation of 2-chloro-1-p-tolylethanone (27). To a suspension of anhydrous aluminum chloride (5.60 g, 40 mmol) in anhydrous toluene (40 mL), chloroacetyl chloride (4.52 g, 40 mmol) was slowly added dropwise. After the aluminum chloride has dissolved, the mixture was heated slowly to 80 °C and held at that temperature for 2 h, then cooled and poured into crushed ice (100 g) containing concentrated hydrochloric acid (10 mL). The aqueous layer was extracted three times with toluene. The combined toluene layers were washed successively with 10% aqueous sodium hydroxide, water and brine, dried over anhydrous sodium sulfate and evaporated under reduced pressure. The residue was purified by crystallization from diethyl ether to give the pure compound 27(5.6 g, yield: 86%). 1H-NMR (CDCl3, 400 MHz), δ: 7.46 (d, 2H, J = 7.5 Hz), 7.01 (d, 2H, J = 7.5 Hz), 4.63 (s, 2H), 2.35 (s, 3H).
Step 2: preparation of 2-(1,3-dioxoisoindolin-2-yl)-1-p-tolylethanone (29). To a solution of compound 27 (5.06 g, 30 mol) in DMF (30 mL), phthalimide potassium salt (5.56 g, 30 mol) was added, and the resulting mixture was stirred at 70 °C for 2 h, cooled, and then poured into ice water (300 mL). The precipitated white solid was filtered and dried to give compound 29 (7.37 g, yield: 88%). 1H-NMR (400 MHz, CDCl3), δ: 7.88–7.83 (m, 4H), 7.73–7.69 (m, 2H), 7.27 (d, J = 7.9 Hz, 2H), 5.08 (s, 2H), 2.40 (s, 3H).
Step 3: preparation of N,N-dimethyl-3-(isoindoline-1,3-dione-2yl)-2-p-tolylprop-1-en-1-amine (31). To a solution of compound 29 (5.58 g, 20 mmol) in DMF (40 mL), N,N-dimethylformamide dimethyl acetal (5.68 g, 24 mmol) was added dropwise. The mixture was heated to 100 °C and kept at this temperature for 24 h, then cooled, poured into ice water (200 mL) and extracted with ethyl acetate. The combined organic phase was dried over anhydrous sodium sulfate, and evaporated in vacuo to provide a residue which was purified by silica gel chromatography using ethyl acetate and petroleum ether (1/3) as eluent to obtain compound 31 as a white solid (4.0 g, yield: 62%).
Step 4: preparation of 1-tert-butyl-3-(p-tolyl)-4-phthalimido-pyrazole (33). A solution of compound 31 (4.0 g, 14.3 mmol) and tert-butyl hydrazine (1.39 g, 15.7 mmol) in 90% ethanol (200 mL) was heated under reflux for 12 h. After cooling to room temperature, the resulting white crystalline solid was filtered, washed with anhydrous ether and dried to give compound 33 as a white solid (3.73 g, yield: 73%).
Step 5: preparation of 1-tert-Butyl-3-(p-tolyl)-4-amine-1H-pyrazole (25p). To a solution of compound 33 (3.59 g, 10 mol) in ethanol (100 mL), 85% hydrazine hydrate solution (2.36 g, 40 mmol) was added dropwise. The mixture was heated to reflux for 2 h, cooled to room temperature, and concentrated, and ether (20 mL) was added. The precipitated white solid was filtered, washed with anhydrous ether, purified by silica gel chromatography using ethyl acetate and petroleum ether (1/1) as eluent to give compound 25p as a white solid (1.89 g, yield 83%). 1H-NMR (CDCl3, 400 MHz) δ: 7.21–7.26 (m, 5H), 2.68 (s, 2H), 2.41 (s, 3H), 1.41 (s, 9H).
3.2.3. 1-(4-Methylphenyl)-2-amino-4-tert-butyl-imidazole (25q)
Step 1: preparation of N-(pivaloylmethyl)-4-methylaniline (36). To a solution of tert-butylchloromethyl ketone (3.23 g, 0.024 mol) and p-toluidine (2.14 g, 0.020 mol) in DMF (20 mL), sodium bicarbonate (2.52 g, 0.030 mol) was added. The mixture was stirred at 75 °C for 48 h, then cooled, and poured into ice water (200 mL). The precipitated white solid was filtered and dried to give compound 36 as a white solid (4.08 g, yield: 99%).
Step 2: preparation of 1-(4-methylphenyl)-2-amino-4-tert-butyl-imidazole (25q). A solution of compound 36 (5.0 g, 20 mmol) and 50% aqueous cyanamide (16.92 g, 20 mmol) in ethanol (200 mL) was refluxed for 12 h and concentrated, water was added to the residue. the mixture was extracted three times with ethyl acetate, then the combined organic layers washed with dilute sodium hydroxide solution, water and saturated brine, and dried. The residue was purified by silica gel chromatography using dichloromethane and methanol (100/1) to obtain a white solid product (2.62 g, yield: 57%). 1H-NMR (CDCl3, 400 MHz), δ: 7.26 (m, 4H), 6.35 (s, 1H), 4.18 (s, 2H), 2.39 (s, 3H), 1.27 (s, 9H).
3-tert-Butyl-5-aminoisoxazole (25r). To an aqueous solution of sodium hydroxide solution (0.84 g, 21 mmol) in water (10 mL), pivaloylacetonitrile (1.25 g, 10 mmol) and hydroxylamine hydrochloride (0.76 g, 11 mmol) were added. The resulting solution was stirred at 50° C for 3 h. The reaction mixture was cooled and the resultant white crystalline solid was filtered, washed with water and dried to provide compound 25r as a white crystalline solid (1.23 g, yield 88%). 1H-NMR (CDCl3, 400 MHz), δ: 5.03 (s, 1H), 4.32 (bs, 2H), 1.27 (s, 9H).
5-(tert-Butyl)-3-aminoisoxazole (25s). To a solution of pivaloylacetonitrile (3 g, 23.97 mmol) in water (20 mL), NaOH (1.06 g, 26.4 mmol) and hydroxylamine hydrochloride (1.83 g, 26.4 mmol) were added continuously with stirring. The resulting solution was stirred for approximately 30 min at room temperature, and the pH adjusted to 10–11 with 1 M NaOH. After stirring for 10 h or more at 50 °C, the mixture was cooled and washed two to three times with carbon tetrachloride. The aqueous layer was acidified with concentrated HC1 until the pH = 4–5, and then further stirred for approximately 3 h at 50 °C. The reaction mixture was cooled to room temperature, and adjusted to pH 12 by adding an aqueous solution of 1 N NaOH. The resulting solid was filtered, washed with distilled water, and dried in air to obtain compound 25s as a white solid (2.0 g, yield: 70%). 1H-NMR (DMSO-d6, 300 MHz), δ: 5.49 (s, 1H), 5.40 (s, 2H), 1.21 (s, 9H).
3.2.4. General Procedure for the Preparation of Chromanylureas (39a–j) and 2H-Chromenylureas (40a–r)
A solution of compounds 10a–d or compounds 17a–d (1.0 mmol) in dichloromethane (10 mL) was slowly added to a stirred solution of triphosgene (109 mg, 0.36 mmol) in dichloromethane (50 mL) over a period of 30 min using a syringe. After stirring for a further 30 min, a solution of compound 25a–r (0.6 mmol) and triethylamine (0.4 mL, 2.77 mmol) in dichloromethane (10 mL) was added in one portion. The reaction mixture was stirred for 2 h at room temperature. After completion of the reaction, the reaction was poured into water (50 mL) and extracted three times with dichloromethane. The organic layer was washed with water (5 mL), sat. NaCl solution (5 mL), and dried over Na2SO4. After evaporation of solvent under vacuum, the residue was purified by silica gel chromatography to give the desired chromanylurea or 2H-chromenylurea compounds.
1-(3-tert-Butyl-1-p-tolyl-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-chroman-8-yl)urea (39a). Yield: 19%, 1H-NMR (CDCl3, 400 MHz), δ: 7.69 (d, 1H, J = 8.4 Hz), 7.29–7.31 (m, 3H), 7.16 (m, 2H), 6.84 (s, 1H), 6.31–6.34 (m, 2H), 4.04–4.07 (m, 4H), 3.72 (t, 4H), 2.80 (t, 2H, J = 5.2 Hz), 2.60 (t, 2H), 2.59 (t, 4H), 2.31 (s, 3H), 1.91 (p, 2H), 1.34 (s, 9H). FAB-MS (m/z): 534.2 [M+H]+.
1-(3-tert-Butyl-1-phenyl-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-chroman-8-yl)urea (39b). White solid, yield: 26%. 1H-NMR (CDCl3, 400 MHz), δ: 7.67 (d, 1H, J = 8.4 Hz), 7.32–7.45 (m, 2H), 7.34 (m, 3H), 7.28 (m, 1H), 7.03 (s, 1H), 6.35 (s, 1H), 6.31 (d, 1H, J = 10.8 Hz), 4.04–4.06 (m, 4H), 3.70 (t, 4H, J = 4.8 Hz), 2.77 (t, 2H, J = 5.6 Hz), 2.56–2.61 (m, 6H), 1.90 (p, 2H), 1.34 (s, 9H). FAB-MS (m/z): 520.2 [M+H]+.
1-(3-tert-Butyl-1-m-tolyl-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-chroman-8-yl)urea (39c). White solid, yield: 18%, 1H-NMR (CDCl3, 400 MHz), δ:7.68 (d, 1H, J = 8.4 Hz), 7.21–7.26 (m, 4H), 7.11 (d, 1H), 6.83 (s, 1H), 6.36 (s, 1H), 6.32 (d, 1H, J = 8.8 Hz), 4.05–4.08 (m, 4H), 3.73 (t, 4H, J = 4.4 Hz), 2.80 (t, 2H, J = 5.6 Hz), 2.57–2.62 (m, 6H), 2.32 (s, 3H), 1.91 (p, 2H), 1.35 (s, 9H). FAB-MS (m/z): 534.2 [M+H]+.
1-(3-tert-Butylisoxazol-5-yl)-3-(5-(2-morpholinoethoxy)-chroman-8-yl)urea (39d). White solid, yield: 19%, 1H-NMR (CDCl3, 400 MHz), δ: 8.59 (s, 1H), 7.67 (d, 1H, J = 8.0 Hz), 6.36 (d, 1H, J = 9.2 Hz), 6.12 (s, 1H), 4.08–4.10 (m, 4H), 3.75 (t, 4H, J = 4.4 Hz), 2.83 (t, 2H, J = 5.6 Hz), 2.60–2.64 (m, 6H), 1.92 (p, 2H), 1.30 (s, 9H). FAB-MS (m/z): 445.1 [M+H]+.
1-(5-tert-Butylisoxazol-3-yl)-3-(5-(2-morpholinoethoxy)-chroman-8-yl)urea (39e). White solid, yield: 31%. 1H-NMR (CDCl3, 400 MHz), δ: 8.75 (s, 2H), 7.86 (d, 1H, J = 8.8 Hz), 6.37 (d, 1H, J = 8.8 Hz), 6.07 (s, 1H), 4.23 (t, 2H, J = 4.8 Hz), 4.10 (t. 2H), 3.74 (t, 4H, J = 4.4 Hz), 2.82 (t, 2H, J = 5.2 Hz), 2.61–2.67 (m, 6H), 1.99 (p, 2H), 1.34 (s, 9H). FAB-MS (m/z): 445.1 [M+H]+.
1-(3-tert-Butyl-1-p-tolyl-1H-pyrazol-5-yl)-3-(5-(2-morpholino-2-oxoethoxy)chroman-8-yl)urea (39f). White solid, yield: 16%, 1H-NMR (CDCl3, 400 MHz), δ: 7.75 (d, 1H, J = 9.2 Hz), 7.34 (d, 2H, J = 8.4 Hz), 7.25 (d, 2H), 6.38 (s, 1H), 6.34 (d, 1H, J = 9.2 Hz), 4.64 (s, 2H), 4.11 (t, 2H, J = 3.6 Hz), 3.63–3.67 (m, 8H), 2.67 (t, 2H, J = 6.4 Hz), 2.37 (s, 3H), 1.95 (p, 2H), 1.36 (s, 9H). FAB-MS (m/z): 548.1 [M+H]+.
1-(3-tert-Butyl-1-p-tolyl-1H-pyrazol-5-yl)-3-(5-(2-(sn-2,6-dimethylmorpholino)ethoxy)chroman-8-yl)-urea (39g). White solid, yield: 21%, 1H-NMR (CDCl3, 400 MHz), δ: 7.71 (d, 1H, J = 9.2 Hz), 7.31 (d, 2H, J = 8.4 Hz), 7.21 (d, 2H), 7.17 (s, 1H), 6.31–6.36 (m, 3H), 4.08–4.10 (m, 4H), 3.71 (m, 2H), 2.80–2.86 (m, 4H), 2.63 (t, 2H, J = 6.4 Hz), 2.36 (s, 3H), 1.92–1.95 (m, 4H), 1.35 (s, 9H), 1.17 (d, 3H), 1.15 (d, 3H). FAB-MS (m/z): 562.2 [M+H]+.
3-(3-tert-Butyl-1-p-tolyl-1H-pyrazol-5-yl)-1-(5-(2-(1H-1,2,4-triazol-1-yl)ethoxy)chroman-8-yl)urea (39h). White solid, yield: 15%, 1H-NMR (CDCl3, 400 MHz), δ: 8.08 (s 1H), 7.82–7.84 (m, 2H), 7.45 (s, 1H), 7.32 (d, 2H, J = 8.4 Hz), 7.17 (d, 2H, J = 8.4 Hz), 6.40 (s, 1H), 6.27 (d, 1H, J = 9.2 Hz), 4.56 (t, 2H, J = 4.8 Hz), 4.23 (t, 2H, J = 4.8 Hz), 3.77 (t, 2H), 2.33–2.36 (m, 5H), 1.69 (p, 2H), 1.35 (s, 9H). FAB-MS (m/z): 516.3 [M+H]+.
3-(3-tert-Butyl-1-p-tolyl-1H-pyrazol-5-yl)-1-(5-(2-(1H-imidazol-1-yl)ethoxy)chroman-8-yl)urea (39i). White solid, yield: 21%, 1H-NMR (CDCl3, 400 MHz), δ: 7.88 (d, 1H, J = 8.8 Hz), 7.73 (s, 1H), 7.53 (s, 1H), 7.32 (d, 2H, J = 8.0 Hz), 7.15 (d, 2H, J = 8.0 Hz), 6.93 (s, 1H), 6.89 (s, 1H), 6.41 (s, 1H), 6.22 (d, 1H, J = 8.8 Hz), 4.32 (t, 2H, J = 4.8 Hz), 4.12 (t, 2H, J = 5.6 Hz), 3.62 (t, 2H), 2.35 (t, 2H), 2.32 (s, 3H), 1.61 (p, 2H), 1.35 (s, 9H). FAB-MS (m/z): 515.2 [M+H]+.
1-(3-tert-Butyl-1-p-tolyl-1H-pyrazol-5-yl)-3-(5-(2-(pyridin-4-yl)ethoxy)chroman-8-yl)urea (39j). White solid, yield: 17%, 1H-NMR (CDCl3, 400 MHz), δ: 7.71 (d, 1H), 7.50–7.53 (m, 2H), 7.31 (d, 2H), 7.21 (d, 2H, J = 8.0 Hz), 6.37 (s, 1H), 6.30 (d, 1H, J = 8.4 Hz), 6.26 (t, 1H), 4.52 (t, 2H, J = 5.2 Hz), 4.28 (t, 2H, J = 5.2 Hz), 4.07 (t, 2H, J = 5.6 Hz), 2.54 (t, 2H, J = 6.4 Hz), 2.35 (s, 3H), 1.91 (p, 2H), 1.36 (s, 9H). FAB-MS (m/z): 515.1 [M+H]+.
1-(3-tert-Butyl-1-p-tolyl-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40a). Yield: 27%, white solid. 1H-NMR (CDCl3, 400 MHz), δ: 7.74 (d, 1H, J = 8.8 Hz), 7.33 (d, 2H, J = 8.4 Hz), 7.21 (d, 2H, J = 8.0 Hz), 7.13 (s, 1H), 6.72–6.74 (dd, 1H, J = 8.4 Hz, 1.6 Hz), 6.39 (d, 1H, J = 9.2 Hz), 6.35 (s, 1H), 6.34 (s, 1H), 5.72 (m, 1H), 4.71 (t, 2H, J = 2.0 Hz), 4.11 (t, 2H), 3.74 (t, 4H), 2.82 (t, 2H), 2.60 (t, 4H), 2.36 (s, 3H), 1.35 (s, 9H). FAB-MS (m/z): 532.2 [M+H]+.
1-(3-tert-Butyl-1-(4-fluorophenyl)-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)-urea (40b). Yield: 22%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.67 (d, 1H, J = 8.4 Hz), 7.43 (m, 2H), 7.07-–7.11 (m, 3H), 6.73 (d, 1H, J = 10.4 Hz), 6.58 (s, 1H), 6.38 (d, 1H, J = 9.2 Hz), 6.35 (s, 1H), 5.73 (m, 1H), 4.71 (t, 2H, J = 2.0 Hz), 4.11 (t, 2H, J = 5.2 Hz), 3.75 (t, 4H), 2.83 (t, 2H), 2.61 (t, 4H), 1.35 (s, 9H). FAB-MS (m/z): 536.2 [M+H]+.
1-(3-tert-Butyl-1-(4-chlorophenyl)-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)-urea (40c). Yield: 29%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.65 (d, 1H, J = 8.8 Hz), 7.43 (d, 2H), 7.35 (d, 2H, J = 8.8 Hz), 7.10 (s, 1H), 6.74 (d, 1H, J = 10.0 Hz), 6.68 (s, 1H), 6.36–6.38 (m, 2H), 5.72 (m, 1H), 4.69 (t, 2H, J = 2.0 Hz), 4.10 (t, 2H, J = 5.2 Hz), 3.74 (t, 4H, J = 4.4 Hz), 2.81 (t, 2H, J = 4.8 Hz), 2.59 (t, 4H), 1.35 (s, 9H). FAB-MS (m/z): 552.2 [M+H]+.
1-(1-(4-Bromophenyl)-3-tert-butyl-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)-urea (40d). Yield: 26%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.65 (d, 1H), 7.50 (d, 2H, J = 8.8 Hz), 7.36 (d, 2H, J = 8.8 Hz), 7.12 (s, 1H), 6.77 (s, 1H), 6.73 (d, 1H, J = 10.4 Hz), 6.38 (d, 1H, J = 7.2 Hz), 6.36 (s, 1H), 5.72 (m, 1H), 4.69 (t, 2H, J = 1.6 Hz), 4.10 (t, 2H, J = 1.6 Hz), 3.74 (t, 4H, J = 4.4 Hz), 2.82 (t, 2H, J = 4.8 Hz), 2.60 (t, 4H), 1.34 (s, 9H). FAB-MS (m/z): 598.0 [M+H]+.
1-(3-tert-Butyl-1-(4-methoxyphenyl)-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)-urea (40e). Yield: 26%, white solid, 1H-NMR (CDCl3, 400 MHz), δ:7.72 (d, 1H, J = 8.8 Hz), 7.33 (d, 2H, J = 9.2 Hz), 7.25 (s, 1H), 6.88 (d, 2H, J = 9.2 Hz), 6.75 (d, 1H), 6.73 (s, 1H), 6.36 (d, 1H), 6.33 (s, 1H), 5.70 (m, 1H), 4.67 (t, 2H, J = 1.6 Hz), 4.10 (t, 2H), 3.77 (s, 3H), 3.74 (t, 4H, J = 4.4 Hz), 2.80 (t, 2H, J = 5.6 Hz), 2.59 (t, 4H), 1.35 (s, 9H). FAB-MS (m/z): 548.1 [M+H]+.
1-(3-tert-Butyl-1-(4-(trifluoromethyl)phenyl)-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40f). Yield: 17%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.65 (m, 5H), 7.07 (s, 1H), 6.76 (s, 1H), 6.73 (d, 1H, J = 9.6 Hz), 6.40 (s, 1H), 6.38 (d, 1H, J = 8.8 Hz), 5.70 (m, 1H), 4.70 (t, 2H, J = 1.6 Hz), 4.10 (t, 2H, J = 5.2 Hz), 3.74 (t, 4H, J = 4.0 Hz), 2.82 (t, 2H), 2.60 (t, 4H), 1.36 (s, 9H). FAB-MS (m/z): 586.1 [M+H]+.
1-(3-tert-Butyl-1-(4-nitrophenyl)-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40g). Yield: 37%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 8.18 (d, 2H, J = 9.2 Hz), 7.74 (d, 2H, J = 8.8 Hz), 7.55 (d, 1H), 7.30 (s, 1H), 6.71 (d, 1H, J = 10.0 Hz), 6.39 (s, 1H), 6.34 (d, 1H, J = 9.2 Hz), 5.70 (m, 1H), 4.68 (t, 2H, J = 1.6 Hz), 4.09 (t, 2H, J = 6.0 Hz), 3.74 (t, 4H, J = 4.4 Hz), 2.81 (t, 2H, J = 6.0 Hz), 2.60 (t, 4H), 1.34 (s, 9H). FAB-MS (m/z): 563.0 [M+H]+.
1-(3-tert-Butyl-1-(4-ethylphenyl)-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40h). Yield: 27%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.74 (d, 1H, J = 8.8 Hz), 7.36 (d, 2H, J = 8.4 Hz), 7.25 (d, 2H), 7.14 (s, 1H), 6.73 (d, 1H, J = 10.0 Hz), 6.35–6.40 (m, 3H), 5.72 (m, 1H), 4.71 (t, 2H, J = 2.0 Hz), 4.12 (t, 2H), 3.75 (t, 4H), 2.84 (t, 2H), 2.62–2.68 (m, 6H), 1.36 (s, 9H), 1.22 (t, 3H). FAB-MS (m/z): 546.0 [M+H]+.
1-(3-tert-Butyl-1-(4-isopropylphenyl)-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)-urea (40i). Yield: 19%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.74 (d, 1H, J = 9.2 Hz), 7.34 (d, 2H), 7.26 (d, 2H), 7.15 (s, 1H), 6.73 (d, 1H, J = 10.0 Hz), 6.43 (s, 1H), 6.39 (d, 1H, J = 9.2 Hz), 6.35 (s, 1H), 5.72 (m, 1H), 4.71 (t, 2H, J = 2.0 Hz), 4.09 (t, 2H, J = 5.2 Hz), 3.73 (t, 4H), 2.81 (t, 2H), 2.59 (t, 4H), 1.36 (s, 9H), 1.24 (d, 3H), 1.22 (d, 3H). FAB-MS (m/z): 560.1 [M+H]+.
1-(3-tert-Butyl-1-(3-chloro-4-fluorophenyl)-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40j). Yield: 27%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.60 (m, 2H), 7.38 (m, 1H), 7.08–7.14 (m, 3H), 6.72 (d, 1H, J = 9.6 Hz), 6.38 (d, 1H, J = 8.8 Hz), 6.35 (s, 1H), 5.72 (m, 1H), 4.70 (t, 2H, J = 1.6 Hz), 4.10 (t, 2H, J = 5.2 Hz), 3.74 (t, 4H, J = 4.4 Hz), 2.83 (t, 2H), 2.61(t, 4H), 1.34 (s, 9H). FAB-MS (m/z): 570.0 [M+H]+.
1-(3-tert-Butyl-1-m-tolyl-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40k). Yield: 21%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.22 (d, 1H, J = 8.8 Hz), 7.26–7.29 (m, 3H), 7.15 (d, 2H), 6.73 (d, 1H, J = 10.0 Hz), 6.58 (s, 1H), 6.38 (d, 1H, J = 9.6 Hz), 6.36 (s, 1H), 5.72 (m, 1H), 4.71 (t, 2H, J = 1.6Hz), 4.12 (t, 2H), 3.76 (t, 4H), 2.84 (t, 2H), 2.63 (t, 4H), 2.36 (s, 3H), 1.36 (s, 9H). FAB-MS (m/z): 532.1 [M+H]+.
1-(3-tert-Butylisoxazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40l). Yield: 39%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 9.27 (s, 1H), 7.74 (d, 1H, J = 8.8 Hz), 7.40 (s, 1H), 6.67 (d, 1H, J = 10.0 Hz), 6.37 (d, 1H, J = 9.2 Hz), 6.13 (s, 1H), 5.63 (m, 1H), 4.62 (t, 2H, J = 1.6 Hz), 4.10 (t, 2H, J = 5.6 Hz), 3.77 (t, 4H, J = 4.4 Hz), 2.83 (t, 2H, J = 5.2 Hz), 2.69 (t, 4H), 1.30 (s, 9H). FAB-MS (m/z): 443.0 [M+H]+.
1-(5-tert-Butylisoxazol-3-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40m). Yield: 33%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 8.77 (s, 1H), 8.27 (s, 1H), 7.86 (d, 1H, J = 8.8 Hz), 6.75 (d, 1H, J = 10.0 Hz), 6.42 (d, 1H, J = 9.6 Hz), 6.02 (s, 1H), 5.76 (m, 1H), 4.84 (t, 2H, J = 2.0 Hz), 4.12 (t, 2H, J = 6.8 Hz), 3.76 (t, 4H), 2.84 (t, 2H), 2.62 (t, 4H), 1.36 (s, 9H). FAB-MS (m/z): 443.0 [M+H]+.
1-(1-tert-Butyl-3-p-tolyl-1H-pyrazol-4-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40n). Yield: 21%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.67–7.70 (m, 2H), 7.18–7.20 (m, 4H), 6.73 (d, 1H, J = 10.0 Hz), 6.68 (s, 1H), 6.36 (d, 1H, J = 9.2 Hz), 5.72 (m, 1H), 5.49 (s, 1H), 4.71 (t, 2H, J = 2.0 Hz), 4.11 (t, 2H), 3.76 (t, 4H), 2.83 (t, 2H), 2.62 (t, 4H), 2.39 (s, 3H), 1.46 (s, 9H). FAB-MS (m/z): 532.1 [M+H]+.
1-(3-tert-Butyl-1-(naphthalen-1-yl)-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)-urea (40o). Yield: 22%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.88–7.90 (m, 2H), 7.48–7.50 (m, 5H), 7.38 (d, 1H), 6.95 (s, 1H), 6.68 (d, 1H, J = 10.0 Hz), 6.48 (s, 1H), 6.24–6.27 (m, 2H), 5.67 (m, 1H), 4.63 (t, 2H, J = 1.6 Hz), 4.07 (t, 2H, J = 5.2 Hz), 3.75 (t, 4H, J = 4.4 Hz), 2.82 (t, 2H), 2.61 (t, 4H), 1.40 (s, 9H). FAB-MS (m/z): 568.1 [M+H]+.
1-(4-tert-Butyl-1-p-tolyl-1H-imidazol-2-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40p). Yield: 27%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 8.03 (d, 1H, J = 9.2 Hz), 7.26–7.30 (m, 5H), 6.78 (d, 1H, J = 10.0 Hz), 6.44 (s, 1H), 6.41 (d, 1H, J = 9.2 Hz), 5.77 (m, 1H), 4.88 (t, 2H), 4.13 (t, 2H, J = 6.8 Hz), 3.76 (t, 4H), 2.84 (t, 2H), 2.62 (t, 4H), 2.42 (s, 3H), 1.34 (s, 9H). FAB-MS (m/z): 532.4 [M+H]+.
1-(3-tert-Butyl-1-p-tolyl-1H-pyrazol-5-yl)-3-(5-(2-(cis-2,6-dimethylmorpholino)ethoxy)-2H-chromen-8-yl)urea (40q). Yield: 28%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.74 (d, 1H, J = 9.2 Hz), 7.33 (d, 2H, J = 8.0 Hz), 7.22 (d, 2H), 7.14 (s, 1H), 6.74 (d, 1H, J = 10.0 Hz), 6.45 (s, 1H), 6.39 (d, 1H, J = 9.2 Hz), 6.35 (s, 1H), 5.72 (m, 1H), 4.71 (t, 2H, J = 1.6 Hz), 4.12 (t, 2H, J = 4.0 Hz), 3.74 (m, 2H), 2.83 (m, 4H), 2.36 (s, 3H), 1.94 (m, 2H), 1.36 (s, 9H), 1.17 (d, 3H), 1.16 (d, 3H). FAB-MS (m/z): 532.4 [M+H]+.
1-(5-(2-(1H-1,2,4-Triazol-1-yl)ethoxy)-2H-chromen-8-yl)-3-(3-tert-butyl-1-p-tolyl-1H-pyrazol-5-yl)-urea (40r). Yield: 16%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 8.10 (s, 1H), 7.86 (s, 1H), 7.83 (d, 1H, J = 8.0 Hz), 7.38 (s, 1H), 7.32 (d, 2H, J = 8.4 Hz), 7.18 (d, 2H), 6.39 (s, 1H), 6.36 (d, 1H), 6.29 (d, 1H, J = 9.2 Hz), 5.47 (m, 1H), 4.55 (t, 2H, J = 4.8 Hz), 4.44 (t, 2H), 4.28 (t, 2H, J = 4.8 Hz), 2.33 (s, 3H), 1.35 (s, 9H). FAB-MS (m/z): 514.2 [M+H]+.






















{kind=link}
{kind=link}
{kind=link}
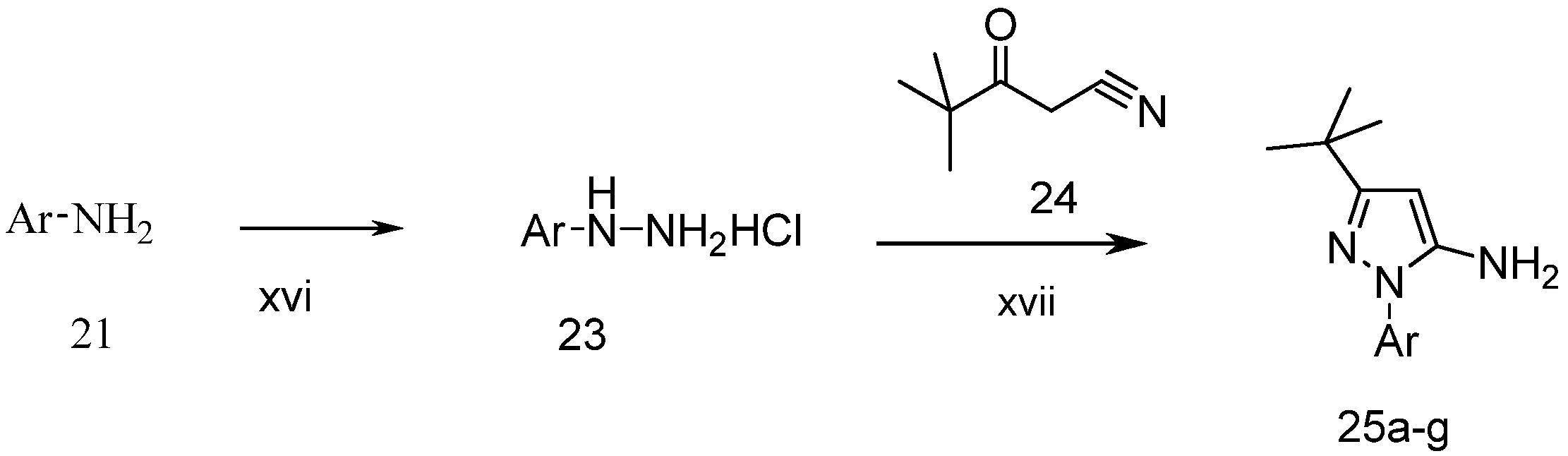{kind=link}
{kind=link}
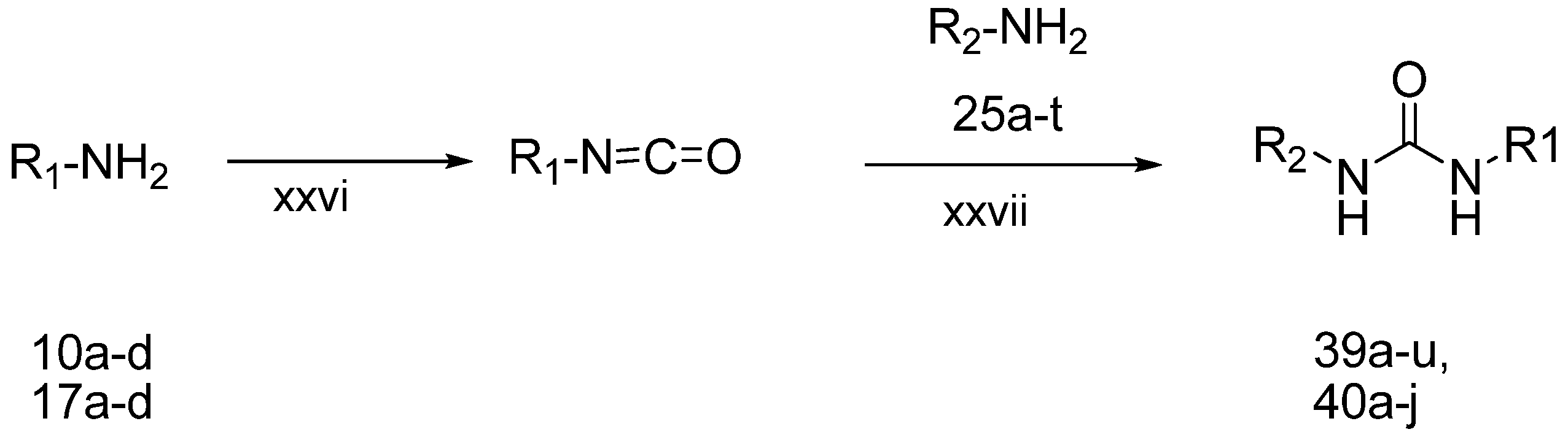{kind=link}














