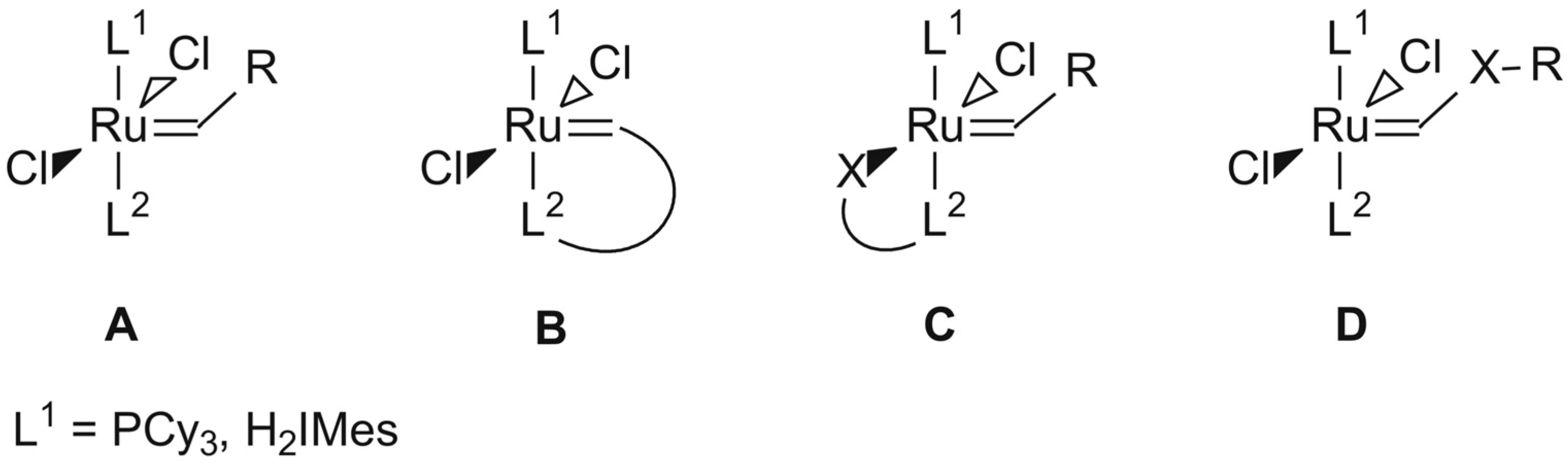
3.4. General Procedure for the Synthesis of the Second Generation Precatalysts [22]
A solution of lithium alcoholate (0.63 mmol, 0.090 g) in THF (5 mL) was added dropwise to a solution of 2 (0.55 mmol, 0.470 g) in THF (5–10 mL) under inert (Ar) and very dry conditions. The reaction was stirred at 30–40 °C for 60min with a colour change from brown to various shades of green. The progress of the reaction was monitored by TLC (Merck Silica gel 60 F254; eluent hexane–ethyl acetate = 1:2) to determine if the starting complex was being consumed. The reaction mixture was stirred until the TLC indicated that the starting complex was not present any more. After removing the solvent under vacuum the residue was dissolved in a minimal amount of toluene. The lithium chloride was then removed by filtration via a syringe filter and the solution concentrated to a volume of ca. 1 mL. The complexes were obtained by adding 10–15 mL cold pentane onto the filtrate and placing it in the fridge for 8 h or by immediate sonification for 10 min resulting in the immediate formation of a precipitate. After removal of the pentane via syringe or filtration, the desired complex was washed with cold pentane to produce analytically pure microcrystalline powders.
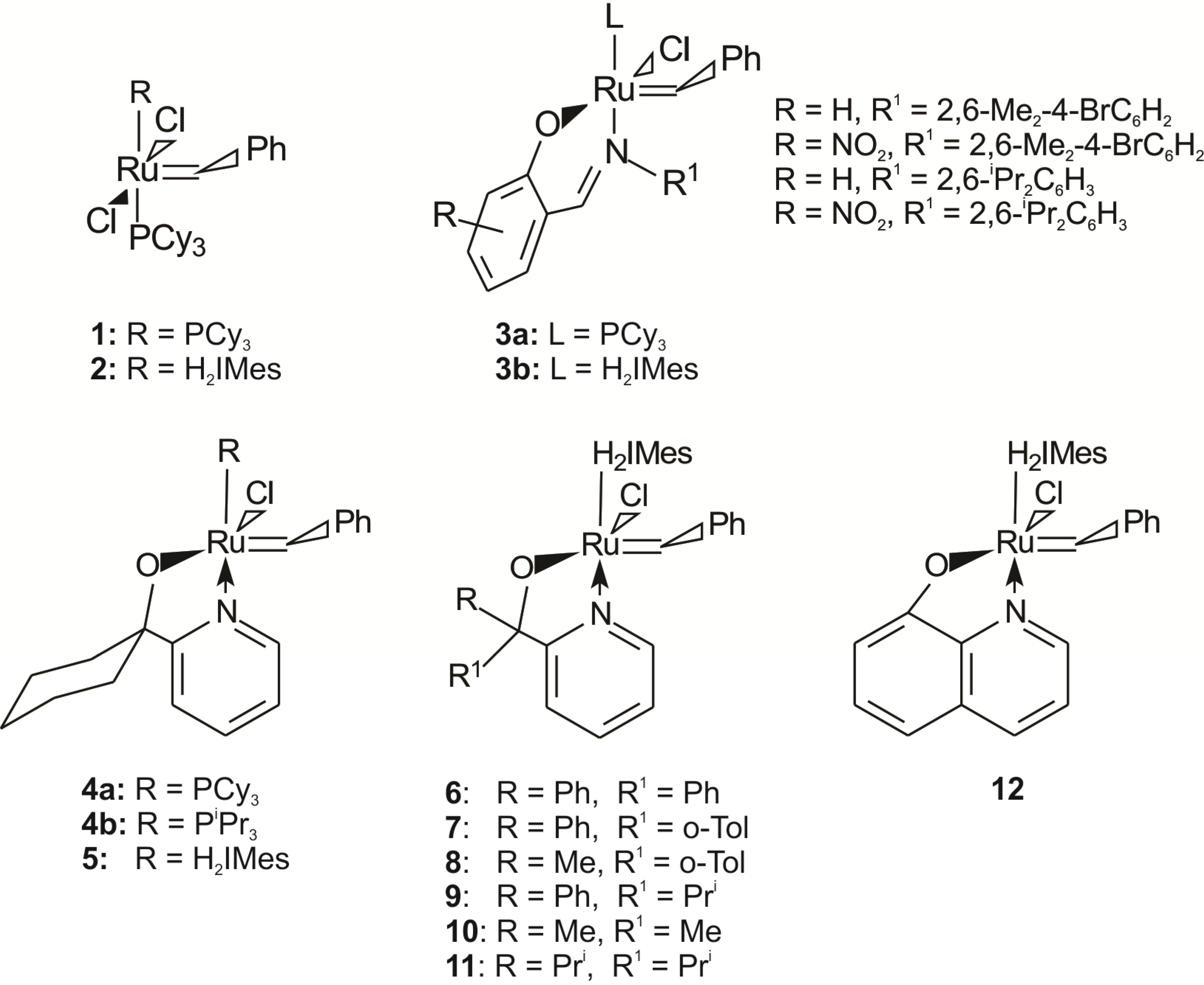
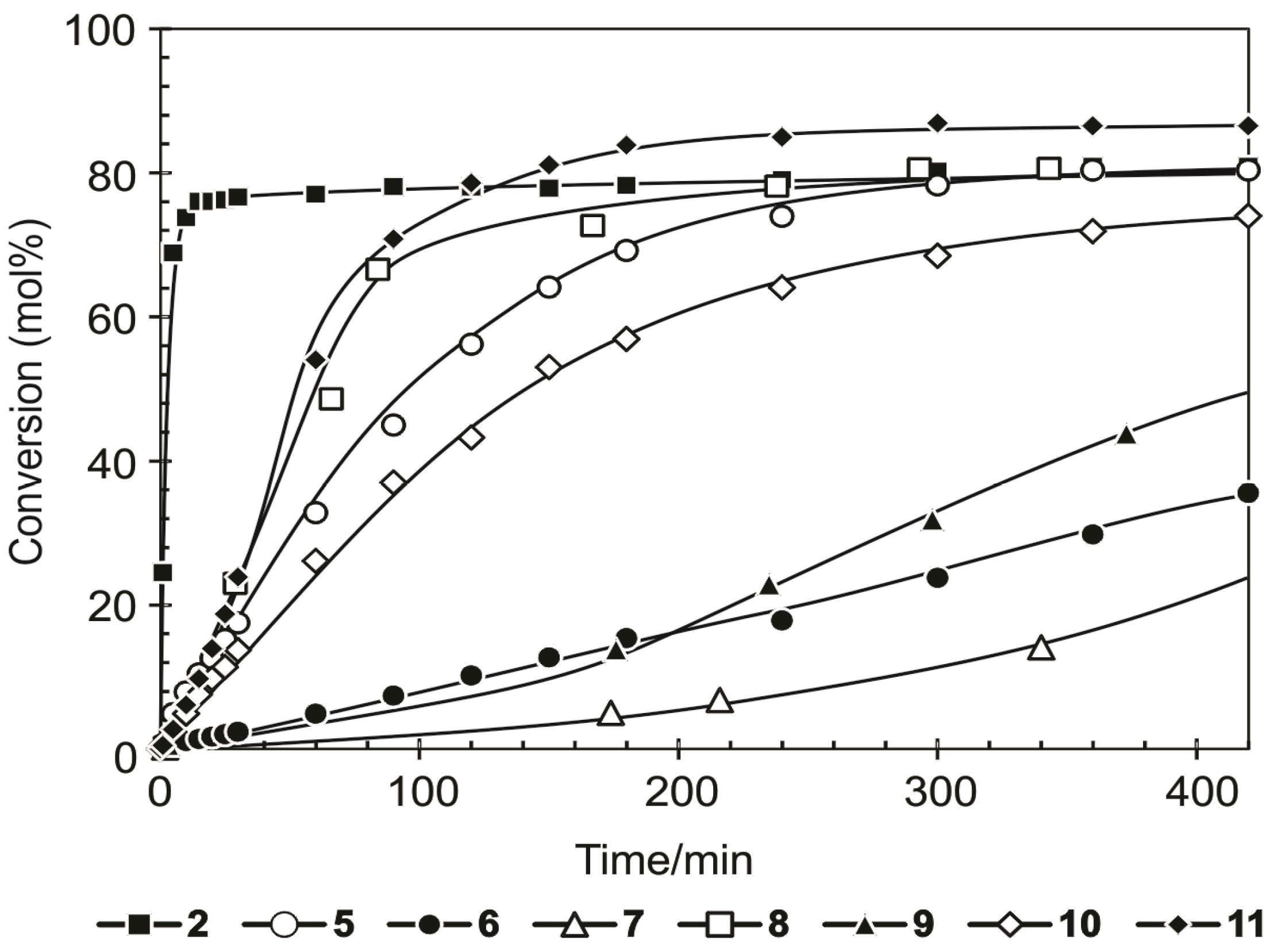
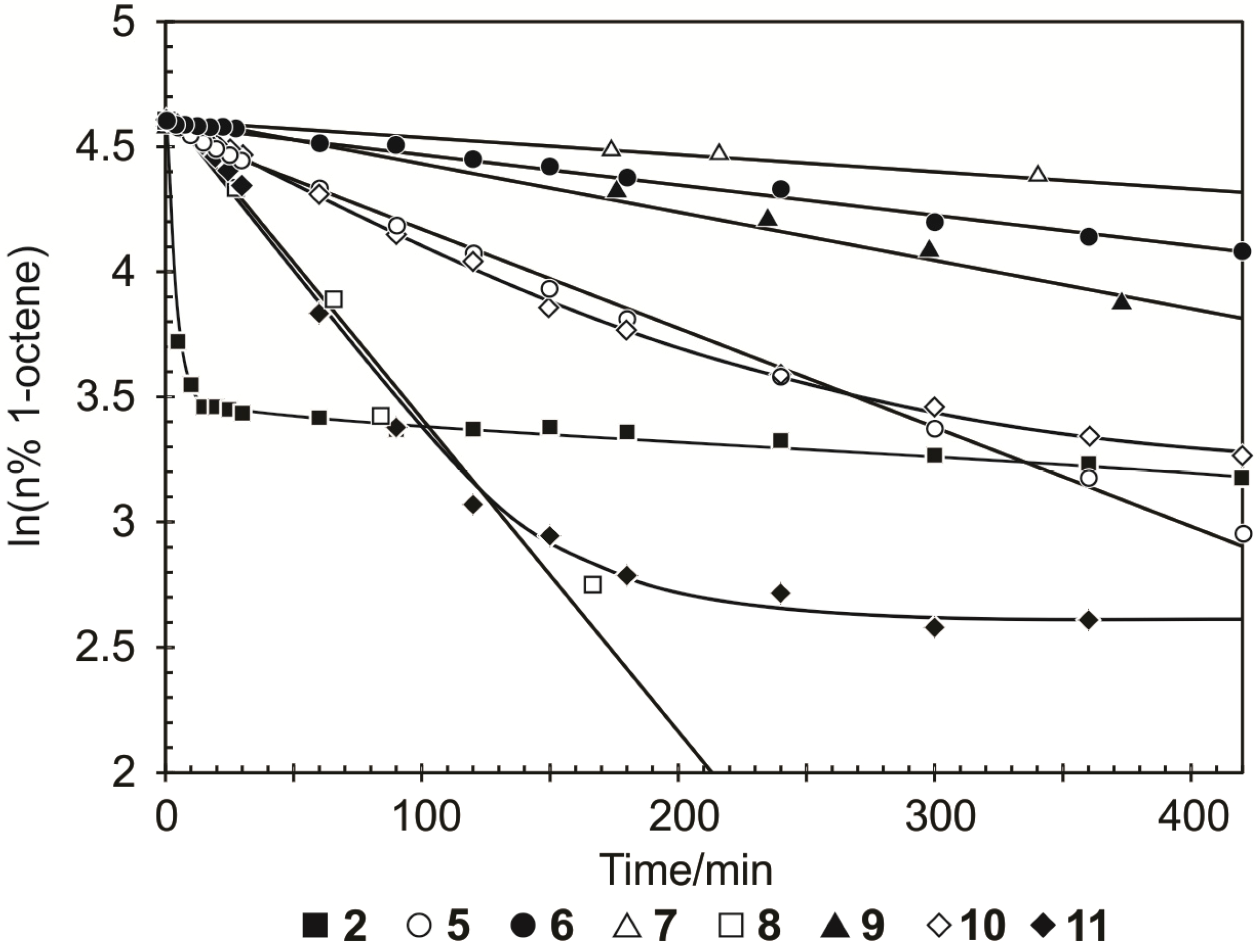
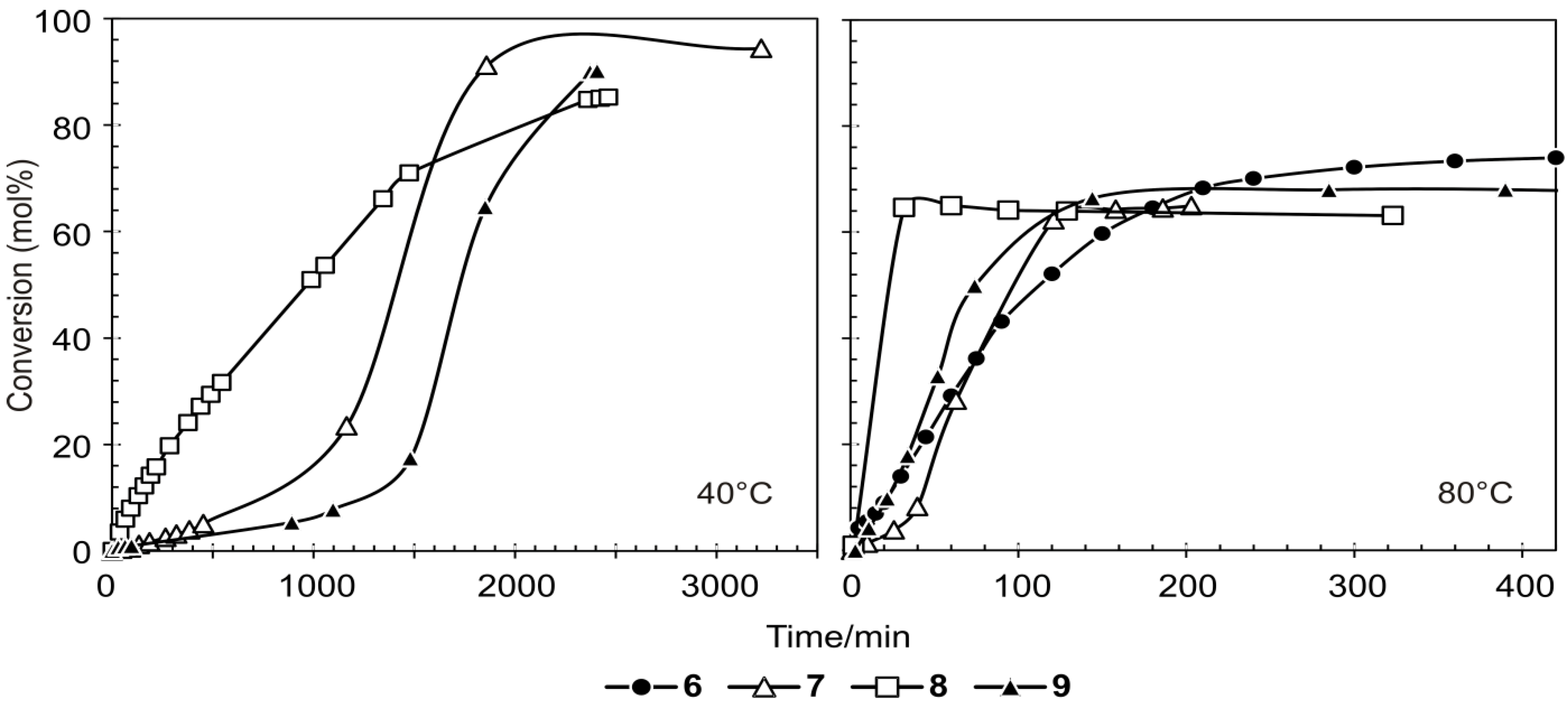
Benzylidene-chloro(1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene)-[1-(2′-pyridinyl)-1,1-diphenylmethanolato]ruthenium (6). Light green microcrystalline powder (91% yield). 1H-NMR (300 MHz, CDCl3): δ = 17.18 (s, 1H, Ru=CHPh), 6.43 (d, 2H, ortho H of C6H5), 7.19 (dd, 1H, para H of C6H5), 6.77 (dd, 2H, meta H of C6H5), 9.67 (d, 1H, ortho H of C5H4N), 7.19 (dd, 1H, para H of C5H4N), 7.04 (dd, 1H, meta H of C5H4N), 6.64 (d, 1H, meta H of C5H4N), 6.71 and 6.99 (s, 4H, meta H mesityl), 4.05 (bs, 4H, CH2 of NHC), 2.20, 2.30 and 2.65 (3s, 18H, CH3 mesityl), 7.12–7.19 (m, 10H, phenyl Hs of O,N-ligand); anal. C46H47ClN3ORu (794.42g/mol): C 69.55, H 5.96, N 5.29; calcd. for C 69.97, H 6.33, N 5.16.
Benzylidene-chloro(1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene)-[1-(2′-pyridinyl)-1-phenyl-1-(2-tolyl)-methanolato]ruthenium (7). Yield: 16%; 1H-NMR (600 MHz, CDCl3): δH 17.33 (s, 1H, Ru=CHPh), 9.76–9.53 (d, 1H, ortho of C4H5N), 7.10–7.08 (dd, 2H, para Hs of C6H5 and C5H4N), 7.08–6.99 (m, 13H, aromatic Hs of O,N ligand and mesityl), 6.68–6.55 (t, 2H, meta Hs of C6H5 and C5H4N), 6.44–6.36 (d, 1H, meta H of C5H4N), 6.36–6.16 (d, 1H, ortho of C6H5), 3.73–3.47 (m, 4H, m, Hs of NHC), 2.49–1.83 (3s, 18H, CH3 mesityl), 1.24 (s, 3H, tolyl CH3 of N,O ligand) ppm, Maldi-TOF: 807 m/z, [M+], HRESIMS m/z 807.2498 (calcd for C47H48ClN3ORu, 807.2533).
Benzylidene-chloro(1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene)-[1-(2′-pyridinyl)-1-(2-tolyl)-1-methylmethanolato]ruthenium (8). Yield: 27%; 1H-NMR (600 MHz, CDCl3): δH 17.32 (s, 1H, Ru=CHPh), 9.87–9.46 (d, 1H, ortho of C4H5N), 7.44–7.26 (d, 1H, ortho H of C6H5), 7.13–6.99 (m, 2H, para Hs of C6H5 and C5H4N), 6.97–6.72 (m, 8H, aromatic Hs of O,N ligand and mesityl), 6.70–6.57 (t, 2H, meta Hs of C6H5 and C5H4N), 6.33–6.00 (d, 1H, meta H of C5H4N), 4.24–3.78 (t, 4H, Hs of NHC), 2.60–2.36 (m, 18H, CH3 mesityl), 1.57 (s, 3H, CH3 of N,O ligand), 1.41–1.09 (m, 3H, tolyl CH3 of N,O ligand) ppm, Maldi-TOF: 745 m/z [M+], HRESIMS m/z 745.2500 (calcd for C43H46ClN3ORu, 745.2498).
Benzylidene-chloro(1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene)-[1-(2′-pyridinyl)-1-phenyl-1-isopropylmethanolato]ruthenium (9). Yield: 49%; 1H-NMR (600 MHz, CDCl3): δH 17.33 (s, 1H, Ru=CHPh), 9.39–9.38 (d, 1H, ortho H of C4H5N), 7.29–7.27 (d, 1H, ortho H of C6H5), 7.03 (m, 3H, aromatic Hs of O,N ligand), 6.94 (m, 2H, para Hs of C6H5 and C5H4N), 6.87–6.845 (m, 2H, meta Hs of mesityl), 6.59–6.57 (t, 2H, meta Hs of C6H5 and C5H4N), 4.08–4.00 (t, 4H, Hs of NHC), 2.72–2.12 (m, 18H, CH3 mesityl), 1.28–1.24 (m, 3H, iso-propyl CH3 of O,N-ligand), 0.65–0.64 (m, 3H, iso-propyl CH3 of O,N-ligand), 1.55–1.34 (t, 1H, iso-propyl CH of O,N-ligand) ppm, Maldi-TOF: 758 m/z [M+], HRESIMS m/z 758.2463 (calcd for C43H47ClN3ORu, 758.2454).
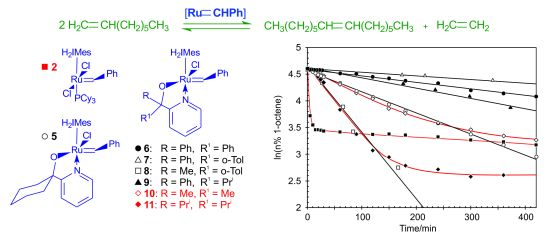
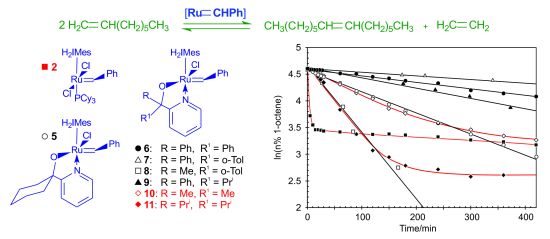
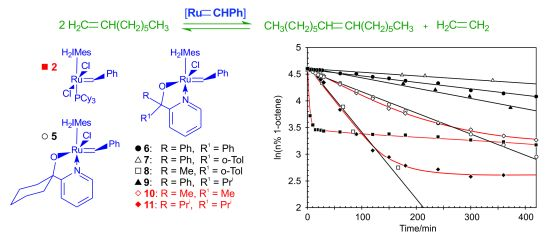
Benzylidene-chloro(1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene)-[1-(2′-pyridinyl)-propane-2-olato]ruthenium (10). Green microcrystalline powder (55% yield). 1H-NMR (300 MHz, CDCl3): δ = 17.82 (s, 1H, Ru=CHPh), 7.15 (d, 2H, ortho H of C6H5), 7.05 (dd, 1H, para H of C6H5), 6.85 (dd, 2H, meta H of C6H5), 9.15 (d, 1H, ortho H of C5H4N), 7.10 (dd, 1H, para H of C5H4N), 6.75 (dd, 1H, meta H of C5H4N), 6.60 (d, 1H, meta H of C5H4N), 6.70 and 6.85 (bs, 4H, meta H mesityl), 4.02 (m, 4H, CH2 of NHC), 2.15, 2.60 and 2.80 (s, CH3 mesityl), 1.20 (s, 6H, CH3 of O,N-ligand); anal. C36H43ClN3ORu (670.28 g/mol): C 64.51, H 6.47, N 6.27; calcd. for C 64.03, H 5.99, N 5.81.
Benzylidene-chloro(1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene)-[1-(2′-pyridinyl)-2,4-dimethylpentan-3-olato]ruthenium (11). Green microcrystalline powder (57% yield). 1H-NMR (300 MHz, CDCl3): δ = 18.52 (s, 1H, Ru=CHPh), 7.65 (d, 2H, ortho H of C6H5), 7.55 (dd, 1H, para H of C6H5), 7.35 (dd, 2H, meta H of C6H5), 9.65 (d, 1H, ortho H of C5H4N), 7.55 (dd, 1H, para H of C5H4N), 7.15 (dd, 1H, meta H of C5H4N), 7.05 (d, 1H, meta H of C5H4N), 7.15 and 7.35 (s, 4H, meta H Mesityl), 4.45 (bs, 4H, CH2 of NHC), 2.59, 2.89 and 3.05 (3s, 18H, CH3 mesityl), 2.15 (dt, 2H, iso-propyl CH of O,N-ligand), 2.15 (dt, 2H, iso-propyl CH of O,N-ligand), 1.01–1.35 (2d, 6H, iso-propyl CH3 of O,N-ligand), 0.10–0.50 (2d, 6H, iso-propyl CH3 of O,N-ligand); anal. C40H51ClN3ORu (726.39 g/mol): C 66.14, H 7.08, N 5.78; calcd. for C 65.68, H 6.73, N 5.08.
Benzylidene-chloro(1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene)-[8-quinolinolate]ruthenium (12). A solution of the lithium salt of quinoline (0.99 mmol, 0.150 g) in THF (5 mL) was added dropwise to a solution of 2 (0.24 mmol, 0.200 g) in THF (5–10 mL). Three carbene complexes formed during the synthesis of 12 of which two were completely soluble in pentane, with the third only partially soluble. As a result of the solubility differences of the three carbenes in pentane, two product layers were formed upon slow vacuum condensation. The bottom analytically pure orange-brown layer contained the partially soluble 12 with a Hα resonance signal at δ 18.25 ppm (42 mg, 26% yield). 1H-NMR (300 MHz, CDCl3): δ = 18.25 (s, 1H, Ru=CHPh), 6.85 (d, 2H, ortho H of C6H5), 7.35 (t, 1H, para H of C6H5), 6.72–6.65 (m, 2H, meta H of C6H5), 7.75 (d, 1H, H-1 of naphthyl), 6.40 (d, 1H, H-3 of naphthyl), 6.72–6.65 (m, H-4 of naphthyl), 7.05–6.89 (m, H-5 of naphthyl), 8.85 (d, H-6 of naphthyl), 6.52 and 6.33 (2 bs, 4H, meta H of mesityl), 3.95 (bs, 4H, CH2 of NHC), 1.95, 2.05 and 2.25 (3s, 18H, CH3 of mesityl); anal. C46H47ClN3ORu (678.26 g/mol): C 65.52, H 5.80, N 6.20; calcd. for C 65.04, H 5.40, N 6.13.

 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



