In Silico Studies of Quinoxaline-2-Carboxamide 1,4-di-N-Oxide Derivatives as Antimycobacterial Agents


Abstract
:1. Introduction
2. Results and Discussion
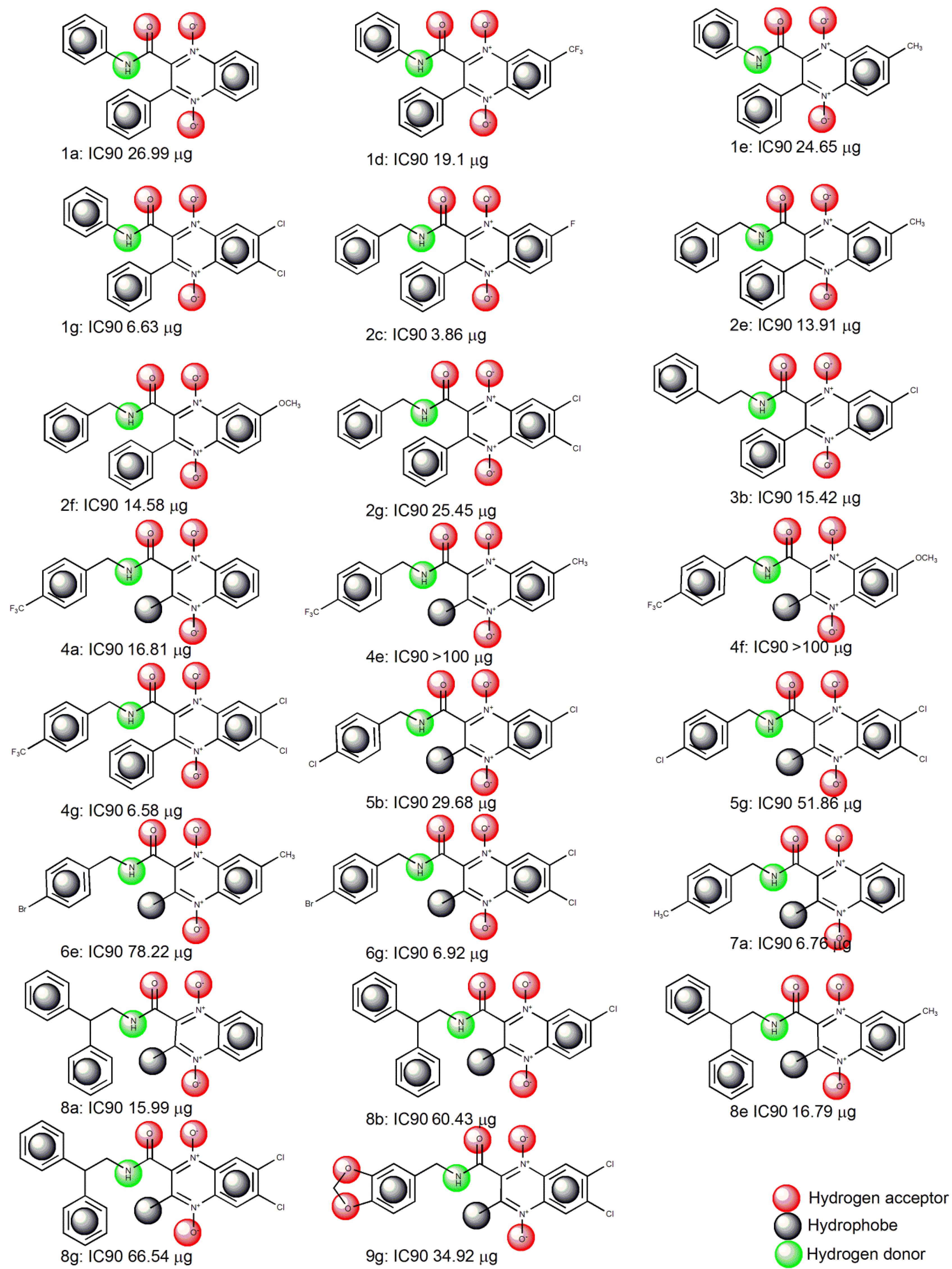
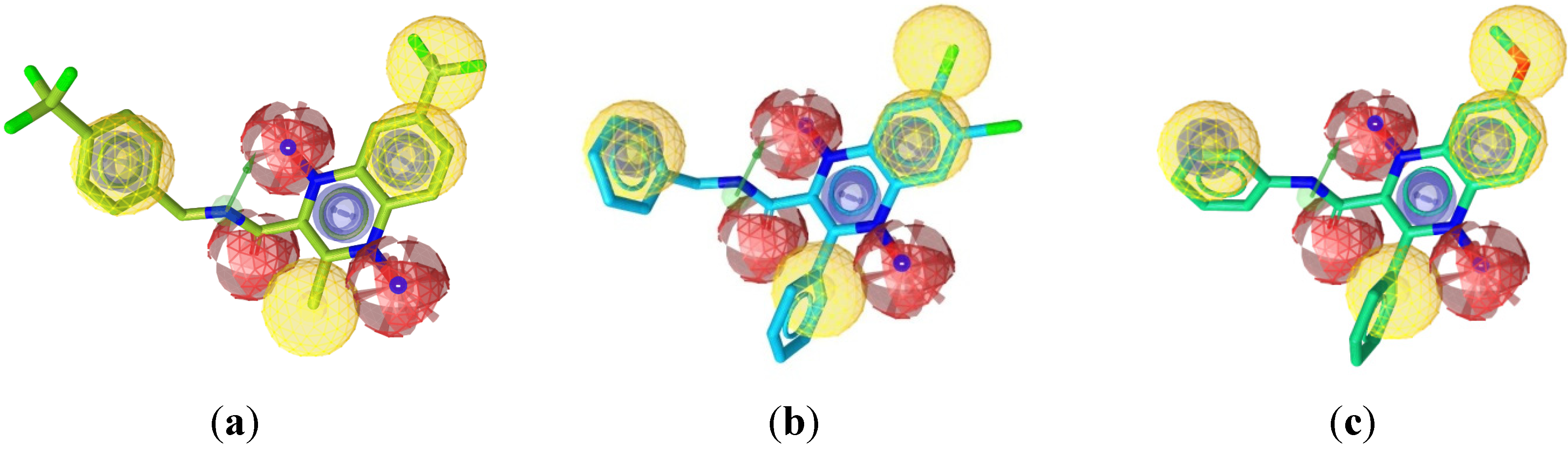
2.1. Pharmacophore Modeling




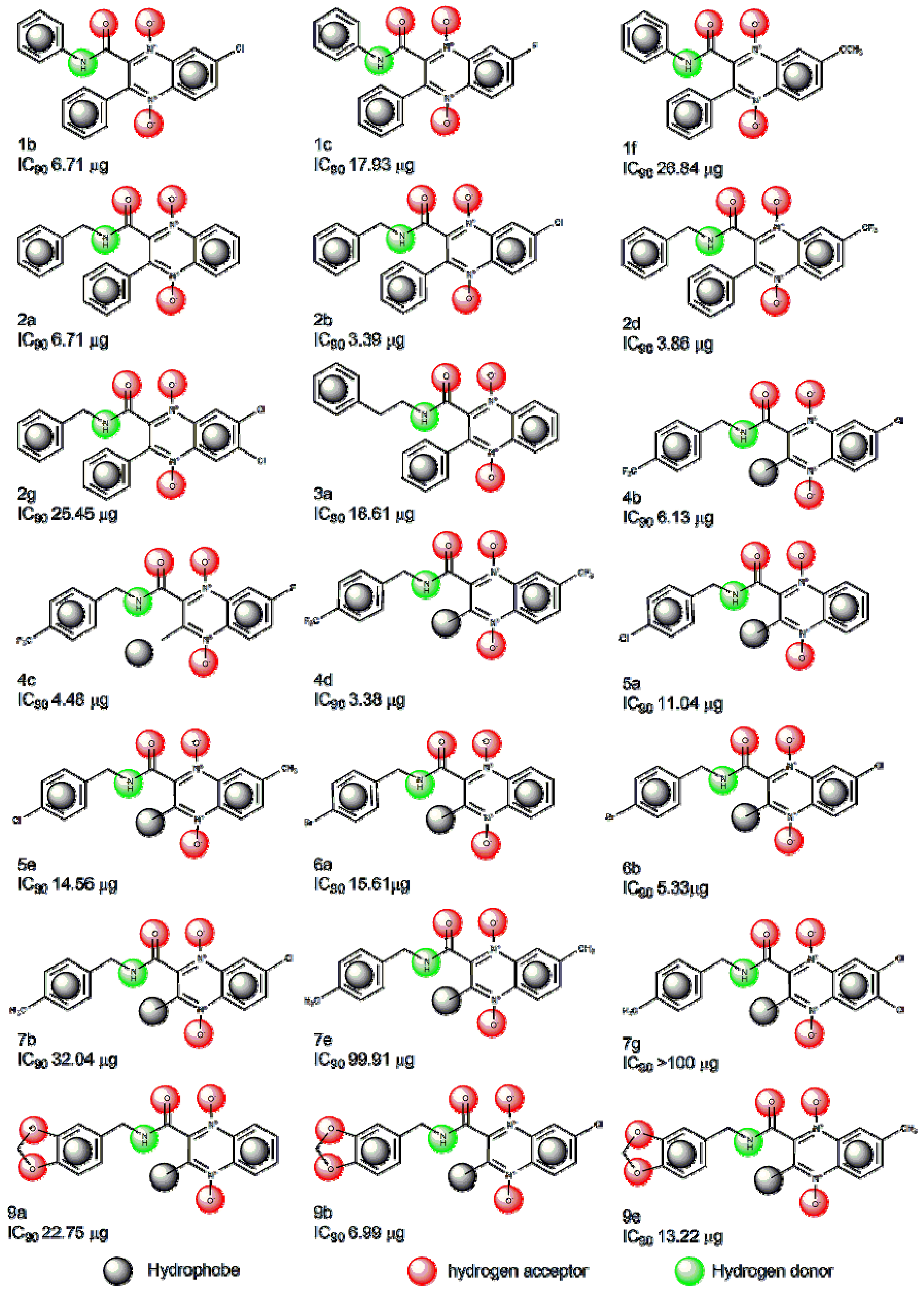{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
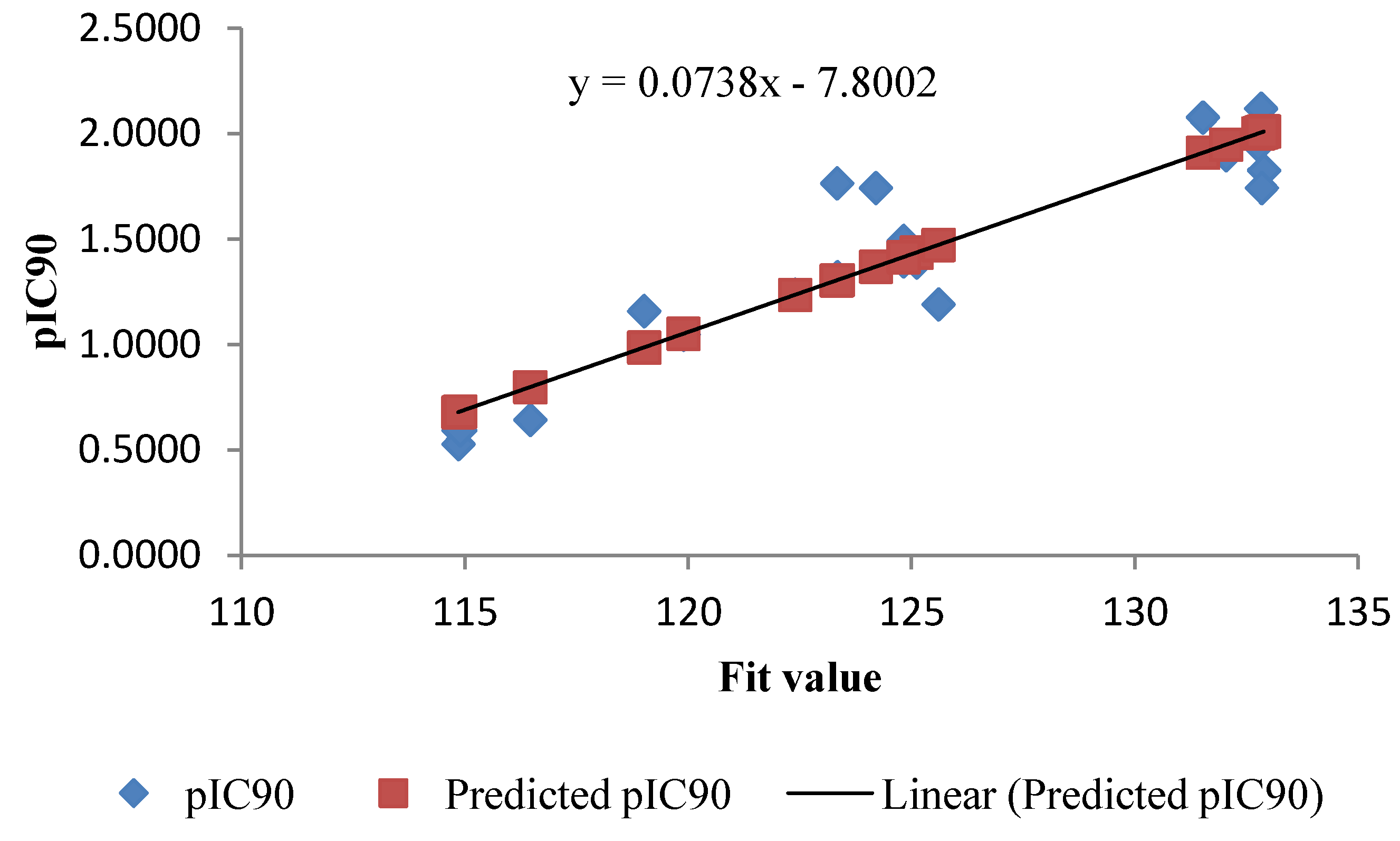
| Compounds | IC90 μM | pIC90 | Fit Value | Predicted pIC90 | Residuals |
|---|---|---|---|---|---|
| 1b | 0.0172 | 1.7655 | 123.33 | 1.3065 | 0.4590 |
| 1c | 0.0478 | 1.3205 | 123.34 | 1.3072 | 0.0133 |
| 1f | 0.0694 | 1.1589 | 119.01 | 0.9875 | 0.1714 |
| 2a | 0.0181 | 1.7427 | 124.2 | 1.3707 | 0.3720 |
| 2b | 0.0084 | 2.0773 | 131.52 | 1.9113 | 0.1660 |
| 2d | 0.2278 | 0.6425 | 116.46 | 0.7992 | –0.1567 |
| 2g | 0.0580 | 1.2368 | 122.39 | 1.2371 | –0.0003 |
| 3a | 0.0483 | 1.3157 | 123.32 | 1.3058 | 0.0099 |
| 4b | 0.0149 | 1.8264 | 132.89 | 2.0125 | –0.1861 |
| 4c | 0.0113 | 1.9453 | 132.76 | 2.0029 | –0.0576 |
| 4d | 0.0076 | 2.1194 | 132.82 | 2.0073 | 0.1121 |
| 4e | 0.2558 | 0.5922 | 114.85 | 0.6803 | –0.0881 |
| 5a | 0.0322 | 1.4923 | 124.82 | 1.4165 | 0.0758 |
| 5e | 0.0408 | 1.3895 | 125.11 | 1.4379 | –0.0484 |
| 6a | 0.0402 | 1.3954 | 124.82 | 1.4165 | –0.0211 |
| 6b | 0.0126 | 1.8986 | 132.04 | 1.9497 | –0.0511 |
| 7b | 0.0897 | 1.0470 | 119.89 | 1.0525 | –0.0055 |
| 7e | 0.2965 | 0.5280 | 114.85 | 0.6803 | –0.1523 |
| 7g | 0.2551 | 0.5933 | 114.89 | 0.6832 | –0.0899 |
| 9a | 0.0644 | 1.1908 | 125.6 | 1.4741 | –0.2833 |
| 9b | 0.0181 | 1.7432 | 132.84 | 2.0088 | –0.2656 |
| 9e | 0.0360 | 1.4434 | 124.81 | 1.4158 | 0.0276 |
| Compounds | IC90 μM | pIC90 | Fit Value | Predicted pIC90 | Residuals |
|---|---|---|---|---|---|
| 1a | 0.0756 | 1.1214 | 122.99 | 1.1506 | –0.0292 |
| 1d | 0.0449 | 1.3473 | 123.31 | 1.176 | 0.1713 |
| 1e | 0.0664 | 1.1775 | 123.34 | 1.1784 | –0.0009 |
| 1g | 0.0156 | 1.8078 | 130.09 | 1.7142 | 0.0936 |
| 2c | 0.0099 | 2.0033 | 131.54 | 1.8293 | 0.174 |
| 2e | 0.0361 | 1.4421 | 122.89 | 1.1427 | 0.2994 |
| 2f | 0.0364 | 1.4393 | 124.24 | 1.2498 | 0.1895 |
| 3b | 0.0368 | 1.4341 | 123.65 | 1.203 | 0.2311 |
| 4a | 0.0446 | 1.3507 | 125.65 | 1.3617 | –0.011 |
| 4f | 0.2457 | 0.6095 | 113.61 | 0.406 | 0.2035 |
| 4g | 0.0148 | 1.8311 | 130.62 | 1.7563 | 0.0748 |
| 5b | 0.0787 | 1.1038 | 125.07 | 1.3157 | –0.2119 |
| 5g | 0.1259 | 0.9 | 122.94 | 1.1466 | –0.2466 |
| 6e | 0.1946 | 0.7109 | 122.94 | 1.1466 | –0.4357 |
| 6g | 0.0151 | 1.8198 | 132.07 | 1.8714 | –0.0516 |
| 7a | 0.0209 | 1.6792 | 130.97 | 1.784 | –0.1048 |
| 8a | 0.0401 | 1.3971 | 124.61 | 1.2792 | 0.1179 |
| 8b | 0.1396 | 0.8552 | 122.09 | 1.0792 | –0.224 |
| 8e | 0.0407 | 1.3908 | 123.83 | 1.2173 | 0.1735 |
| 8g | 0.1422 | 0.8471 | 122.7 | 1.1276 | –0.2805 |
| 9g | 0.0827 | 1.0822 | 123.8 | 1.2149 | –0.1327 |



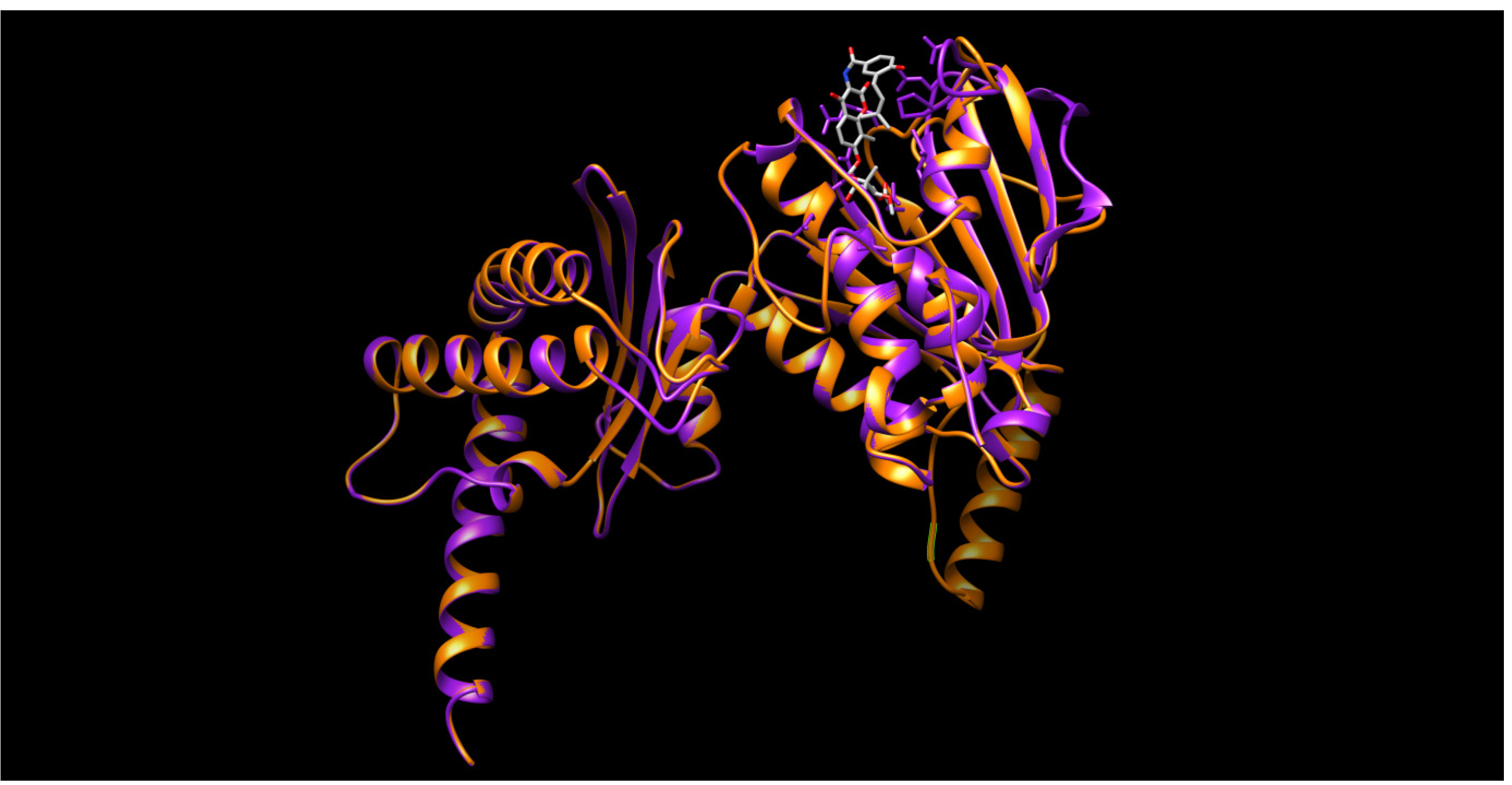
2.2. Homology Modeling

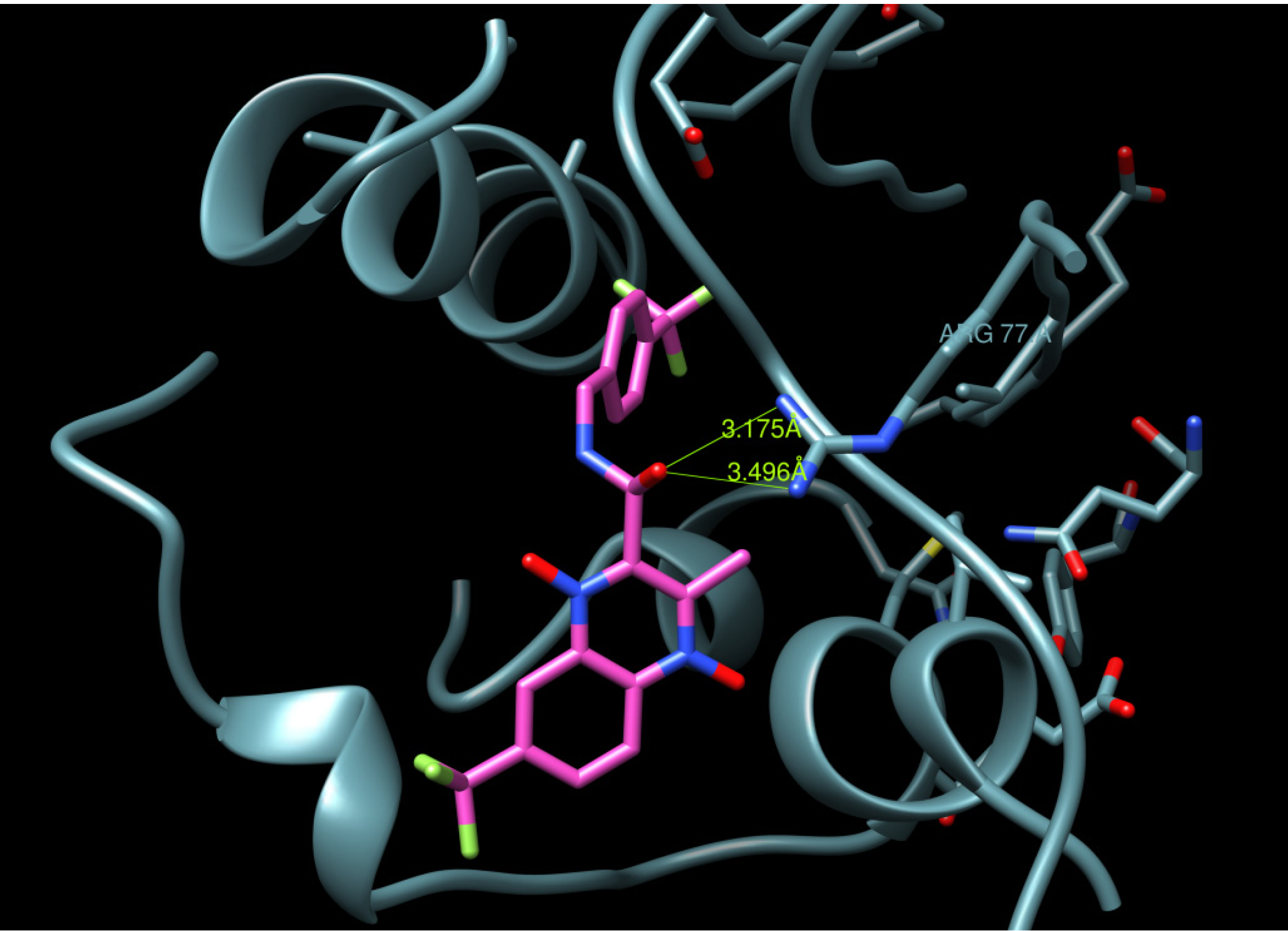
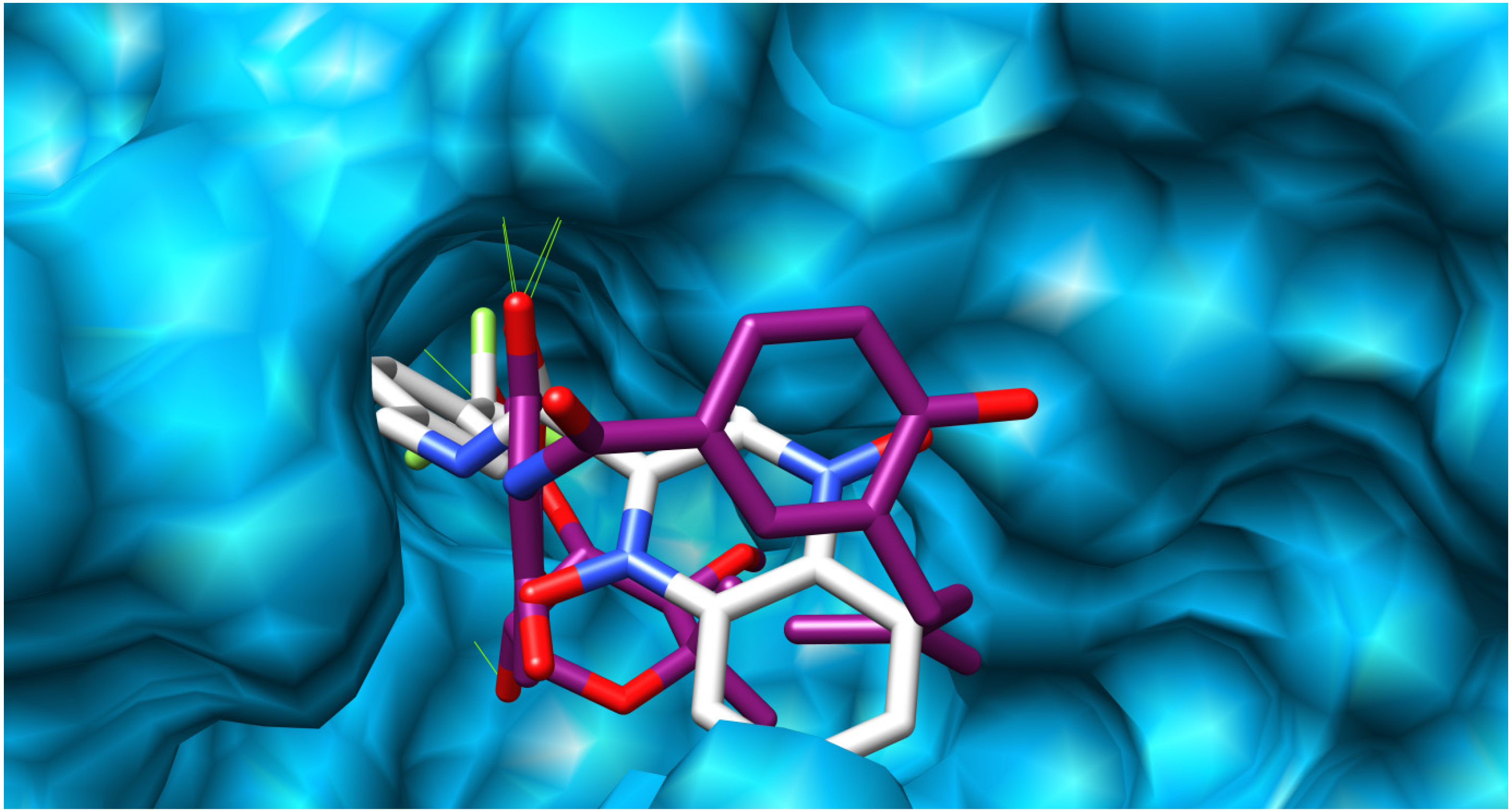
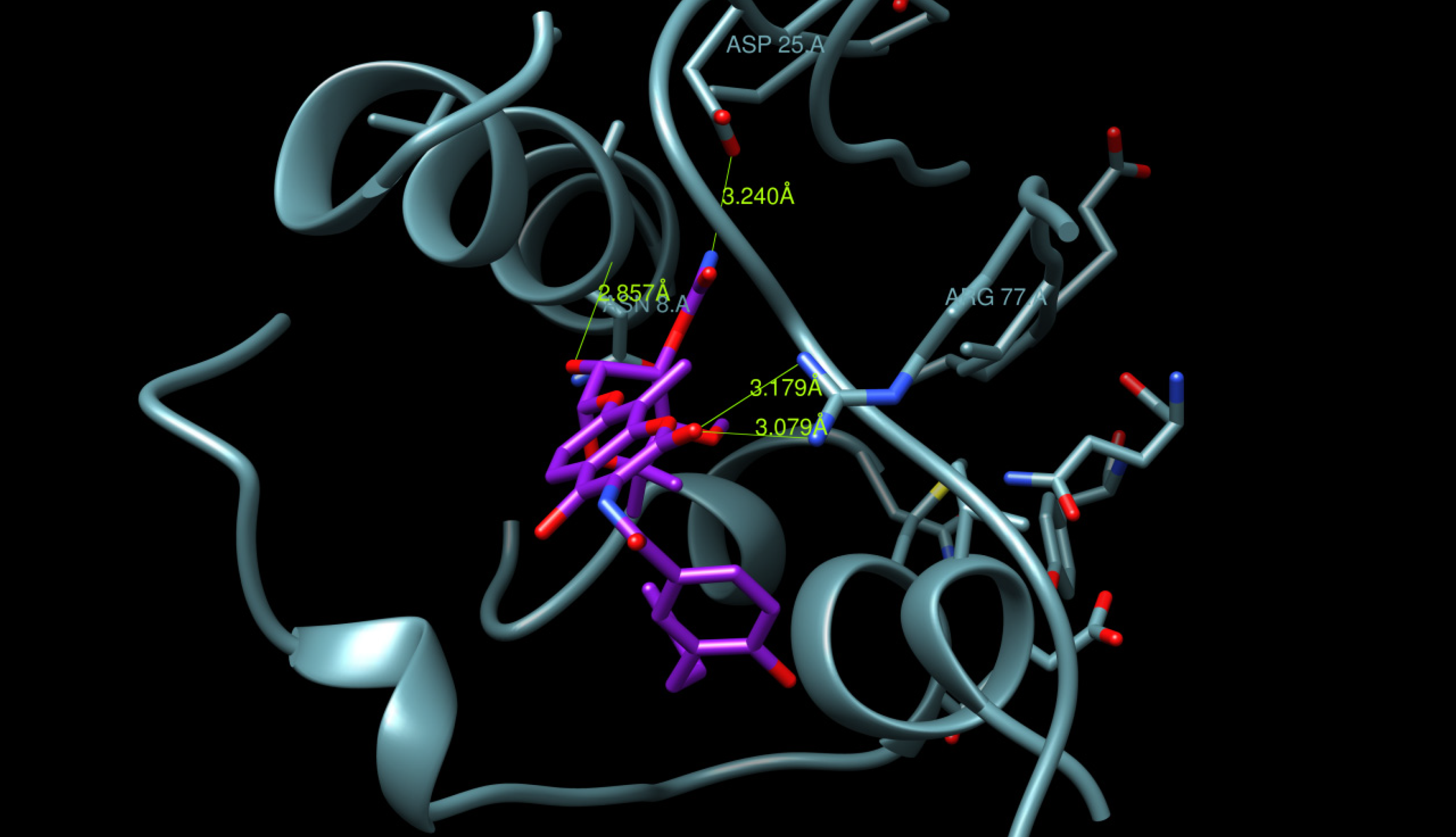
2.3. Docking Procedure




3. Experimental
3.1. General
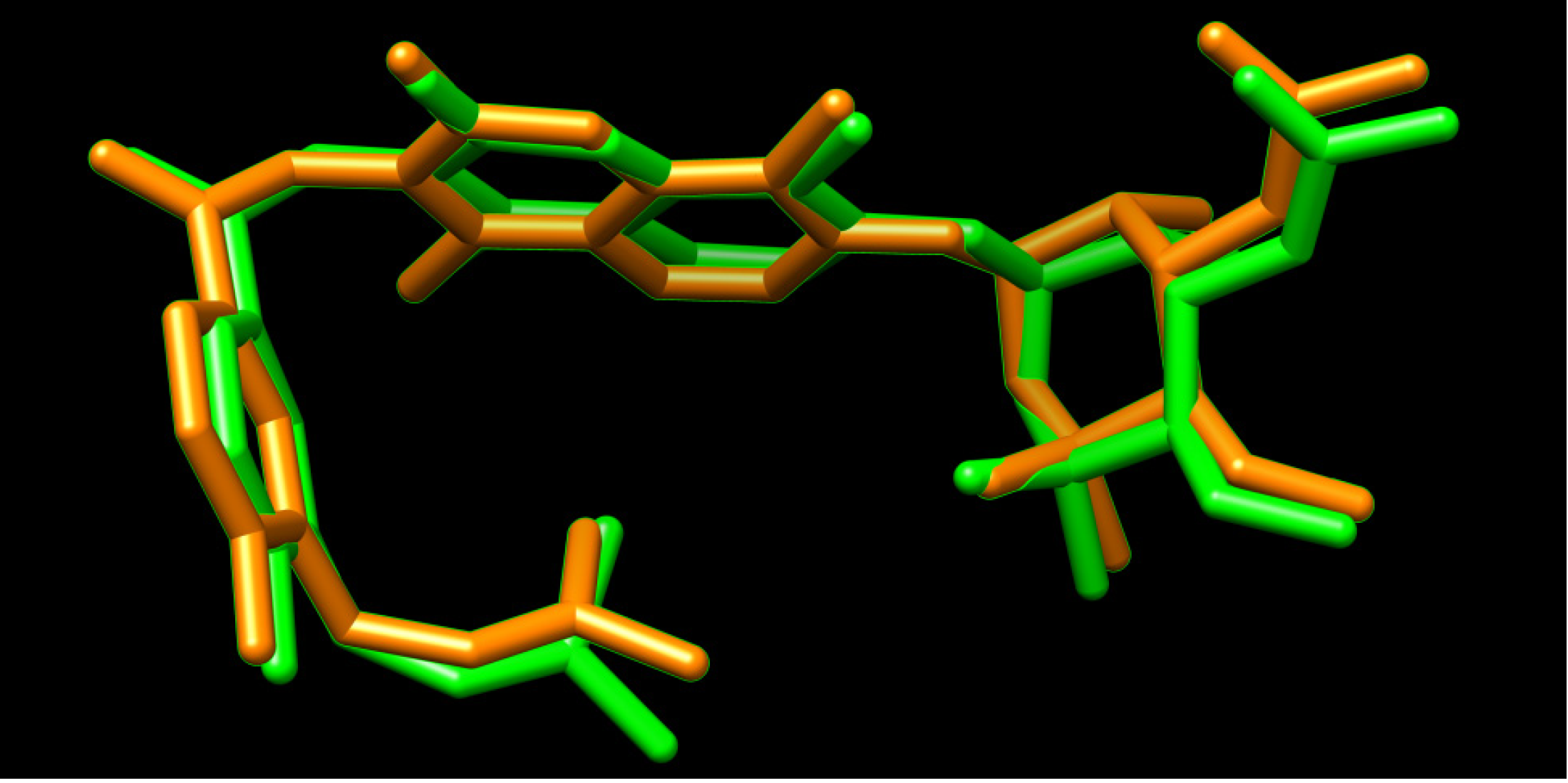
3.2. Ligand Based Pharmacophore Modelling
3.2.1. Pharmacophore Validation
3.2.2. Leave-One-Out Method
3.2.3 Pharmacophore Validation using Test Set
3.3. Homology Modeling
4. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Global Tuberculosis Control WHO Report 2010. Available online: http://reliefweb.int/sites/reliefweb.int/files/resources/F530290AD0279399C12577D8003E9D65-Full_Report.pdf (accessed on 8 March 2010).
- NIAID MDR/XDR TB Research Agenda June 6, 2007. Available online: http://www3.niaid.nih.gov/topics/tuberculosis/ (accessed on 8 March 2010).
- Young, D.B.; Perkins, M.D.; Duncan, K.; Barry, C.E. Confronting the scientific obstacles to global control of tuberculosis. J. Clin. Invest. 2008, 118, 1255–1265. [Google Scholar] [CrossRef]
- Junior, I.N.; Lourenço, M.C.S.; Henriques, M.G.M.O.; Ferreira, B.; Vasconcelos, T.R.A.; Peralta, M.A.; de Oliveira, P.S.M.; Wardell, S.M.S.V.; de Souza, M.V.N. Synthesis and anti-Mycobacterial activity of N'-[(E)-(disubstituted-phenyl)methylidene]isoni-cotino-hydrazide derivatives. Lett. Drug Des. Dis. 2005, 2, 563–566. [Google Scholar] [CrossRef]
- Vicente, E.; Villar, R.; Solano, B.; Burguete, A.; Ancizu, S.; Pérez-Silanes, S.; Aldana, I.; Monge, A. Derivados de 1,4-di-N-óxido de quinoxalina y enfermedades olvidadas. An. R. Acad. Nac. Farm. 2007, 73, 927–945. [Google Scholar]
- Aguirre, G.; Cerecetto, H.; di Maio, R.; Gonzalez, M.; Alfaro, M.E.M.; Jaso, A.; Zarranz, B.; Ortega, M.A.; Aldana, I.; Monge-Vega, A. Quinoxaline N,N'-dioxide derivatives and related compounds as growth inhibitors of Trypanosoma cruzi. Structure-activity relationships. Bioorg. Med. Chem. Lett. 2004, 14, 3835–3839. [Google Scholar] [CrossRef]
- Urquiola, C.; Vieites, M.; Aguirre, G.; Marin, A.; Solano, B.; Arrambide, G.; Noblia, P.; Lavaggi, M.L.; Torre, M.H.; Gonzalez, M.; et al. Improving anti-trypanosomal activity of 3-aminoquinoxaline-2-carbonitrile N1,N4-dioxide derivatives by complexation with vanadium. Bioorg. Med. Chem. 2006, 14, 5503–5509. [Google Scholar] [CrossRef]
- Carta, A.; Loriga, M.; Paglietti, G.; Mattana, A.; Fiori, P.L.; Mollicotti, P.; Sechi, L.; Zanetti, S. Synthesis, anti-mycobacterial, anti-trichomonas and anti-candida in vitro activities of 2-substituted-6,7-difluoro-3-methylquinoxaline 1,4-dioxides. Eur. J. Med. Chem. 2004, 39, 195–203. [Google Scholar] [CrossRef]
- Ganley, B.; Chowdhury, G.; Bhansali, J.; Daniels, J.S.; Gates, K.S. Redox-activated, hypoxia-selective DNA cleavage by quinoxaline 1,4-di-N-oxide. Bioorg. Med. Chem. 2001, 9, 2395–2401. [Google Scholar] [CrossRef]
- Ortega, M.A.; Montoya, M.E.; Jaso, A.; Zarranz, B.; Tirapu, I.; Aldana, I.; Monge, A. Anti-mycobacterial activity of new quinoxaline-2-carbonitrile and quinoxaline-2-carbonitrile 1,4-di-N-oxide derivatives. Pharmazie 2001, 56, 205–207. [Google Scholar]
- Ortega, M.A.; Sainz, Y.; Montoya, M.E.; Jaso, A.; Zarranz, B.; Aldana, I.; Monge, A. Anti-Mycobacterium tuberculosis agents derived from quinoxaline-2-carbonitrile and quinoxaline-2-carbonitrile 1,4-di-N-oxide. Arzneim.-Forsch. 2002, 52, 113–119. [Google Scholar]
- Jaso, A.; Zarranz, B.; Aldana, I.; Monge, A. Synthesis of new 2-acetyl and 2-benzoyl quinoxaline 1,4-di-N-oxide derivatives as anti-Mycobacterium tuberculosis agents. Eur. J. Med. Chem. 2003, 38, 791–800. [Google Scholar]
- Jaso, A.; Zarranz, B.; Aldana, I.; Monge, A. Synthesis of new quinoxaline-2-carboxylate 1,4-dioxide derivatives as anti-Mycobacterium tuberculosis agents. J. Med. Chem. 2005, 48, 2019–2025. [Google Scholar] [CrossRef]
- Zarranz, B.; Jaso, A.; Aldana, I.; Monge, A. Synthesis and antimycobacterial activity of new quinoxaline-2-carboxamide 1,4-di-N-oxide derivatives. Bioorg. Med. Chem. 2003, 11, 2149–2156. [Google Scholar] [CrossRef]
- Ancizu, S.; Moreno, E.; Solano, B.; Villar, R.; Burguete, A.; Torres, E.; Pérez-Silanes, S.; Aldana, I.; Monge, A. New 3-methylquinoxaline-2-carboxamide 1,4-di-N-oxide derivatives as anti-Mycobacterium tuberculosis agents. Bioorg. Med. Chem. 2010, 18, 2713–2719. [Google Scholar] [CrossRef]
- Moreno, E.; Ancizu, S.; Pérez-Silanes, S.; Torres, E.; Aldana, I.; Monge, A. Synthesis and antimycobacterial activity of new quinoxaline-2-carboxamide 1,4-di-N-oxide derivatives. Eur. J. Med. Chem. 2010, 45, 4418–4426. [Google Scholar] [CrossRef]
- Ancizu, S.; Moreno, E.; Torres, E.; Burguete, A.; Pérez-Silanes, S.; Benítez, D.; Villar, R.; Solano, B.; Marín, A.; Aldana, I.; et al. Heterocyclic-2-carboxylic acid (3-cyano-1,4-di-N-oxidequinoxalin-2-yl)amide derivatives as hits for the development of neglected disease drugs. Molecules 2009, 14, 2256–2272. [Google Scholar] [CrossRef]
- Torres, E.; Moreno, E.; Ancizu, S.; Barea, C.; Galiano, S.; Aldana, I.; Monge, A.; Pérez-Silanes, S. New 1,4-di-N-oxide-quinoxaline-2-ylmethylene isonicotinic acid hydrazide derivatives as anti-Mycobacterium tuberculosis agents. Bioorg. Med. Chem. Lett. 2011, 21, 3699–3703. [Google Scholar] [CrossRef]
- Vicente, E.; Duchowicz, P.R.; Benítez, D.; Castro, E.A.; Cerecetto, H.; González, M.; Monge, A. Anti-T. cruzi activities and QSAR studies of 3-arylquinoxaline-2-carbonitrile di-N-oxides. Bioorg. Med. Chem. Lett. 2010, 20, 4831–4835. [Google Scholar]
- Vicente, E.; Duchowicz, P.R.; Castro, E. A.; Monge, A. QSAR analysis for quinoxaline-2-carboxylate 1,4-di-N-oxides as anti-mycobacterial agents. J. Mol. Graph. Model. 2009, 28, 28–36. [Google Scholar]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Lamour, V.; Hoermann, L.; Jeltsch, J.M.; Oudet, P.; Moras, D. An open conformation of the Thermus thermophilus gyrase B ATP-binding domain. J. Biol. Chem. 2002, 277, 18947–18953. [Google Scholar]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.T.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. Dock 6: Combining technique to model RNA-small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef]
- Aboul-Fadl, T.; Radwan, A.A.; Attia, M.I.; Al-Dhfyan, A.; Abdel-Aziz, H.A. Schiff bases of indoline-2,3-dione (isatin) with potential antiproliferative activity. Chem. Cent. J. 2012, 6, 49–59. [Google Scholar] [CrossRef]
- Camus, J.C.; Pryor, M.J.; Medigue, C. Re-annotation of the genome sequence of Mycobacterium tuberculosis H37Rv. Microbiology 2002, 148, 2967–2973. [Google Scholar]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Radwan, A.A.; Abdel-Mageed, W.M. In Silico Studies of Quinoxaline-2-Carboxamide 1,4-di-N-Oxide Derivatives as Antimycobacterial Agents. Molecules 2014, 19, 2247-2260. https://doi.org/10.3390/molecules19022247
Radwan AA, Abdel-Mageed WM. In Silico Studies of Quinoxaline-2-Carboxamide 1,4-di-N-Oxide Derivatives as Antimycobacterial Agents. Molecules. 2014; 19(2):2247-2260. https://doi.org/10.3390/molecules19022247
Chicago/Turabian StyleRadwan, Awwad A., and Wael M. Abdel-Mageed. 2014. "In Silico Studies of Quinoxaline-2-Carboxamide 1,4-di-N-Oxide Derivatives as Antimycobacterial Agents" Molecules 19, no. 2: 2247-2260. https://doi.org/10.3390/molecules19022247