Five New Alkaloids from the Stem Bark of Daphniphyllum macropodum

Abstract
:1. Introduction

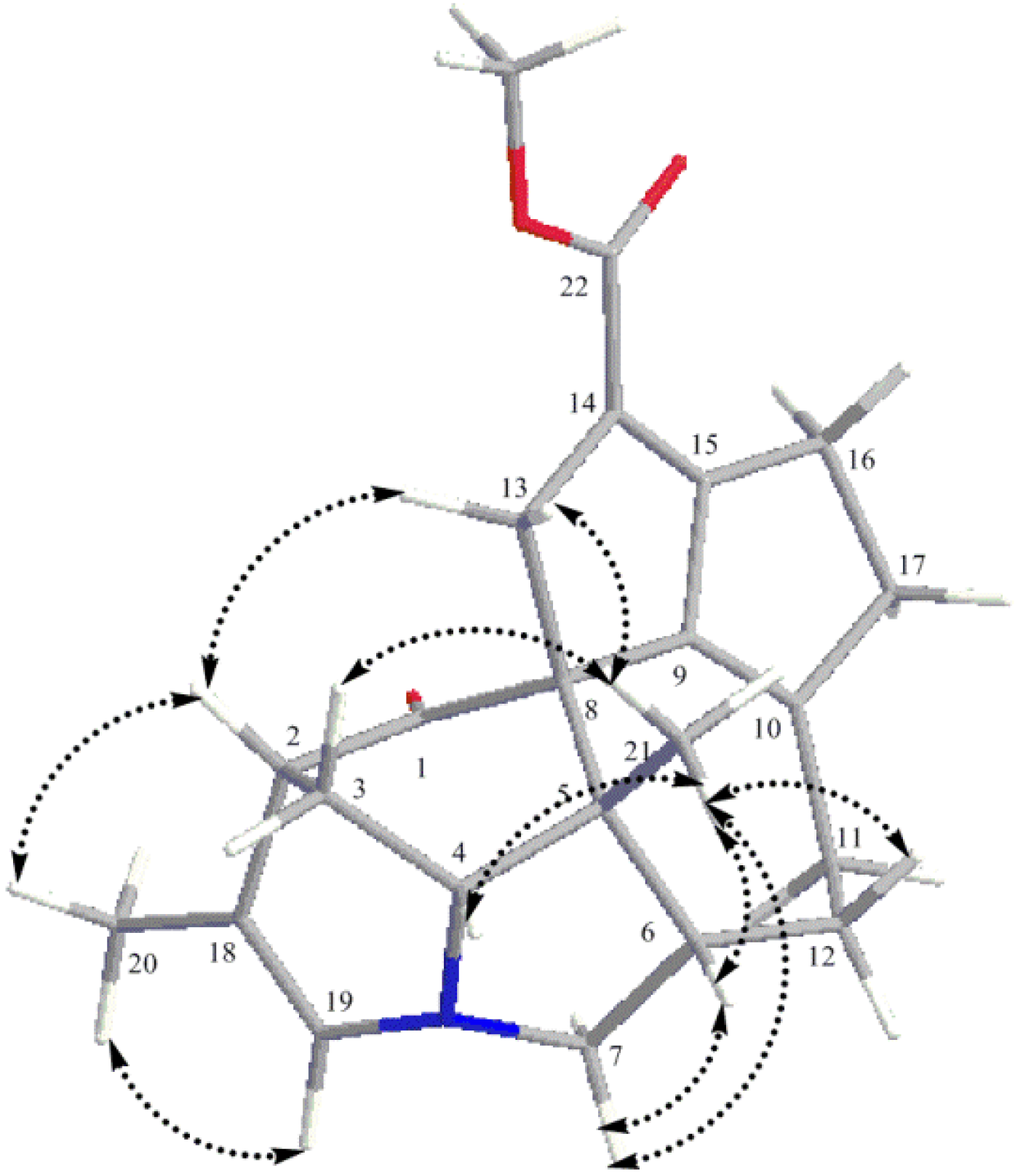
2. Results and Discussion

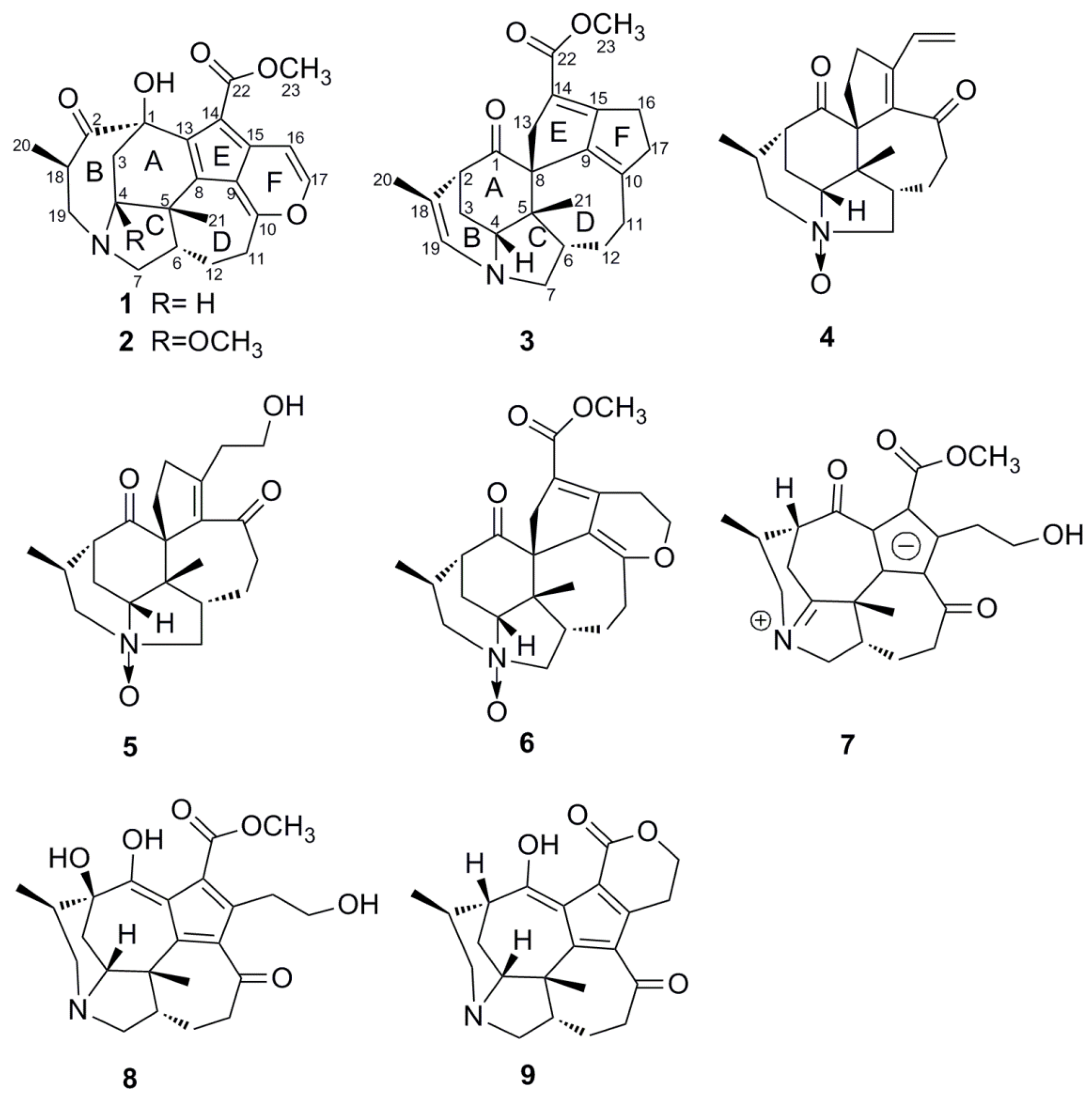{kind=link}
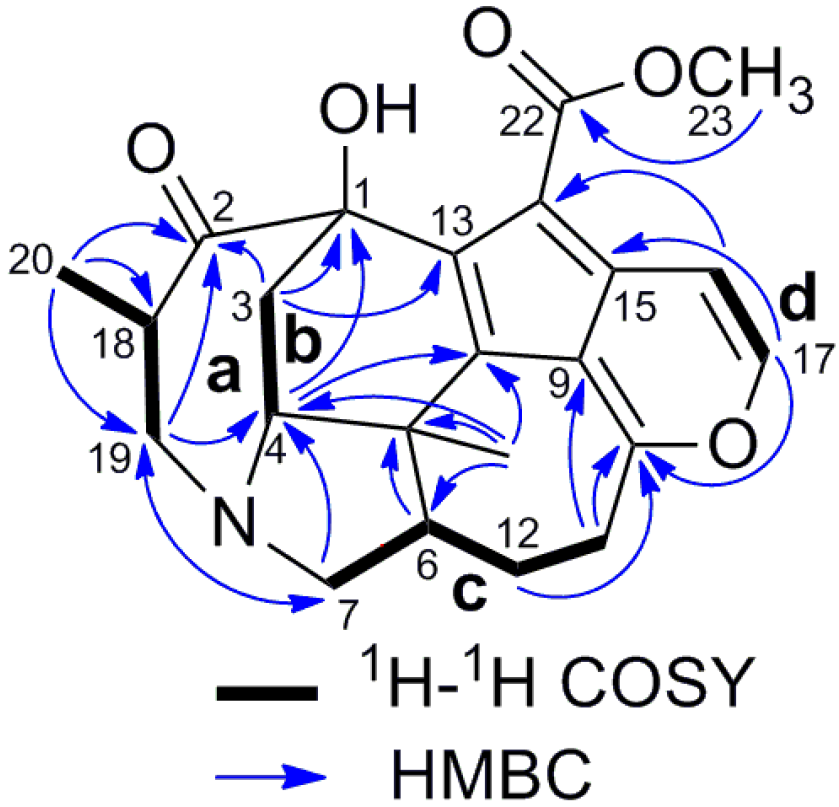{kind=link}
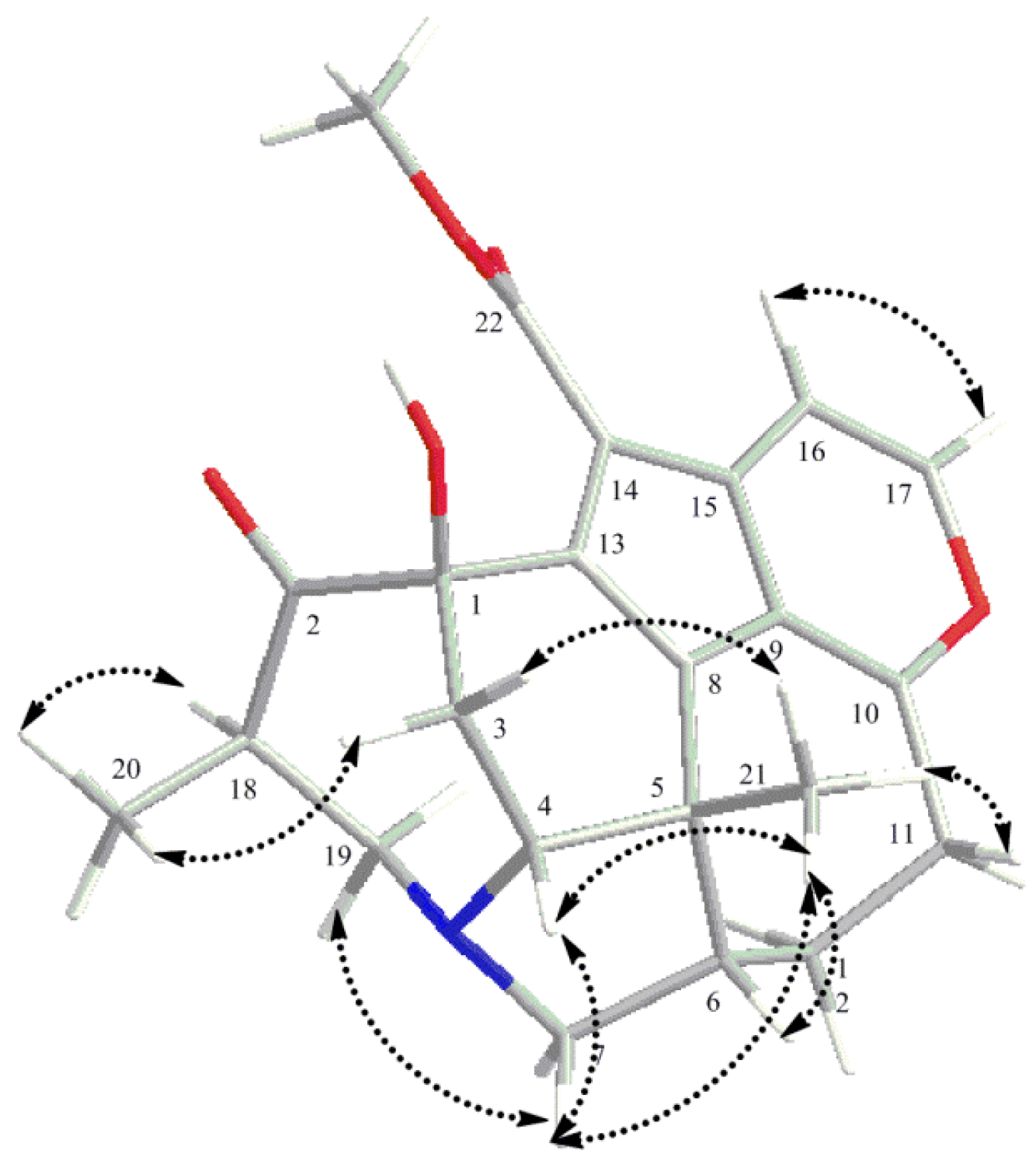{kind=link}
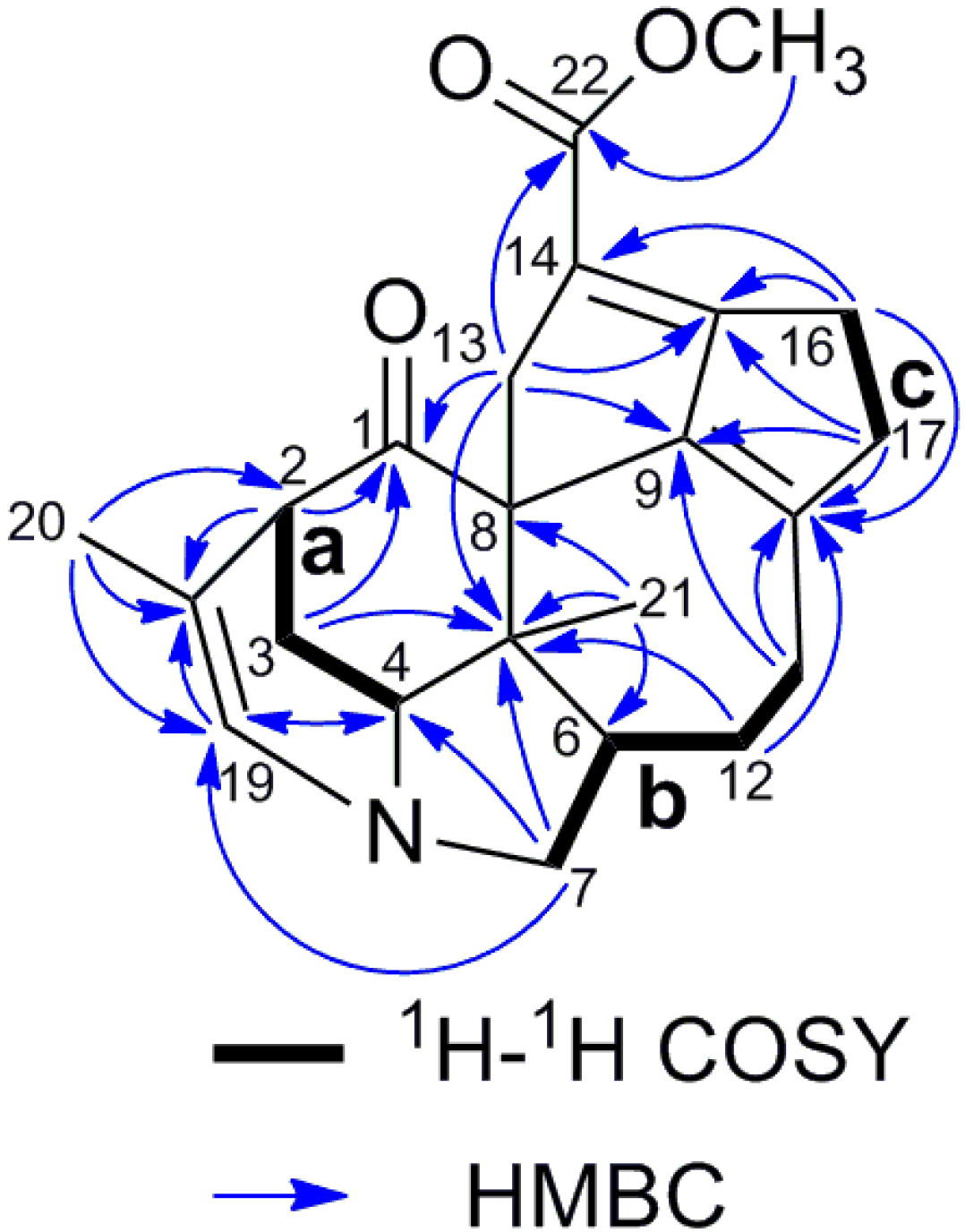{kind=link}
{kind=link}
| C | 1 a | 2 a | 3 a | 4 a | 5 b | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δC | δH | δC | δH | δC | δH | δC | δH | δC | δH | |
| 1 | 77.8 | - | 78.9 | - | 209.2 | - | 217.6 | - | 216.4 | - |
| 2 | 213.1 | - | 212.4 | - | 44.4 | 2.93 (t, 3.1) | 43.3 | 2.38 (brd, 5.0) | 41.5 | 2.28–2.31 (m) |
| 3a 3b | 31.4 | 2.26 (d, 3.4) 2.26 (d, 3.4) | 33.9 | 2.18 (d, 14.8) 2.35 (d, 14.8) | 24.4 | 1.97 (dt, 14.1, 2.7) 2.26 (dt, 14.2, 3.1) | 34.6 | 1.86–1.92 (m) 2.76–2.78 (m) | 18.6 | 2.38–2.40 (m) 2.64–2.66 (m) |
| 4 | 67.9 | 3.50 (t, 3.3) | 98.0 | - | 64.6 | 3.07–3.09 (m) | 90.4 | 4.00 (t, 2.2) | 87.9 | 3.93 (br s) |
| 5 | 46.8 | - | 53.3 | - | 55.9 | - | 54.7 | - | 52.5 | - |
| 6 | 46.7 | 2.56–2.58 (m) | 42.3 | 2.85–2.88 (m) | 58.0 | 1.78–1.85 (m) | 47.2 | 3.01–3.07 c | 45.2 | 2.89 (t, 7.6) |
| 7a 7b | 60.1 | 2.37 (t, 9.6) 3.06–3.08 (m) | 59.4 | 2.31 (dd, 8.2, 2.8) 3.13–3.16 (m) | 61.9 | 2.92 (d, 9.4) 3.28–3.31 (m) | 69.6 | 3.12 (t, 6.3) 3.51 (t, 13.2) | 67.4 | 3.04–3.09 (m) 3.26–3.32 (m) |
| 8 | 126.7 | - | 128.5 | - | 57.3 | - | 72.3 | - | 69.7 | - |
| 9 | 108.8 | - | 108.2 | - | 150.7 | - | 139.7 | - | 136.9 | - |
| 10 | 168.4 | - | 168.1 | - | 151.7 | - | 206.1 | - | 202.8 | - |
| 11a 11b | 31.0 | 2.98–3.01 (m) 3.67–3.73 (m) | 31.1 | 2.94–2.98 (m) 3.65–3.69 (m) | 34.5 | 2.06–2.14 (m) 2.36 (d, 18.5) | 37.2 | 2.32–2.35 (m) 2.32–2.35 (m) | 36.0 | 2.12–2.15 (m) 2.12–2.15 (m) |
| 12a 12b | 28.2 | 1.69–1.73 (m) 2.57–2.59 (m) | 27.6 | 1.67–1.75 (m) 2.54–2.58 (m) | 32.1 | 1.32–1.37 (m) 1.78–1.85 (m) | 19.4 | 1.86–1.92 (m) 2.01–2.08 (m) | 17.8 | 1.71–1.74 (m) 1.91–1.98 (m) |
| 13a 13b | 142.0 | - - | 141.5 | - - | 47.6 | 2.81 (d, 16.5) 3.40 (d, 17.7) | 20.2 | 2.44–2.50 (m) 2.78–2.82 (m) | 32.9 | 1.68–1.71 (m) 2.62–2.65 (m) |
| 14a 14b | 123.2 | - - | 123.3 | - - | 115.9 | - - | 33.2 | 2.64 (t, 8.7) 2.74–2.77 (m) | 35.9 | 2.32–2.37 (m) 2.50–2.52 (m) |
| 15 | 134.4 | - | 134.7 | - | 173.5 | - | 154.6 | - | 156.4 | - |
| 16a 16b | 111.8 | 7.70 (d, 5.3) - | 117.7 | 7.69 (d, 5.3) - | 26.8 | 2.74–2.78 (m) 2.74–2.74 (m) | 132.5 | 6.89 (dd, 10.8, 6.7) - | 33.3 | 2.18–2.23 (m) 2.66–2.70 (m) |
| 17a 17b | 146.1 | 7.93 (d, 5.3) - | 145.9 | 7.92 (d, 5.3) - | 41.7 | 2.78–2.80 (m) 2.85–2.87 (m) | 122.2 | 5.42 (d, 10.8) 5.52 (d, 17.5) | 59.1 | 3.43–3.45 (m) 3.50–3.53 (m) |
| 18 | 36.9 | 2.66–2.70 (m) | 39.1 | 2.49–2.53(m) | 112.8 | - | 33.0 | 2.57–2.62(m) | 31.0 | 2.43–2.47 (m) |
| 19a 19b | 53.1 | 2.61–2.65 (m) 3.02–3.06 (m) | 53.8 | 2.91 (dd, 15.6, 2.8) 3.17 (dd, 15.2, 2.3) | 135.8 | 5.77(s) - | 68.2 | 3.01–3.07 c 3.63 (dd, 13.3, 7.1) | 66.1 | 2.99–3.04 (m) 3.55–3.57 (m) |
| 20 | 14.0 | 0.81 (3H, d, 6.7) | 13.3 | 0.83 (3H, d, 6.7) | 19.9 | 1.70 (3H, s) | 19.6 | 1.14 (3H, d, 6.7) | 18.9 | 1.03 (3H, d, 6.7) |
| 21 | 28.8 | 1.48 (3H, s) | 25.6 | 1.46 (3H, s) | 27.2 | 1.20 (3H, s) | 23.2 | 1.50 (3H, s) | 22.1 | 1.39 (3H, s) |
| 22 | 169.0 | - | 169.0 | - | 168.1 | - | - | - | - | - |
| 23 | 51.9 | 3.83 (3H, s) | 51.8 | 3.82 (3H, s) | 51.8 | 3.70 (3H, s) | - | - | - | - |
| 4-OMe | - | - | 50.0 | 3.35 (3H, s) | - | - | - | - | - | - |




| Compounds a | Cytotoxic activity (IC50, µM) | |||
|---|---|---|---|---|
| P-388 | A-549 | SGC-7901 | HL-60 | |
| 1 | 5.7 | >50 | 22.4 | >50 |
| 2 | 6.5 | >50 | 25.6 | >50 |
| 8 | 10.3 | >50 | >50 | >50 |
| 9 | 13.8 | >50 | >50 | >50 |
| Cisplatin b | 0.3 | 0.9 | 3.2 | 1.1 |
3. Experimental
3.1. General Information
3.2. Plant Material
3.3. Extraction and Isolation
 −40.3 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 274 (1.36), 221 (1.26) nm; IR (KBr) νmax 3423, 2915, 1690, 1643, 1616, 1522, 1454, 1375, 1127, 984 cm−1; 1H-NMR and 13C-NMR, see Table 1; positive ESIMS m/z 418 [M+Na]+; positive HRESIMS [M+Na]+ m/z 418.1632 (calcd for C23H25NO5Na, 418.1630). −65.5 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 281 (1.14), 204 (5.00) nm; IR (KBr) νmax 3425, 2920, 1686, 1643, 1616, 1525, 1456, 1385, 1126, 991 cm−1; 1H-NMR and 13C-NMR, see Table 1; positive ESIMS m/z 448 [M+Na]+; positive HRESIMS [M+Na]+ m/z 448.1738 (calcd for C24H27NO6Na, 448.1736). −16.8 (c 0.08, MeOH); UV (MeOH) λmax (log ε) 280 (4.96) nm; IR (KBr) νmax 2925, 1709, 1685, 1615, 1440, 1378, 1096 cm−1; 1H-NMR and 13C-NMR, see Table 1; positive ESIMS m/z 388 [M+Na]+; positive HRESIMS [M+Na]+ m/z 388.1890 (calcd for C23H27NO3Na, 388.1889). −44.7 (c 0.15, MeOH); UV (MeOH) λmax (log ε) 270.5 (4.23), 204.5 (4.88) nm; IR (KBr) νmax 2920, 1745, 1695, 1575, 1445, 1380 cm−1; 1H-NMR and 13C-NMR, see Table 1; positive ESIMS m/z 342 [M+H]+; positive HRESIMS [M+H]+ m/z 342.2068 (calcd for C21H28NO3, 342.2069). −81.2 (c 0.13, DMSO); UV (MeOH) λmax (log ε) 247.5 (4.54) nm; IR (KBr) νmax 3425, 2920, 1701, 1675, 1611, 1438, 1380, 1226, 995, 565 cm−1; 1H-NMR and 13C-NMR, see Table 1; positive ESIMS m/z 360 [M+H]+; positive HRESIMS m/z [M+H]+ 360.2172 (calcd for C21H30NO4, 360.2175).
−40.3 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 274 (1.36), 221 (1.26) nm; IR (KBr) νmax 3423, 2915, 1690, 1643, 1616, 1522, 1454, 1375, 1127, 984 cm−1; 1H-NMR and 13C-NMR, see Table 1; positive ESIMS m/z 418 [M+Na]+; positive HRESIMS [M+Na]+ m/z 418.1632 (calcd for C23H25NO5Na, 418.1630). −65.5 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 281 (1.14), 204 (5.00) nm; IR (KBr) νmax 3425, 2920, 1686, 1643, 1616, 1525, 1456, 1385, 1126, 991 cm−1; 1H-NMR and 13C-NMR, see Table 1; positive ESIMS m/z 448 [M+Na]+; positive HRESIMS [M+Na]+ m/z 448.1738 (calcd for C24H27NO6Na, 448.1736). −16.8 (c 0.08, MeOH); UV (MeOH) λmax (log ε) 280 (4.96) nm; IR (KBr) νmax 2925, 1709, 1685, 1615, 1440, 1378, 1096 cm−1; 1H-NMR and 13C-NMR, see Table 1; positive ESIMS m/z 388 [M+Na]+; positive HRESIMS [M+Na]+ m/z 388.1890 (calcd for C23H27NO3Na, 388.1889). −44.7 (c 0.15, MeOH); UV (MeOH) λmax (log ε) 270.5 (4.23), 204.5 (4.88) nm; IR (KBr) νmax 2920, 1745, 1695, 1575, 1445, 1380 cm−1; 1H-NMR and 13C-NMR, see Table 1; positive ESIMS m/z 342 [M+H]+; positive HRESIMS [M+H]+ m/z 342.2068 (calcd for C21H28NO3, 342.2069). −81.2 (c 0.13, DMSO); UV (MeOH) λmax (log ε) 247.5 (4.54) nm; IR (KBr) νmax 3425, 2920, 1701, 1675, 1611, 1438, 1380, 1226, 995, 565 cm−1; 1H-NMR and 13C-NMR, see Table 1; positive ESIMS m/z 360 [M+H]+; positive HRESIMS m/z [M+H]+ 360.2172 (calcd for C21H30NO4, 360.2175).3.4. Assays for In Vitro Antitumor Activity
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflictts of Interest
References
- Wu, H.F.; Zhang, X.P.; Ding, L.S.; Chen, S.L.; Yang, J.S.; Xu, X.D. Daphniphyllum alkaloids: Recent findings on chemistry and pharmacology. Planta Med. 2013, 79, 1589–1598. [Google Scholar] [CrossRef]
- Kobayashi, J.; Kubota, T. The Daphniphyllum alkaloids. Nat. Prod. Rep. 2009, 26, 936–962. [Google Scholar] [CrossRef]
- Wallence, G.A.; Heathcock, C.H. Further studies of the Daphinphyllum alkaloid polycyclization cascade. J. Org. Chem. 2011, 66, 450–454. [Google Scholar]
- Zhang, M.; Min, T.L. Frola Republicae Popularis Sinicae (Zhongguo Zhiwu Zhi); Science Press: Beijing, China, 1980; Volume 45, pp. 3–4. [Google Scholar]
- Matsuno, Y.; Okamoto, M.; Hirasawa, Y.; Kawahara, N.; Goda, Y.; Shiro, M.; Morita, H. Pordamacrines A and B, alkaloids from Daphniphyllum macropodum. J. Nat. Prod. 2007, 70, 1516–1518. [Google Scholar] [CrossRef]
- Kong, N.C.; He, H.P.; Wang, Y.H.; Mu, S.Z.; Di, Y.T.; Hao, X.J. Daphnimacropodines A–D, alkaloids from Daphniphyllum macropodum. J. Nat. Prod. 2007, 70, 1348–1351. [Google Scholar] [CrossRef]
- Kong, N.C.; He, H.P.; Wang, Y.H.; Gao, S.; Di, Y.T.; Hao, X.J. Two new iridoid alkaloids, daphmacropodosidines A and B, from Daphniphyllum macropodum. Helv. Chim. Acta 2007, 90, 972–976. [Google Scholar] [CrossRef]
- Li, Z.Y.; Chen, P.; Xu, H.G.; Peng, S.Y.; Yang, Y.M.; Zhao, Z.Z.; Guo, Y.W. Further Daphniphyllum alkaloids from the leaves of Daphniphyllum macropodum Miq. Helv. Chim. Acta 2007, 90, 1353–1359. [Google Scholar] [CrossRef]
- Cao, M.; Zhang, Y.; He, H.; Li, S.; Huang, S.; Chen, D.; Tang, G.; Li, S.; Di, Y.; Hao, X.J. Daphmacromines A–J, alkaloids from Daphniphyllum macropodum. J. Nat. Prod. 2012, 75, 1076–1082. [Google Scholar] [CrossRef]
- Zhang, C.R.; Fan, C.Q.; Dong, S.H.; Liu, H.B.; Zhou, W.B.; Wu, Y.; Yue, J.M. Angustimine and angustifolimine: Two new alkaloids from Daphniphyllum angustifolium. Org. Lett. 2011, 13, 2440–2443. [Google Scholar] [CrossRef]
- Zhang, Y.; Di, Y.T.; Zhang, Q.; Mu, S.Z.; Tan, C.J.; Fang, X.; He, H.P.; Li, S.L.; Hao, X.J. Daphhimalenine A, a new alkaloid with an unprecedented skeleton, from Daphniphyllum himalense. Org. Lett. 2009, 11, 5414–5417. [Google Scholar] [CrossRef]
- Mu, S.Z.; Li, C.S.; He, H.P.; Di, Y.T.; Wang, Y.; Wang, Y.H.; Zhang, Z.; Lu, Y.; Zhang, L.; Hao, X.J. Daphnipaxianines A–D, alkaloids from Daphniphyllum paxianum. J. Nat. Prod. 2007, 70, 1628–1631. [Google Scholar] [CrossRef]
- Li, Z.Y.; Chen, P.; Xu, H.G.; Yang, Y.M.; Peng, S.Y.; Zhao, Z.Z.; Guo, Y.W. Macropodumines D and E, two new alkaloids with unusual skeletons from Daphniphyllum macropodum Miq. Org. Lett. 2007, 9, 477–480. [Google Scholar] [CrossRef]
- Di, Y.T.; He, H.P.; Li, C.S.; Tian, J.M.; Mu, S.Z.; Li, S.L.; Gao, S.; Hao, X.J. Yuzurimine-type alkaloids from Daphniphyllum yunnanense. J. Nat. Prod. 2006, 69, 1745–1748. [Google Scholar] [CrossRef]
- Morita, H.; Ishioka, N.; Takatsu, H.; Shinzato, T.; Obara, Y.; Nakahata, N.; Kobayashi, J. Daphmanidins C and D, novel pentacyclic alkaloids from Daphniphyllum teijsmanii. Org. Lett. 2005, 7, 459–462. [Google Scholar] [CrossRef]
- Chen, X.; Zhan, Z.J.; Yue, J.M. Oldhamiphylline A: A novel hexacyclic alkaloid from Daphniphyllum oldhami. Chem. Biodivers. 2004, 1, 1513–1518. [Google Scholar] [CrossRef]
- Yang, S.P.; Yue, J.M. Two novel alkaloids with a unique fused hexacyclic skeleton from Daphniphyllym subverticillatum. J. Org. Chem. 2003, 68, 7961–7966. [Google Scholar] [CrossRef]
- Kobayashi, J.; Ueno, S.; Morita, H. Daphmanidin A, a novel hexacyclic alkaloid from Daphniphyllum teijsmanii. J. Org. Chem. 2002, 67, 6546–6549. [Google Scholar] [CrossRef]
- Morita, H.; Yoshida, N.; Kobayashi, J. Daphnezomines F and G: Novel alkaloids with 1-azabicyclo[5.2.2]undecane moiety from Daphniphyllum humile. J. Org. Chem. 2000, 65, 3558–3562. [Google Scholar] [CrossRef]
- Ma, N.; Tang, H.F.; Qiu, F.; Lin, H.W.; Tian, X.R.; Yao, M.N. Polyhydroxysteroidal glycosides from the starfish Anthenea chinensis. J. Nat. Prod. 2010, 73, 590–597. [Google Scholar] [CrossRef]
- Tian, X.R.; Tang, H.F.; Li, Y.S.; Lin, H.W.; Chen, X.L.; Ma, N.; Yao, M.N.; Zhang, P.H. New cytotoxic oxygenated sterols from the marine bryozoan Cryptosula pallasiana. Mar. Drugs. 2011, 9, 162–183. [Google Scholar] [CrossRef]
- Wang, X.Y.; Zhang, W.; Gao, K.; Lu, Y.Y.; Tang, H.F.; Sun, X.L. Oleanane-type saponins from the rhizomes of Anemone taipaiensis and their cytotoxic activities. Fitoterapia 2013, 89, 224–230. [Google Scholar] [CrossRef]
- Ding, Y.; Tang, H.F.; Wang, J.B.; Liu, D.; Tian, X.R.; Wang, X.Y.; Zhou, X.M. Triterpenoid saponins from Anemone rivularis var. flore-minore. Biochem. Syst. Ecol. 2011, 39, 236–239. [Google Scholar] [CrossRef]
- Hai, W.L.; Chen, H.; Zhao, M.; Wang, Y.; Hong, L.J.; Tang, H.F.; Tian, X.R. Two new cytotoxic triterpenoid saponins from the roots of Clematis argentilucida. Fitoterapia 2012, 83, 759–764. [Google Scholar] [CrossRef]
- Li, Z.Y.; Gu, Y.C.; Irwin, D.; Sheridan, J.; Clough, J.; Chen, P.; Peng, S.Y.; Yang, Y.M.; Guo, Y.W. Further Daphniphyllum alkaloids with insecticidal activity from the bark of Daphniphyllum macropodum Miq. Chem. Biodivers. 2009, 6, 1744–1750. [Google Scholar] [CrossRef]
- Li, Z.Y.; Xu, H.G.; Zhao, Z.Z.; Guo, Y.W. Two new Daphniphyllum alkalids from Daphniphyllum macropodum Miq. J. Asian Nat. Prod. Res. 2009, 11, 153–158. [Google Scholar] [CrossRef]
- Li, Z.Y.; Chen, P.; Xu, H.G.; Peng, S.Y.; Yang, Y.M.; Zhao, Z.Z.; Guo, Y.W. Further Daphniphyllum alkaloids from the bark of Daphniphyllum macropodum Miq. Chin. J. Chem. 2008, 26, 348–352. [Google Scholar] [CrossRef]
- Gan, X.W.; Bai, H.Y.; Chen, Q.G.; Ma, L.; Hu, L.H. A zwitterionic alkaloid, containing a rare cyclopentadienyl anion unit, from the stem barks of Daphniphyllum macropodum Miq. Chem. Biodivers. 2006, 3, 1255–1259. [Google Scholar] [CrossRef]
- Morita, H.; Yoshida, N.; Kobayashi, J. Daphnicyclidins J and K, unique polycyclic alkaloids from Daphniphyllum humile. J. Org. Chem. 2002, 67, 2278–2282. [Google Scholar] [CrossRef]
- Kobayashi, J.; Inaba, Y.; Shiro, M.; Yoshida, N.; Morita, H. Daphnicyclidins A–H, novel hexa- or pentacyclic alkaloids from two species of Daphniphyllum. J. Am. Chem. Soc. 2001, 123, 11402–11408. [Google Scholar] [CrossRef]
- Zhang, Y.; Di, Y.T.; Mu, S.Z.; Li, C.S.; Zhang, Q.; Tan, C.J.; Zhang, Z.; Fang, X.; Hao, X.J. Dapholdhamines A–D, alkaloids from Daphniphyllum oldhami. J. Nat. Prod. 2009, 72, 1325–1327. [Google Scholar]
- Zhang, H.; Yang, S.P.; Fan, C.Q.; Ding, J.; Yue, J.M. Daphniyunnines A–E, alkaloids from Daphniphyllum yunnanense. J. Nat. Prod. 2006, 69, 553–557. [Google Scholar] [CrossRef]
- Li, C.S.; Di, Y.T.; Zhang, Q.; Zhang, Y.; Tan, C.J.; Hao, X.J. Alkaloids from the leaves of Daphniphyllum longeracemosum Rosenth. Helv. Chim. Acta 2009, 92, 653–659. [Google Scholar] [CrossRef]
- Morita, H.; Yoshida, N.; Kobayashi, J. Daphnezomines C, D, and E, new alkaloids with an N-oxide moiety from Daphniphyllum humile. Tetrahedron 1999, 55, 12549–12556. [Google Scholar] [CrossRef]
- Di, Y.T.; He, H.P.; Lu, Y.; Yi, P.; Li, L.; Wu, L.; Hao, X.J. Alkaloids from the leaves of Daphniphyllum longeracemosum. J. Nat. Prod. 2006, 69, 1074–1076. [Google Scholar] [CrossRef]
- Zhang, Y.; Di, Y.T.; Liu, H.Y.; Li, C.S.; Tan, C.J.; Zhang, Q.; Fang, X.; Li, S.L.; Hao, X.J. Three new alkaloids, paxiphyllines C–E, from Daphniphyllum paxianum. Helv. Chim. Acta 2008, 91, 2153–2158. [Google Scholar] [CrossRef]
- Zhang, W.; Guo, Y.W.; Krohn, K. Macropodumines A–C: Novel pentacyclic alkaloids with unusual skeleton or zwitterion moiety from Daphniphyllum macropodum Miq. Chem. Eur. J. 2006, 12, 5122–5127. [Google Scholar] [CrossRef]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988, 48, 589–601. [Google Scholar]
- Sample Availability: Samples of the compounds 1–9 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lu, Y.; Gao, K.; Wang, X.; Zhang, W.; Ma, N.; Tang, H. Five New Alkaloids from the Stem Bark of Daphniphyllum macropodum. Molecules 2014, 19, 3055-3067. https://doi.org/10.3390/molecules19033055
Lu Y, Gao K, Wang X, Zhang W, Ma N, Tang H. Five New Alkaloids from the Stem Bark of Daphniphyllum macropodum. Molecules. 2014; 19(3):3055-3067. https://doi.org/10.3390/molecules19033055
Chicago/Turabian StyleLu, Yunyang, Kai Gao, Xiaoyang Wang, Wei Zhang, Ning Ma, and Haifeng Tang. 2014. "Five New Alkaloids from the Stem Bark of Daphniphyllum macropodum" Molecules 19, no. 3: 3055-3067. https://doi.org/10.3390/molecules19033055