2.3. G–A Plasma Levels and Brain
The data obtained from the determination of G–A concentration in plasma and brain tissue by HPLC (
Table 4), were used in the PKSolver program [
8]. After a single oral administration of 200 mg/kg of G–A, the behavior of the concentration-time data allowed to determine the main pharmacokinetic parameters under the two models, one- and two-compartment (
Table 5). C
max values were 3.33 and 3.42 μg/mL respectively, likewise AUC
0→1440min were 1951.58 y 1824.95 μg/mL·min. G–A was fast absorbed (
Table 3), its presence was detectable in plasma 5 min after oral administration and reached a concentration of 2.47 μg/mL. The fall in G–A plasma concentration continued for 24 h (corresponding to the monitoring time). Regarding the performance of G–A in the brain compartment, it reached C
max of 2.74 μg/mL, T
max 81.6 min (
Table 6), and then dropped gradually to 0.32 μg/mL detected at 24 h. Such a good distribution of G–A in the brain is certainly a result of the lipophilic properties of the molecule, which allows it to easily cross the blood-brain barrier.
Table 4.
G–A concentration variations in ICR mouse plasma and brain in the first 24 h after oral administration of a single dose of 200 mg/kg of G–A.
Table 4.
G–A concentration variations in ICR mouse plasma and brain in the first 24 h after oral administration of a single dose of 200 mg/kg of G–A.
| | G–A Concentration (μg/mL) |
|---|
| Time (min) | Plasma | Brain | |
|---|
| 5 | 2.47 ± 0.43 | 0.00 | |
| 10 | 2.57 ± 0.52 | 0.88 ± 0.33 * | |
| 15 | 2.79 ± 0.79 | 1.48 ± 0.58 * | |
| 30 | 3.70 ± 0.70 | 3.44 ± 0.50 | |
| 60 | 3.44 ± 0.35 | 3.01 ± 0.59 | |
| 120 | 2.95 ± 0.43 | 2.50 ± 0.58 | |
| 240 | 2.33 ± 0.42 | 2.18 ± 0.48 | |
| 480 | 1.44 ± 0.37 | 1.78 ± 0.37 | |
| 720 | 0.96 ± 0.21 | 0.55 ± 0.18 * | |
| 1440 | 0.31 ± 0.02 | 0.32 ± 0.14 | |
The data were fit both to one compartment and two compartment models (
Figure 3). The equations describing the compartments are as follows:
where C
p is the plasma concentration (μg/mL);
kd, β, disposition rate constant (min
−1);
ka, absorption rate constant (min
−1); C
1, C
2 and C
3, coefficients (µg/mL). The goodness of fit were assessed with Sum of Squares (SS), Akaike Information Criteria (AIC), Schwarz Criteria (SC) and Determination coefficient (R
2) (
Table 4), to establish the pharmacokinetics of GA after oral dosage of 200 mg/kg. Data from the plasma concentration
versus time were analyzed under the two proposed model (one and two-compartment). It was established that the one-compartment model was fitted best according to the value of AIC and SC (
Table 5). However, the value of R
2 and the behavior of the data in
Figure 3, suggest that the two-compartment model can be considered.
The
Table 5 shows
k10 values of G–A was 0.0016 and 0.0017 min
−1 one- and two-compartment, respectively.
Table 5.
Pharmacokinetic parameters of G–A in plasma ICR mice, n = 8.
Table 5.
Pharmacokinetic parameters of G–A in plasma ICR mice, n = 8.
| Parameter | One-Compartament Value | Two-Compartament Value | Units |
|---|
| A | 3.50 | 1.83 | μg/mL |
| B | --- | 1.82 | μg/mL |
| ka | 0.182 | 0.164 | 1/min |
| k10 | 0.0016 | 0.0017 | 1/min |
| k12 | --- | 0.00017 | 1/min |
| k21 | --- | 0.00195 | 1/min |
| t1/2ka | 3.79 | 4.22 | 1/min |
| α | --- | 0.0025 | 1/min |
| β | --- | 0.0014 | 1/min |
| V/F | 57.59 | 55.49 | (mg/kg)/(μg/mL) |
| CL/F | 0.092 | 0.098 | (mg/kg)/(μg/mL)/min |
| Tmax | 26.16 | 27.35 | min |
| Cmax | 3.33 | 3.42 | μg/mL |
| AUC0→∞ | 2170.15 | 2030.23 | μg/mL·min |
| AUC0-1440 min | 1951.58 | 1824.95 | μg/mL·min |
| MRT | 630.40 | 620.45 | min |
| Diagnostics | |
| SS | 0.7217 | 0.6331 | |
| R2 | 0.9885 | 0.9899 | |
| AIC | −6.1575 | −4.6565 | |
| SC | −5.2498 | −3.1436 | |
Table 6.
Pharmacokinetic parameters of G–A in brain tissue ICR mice, n = 8.
Table 6.
Pharmacokinetic parameters of G–A in brain tissue ICR mice, n = 8.
| Parameter | Value | Unit |
|---|
| A | 2.98 | μg/mL |
| ka | 0.051 | 1/min |
| k10 | 0.0008 | 1/min |
| t1/2 ka | 13.50 | 1/min |
| V/F | 68.16 | (mg/kg)/(μg/mL) |
| CL/F | 0.057 | (mg/kg)/(μg/mL)/min |
| Tmax | 81.59 | min |
| Cmax | 2.74 | μg/mL |
| AUC0→∞ | 3519.23 | μg/mL·min |
| AUC0-1440 min | 1912.49 | μg/mL·min |
| MRT | 1218.86 | min |
The concentration-time data after a single oral administration of 200 mg/kg of G–A conformed to one- and two-compartment models. A and B: G–A plasma concentration in central and peripheral compartment, respectively; ka: apparent first-order absorption rate constant; k10: apparent first-order elimination rate constant from central compartment; k12: apparent first-order transfer rate constant from central compartment to peripheral compartment; k21: apparent first-order transfer rate constant from peripheral compartment to central compartment; t1/2ka: absorption half time; α and β: empirical constants corresponding to the coefficients of the exponents of the values of A and B; V/F: apparent volume of distribution; CL/F:apparent clearance; Tmax: time to maximal concentration; Cmax: maximal concentration; AUC0→∞ y AUC0-1440 min: Area under the plasma concentration curve from 0 to infinity and form 0 to 1440 min, respectively; MRT: mean residence time; SS: sum of square; R2: correlation index; AIC: Akaike information criteria; SC: Schwarz criterion.
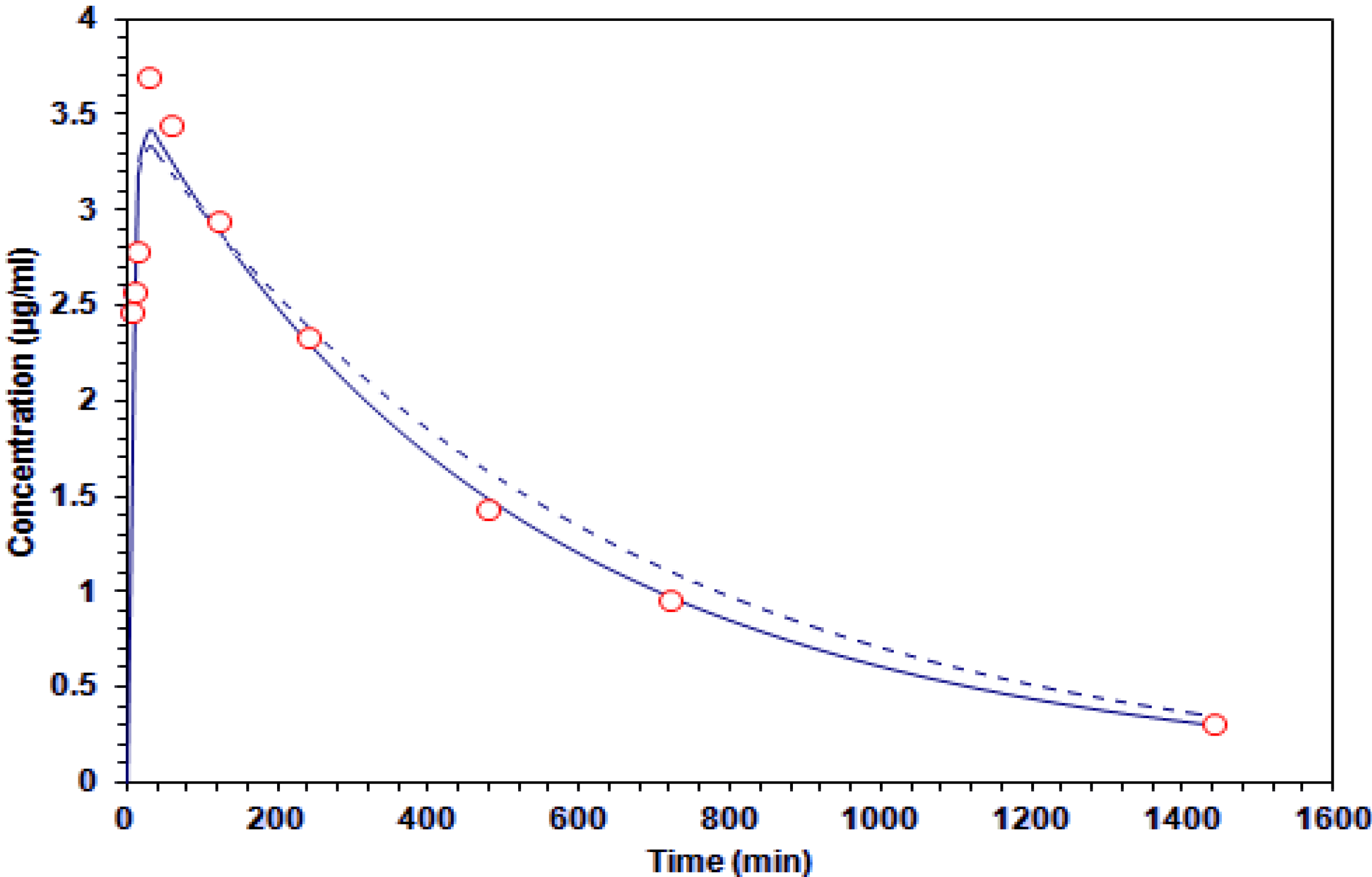
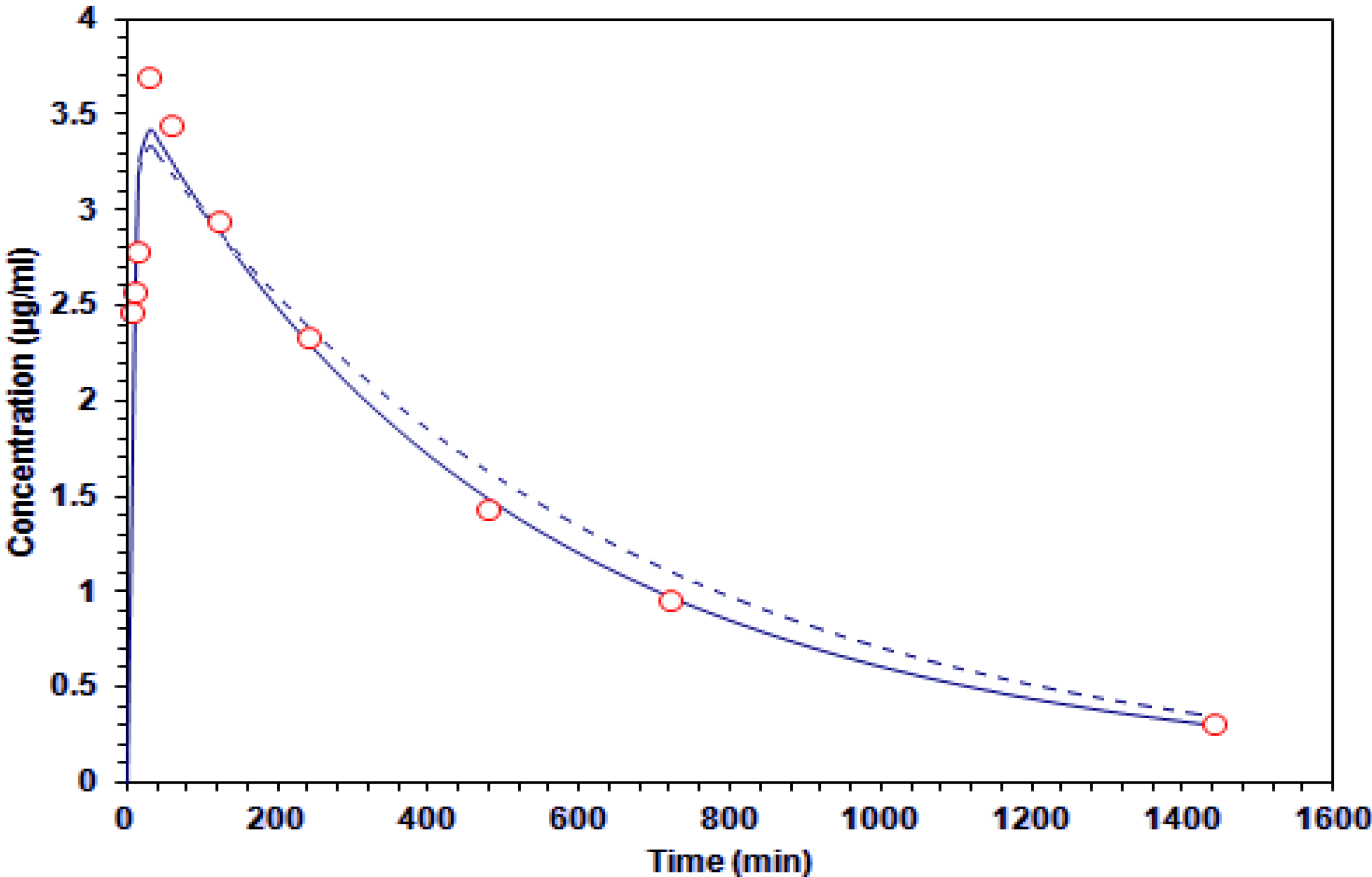
Figure 3.
Concentration-time curve of galphimine A in plasma. The experimental data (○) and estimates of the application of one-compartment (
![Molecules 19 03120 i001]()
) and two-compartment (-----) models.
Figure 3.
Concentration-time curve of galphimine A in plasma. The experimental data (○) and estimates of the application of one-compartment (
![Molecules 19 03120 i001]()
) and two-compartment (-----) models.
The plasma area under the G–A concentration time curve throughout the measurement period reached values of 2,170 and 2,030 μg·min·mL−1.
The anxiolytic effect of galphimines already had been reported [
3], indicating that G–A and G–B are the most active pharmacologically [
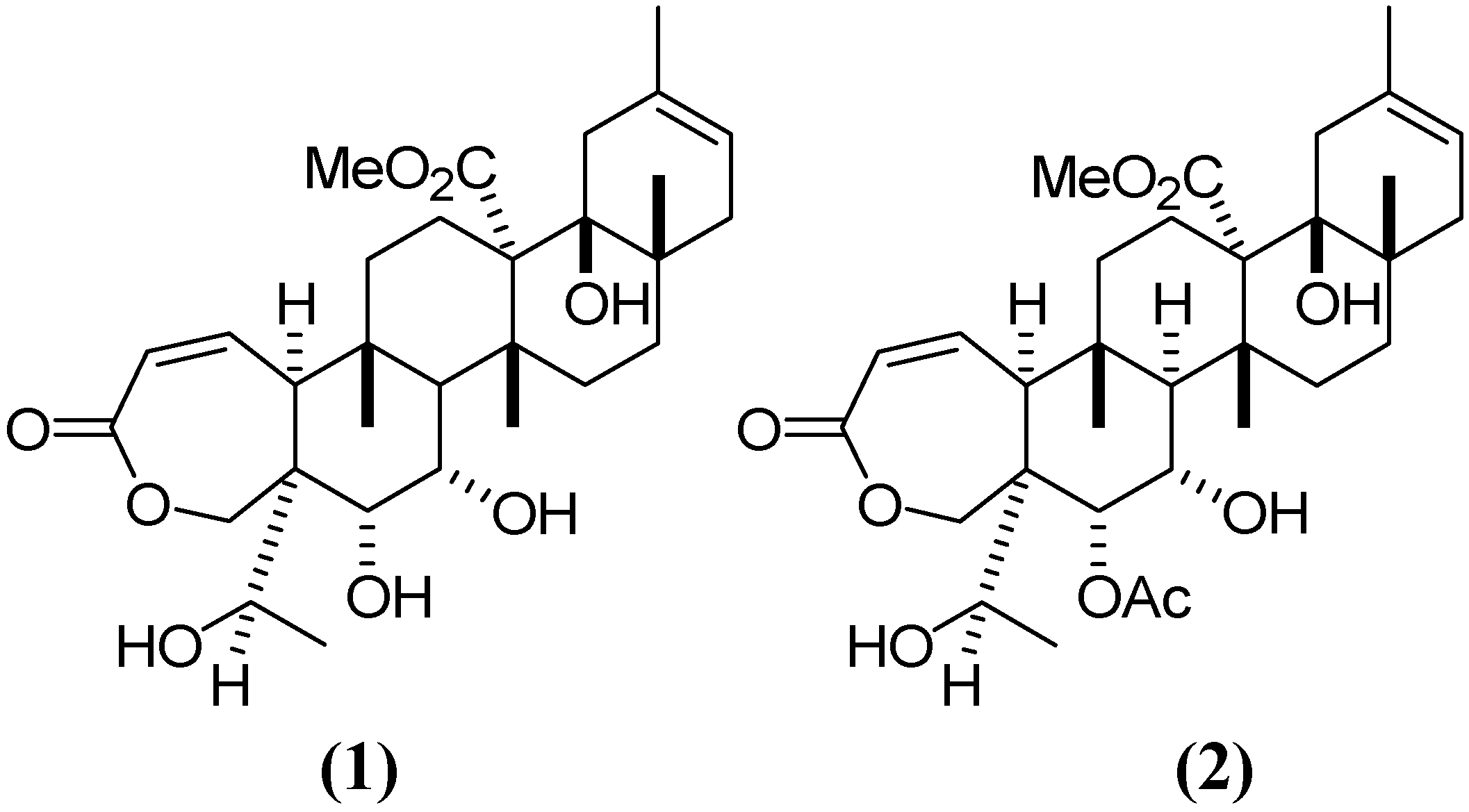
4]. G–E is much more abundant that the other galphimines, although inactive [
6]. In this report we show that deacetylation of G–E converts it into GA, with a conversion rate higher than 80%. This simple reaction may represent an important improvement in the production process of an anxiolytic phyto-medicine, standardized concentration of active galphimines. G–A had anxiolytic effects with a 15 mg/kg dose administered intraperitoneally [
4], and in the present work the dose chosen was 200 mg/kg, due to the fact the administration pathway was oral and to make sure it would be detected when monitored in the plasma and brain samples. Regarding the pharmacokinetic behavior of G–A, and with the intention of measuring the concentration of this triterpene in the brain this organ was lyophilized and the water removed was taken into account to infer the amount of G–A in this system and report it in concentration units (µg·mL
−1).
The data analysis shows that G–A its absorbed quickly, as it was detected in plasma within 5 min of its oral administration, with a slow decay of plasma concentration, since it was still detected at 24 h after that the test started. This suggests slow plasma clearance, similar to other triterpenes like glycyrrhetinic acid, which is the active compound in licorice [
9].
The anxiolytic activity of galphimines implies that they can access the brain compartment, however in this work evidence that galphimines cross the blood-brain barrier is provided. This is based on the fact the G–A concentration in the brain compartment reached 2.74 µg/mL within 81.6 min, and gradually decays within 24 h. These parameters indicate a good distribution of G–A in the brain that is certainly a result of the lipophilic properties of the molecule, which allow it to easily cross the blood-brain barrier.
There are no antecedents concerning the pharmacokinetic behavior of the galphimines, not even of similar compounds. There are reports about another triterpene, glycyrrhetinic acid. Considering the mass exchange between the compartments, it was observed that the value of
k10 (elimination rate constant) obtained for G–A, in a model of one-compartment was of 0.0016 min
−1, and for the two-compartment model it was 0.0017 min
−1, while for the triterpene glycyrrhetinic acid in mice (administered with 100 mg/kg, orally) and the one-compartment model the corresponding value of
k10 was of 0.0006 min
−1 [
10], which is evidently different. In contrast, the
k10 value for G–A in mice and the
k10 value for glycyrrhetinic acid (0.0012 min
−1) in humans, show the same order of magnitude [
11].
The area under the curve for G–A was comparable in the two models analyzed compared to the AUC
0→∞ found for oral administration of glycyrrhetinic acid in mice: 2,454 µg·min·mL
−1 [
10]. Finally, the G–A concentration decay rate was determined as a function of time in the brain compartment. According to AIC values, the model that best fits these pharmacokinetic parameters is the one-compartment one.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
 ) and two-compartment (-----) models.
) and two-compartment (-----) models.