Non-Conventional Methodologies in the Synthesis of 1-Indanones

 ,
,  , ,
, ,  , and
, and
Abstract
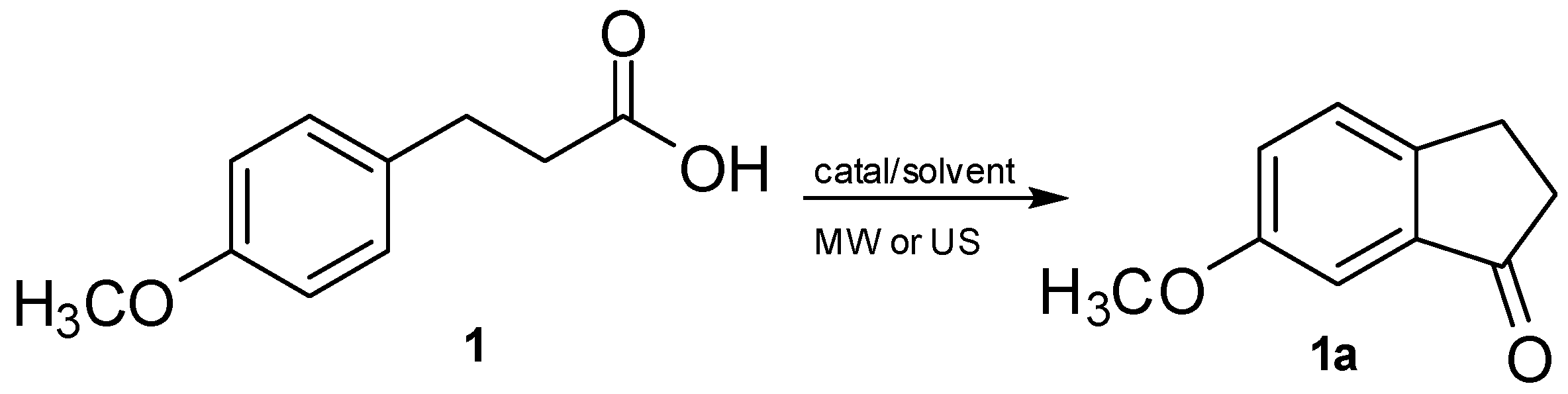
:
1. Introduction
2. Results and Discussion


{kind=link}
{kind=link}
{kind=link}
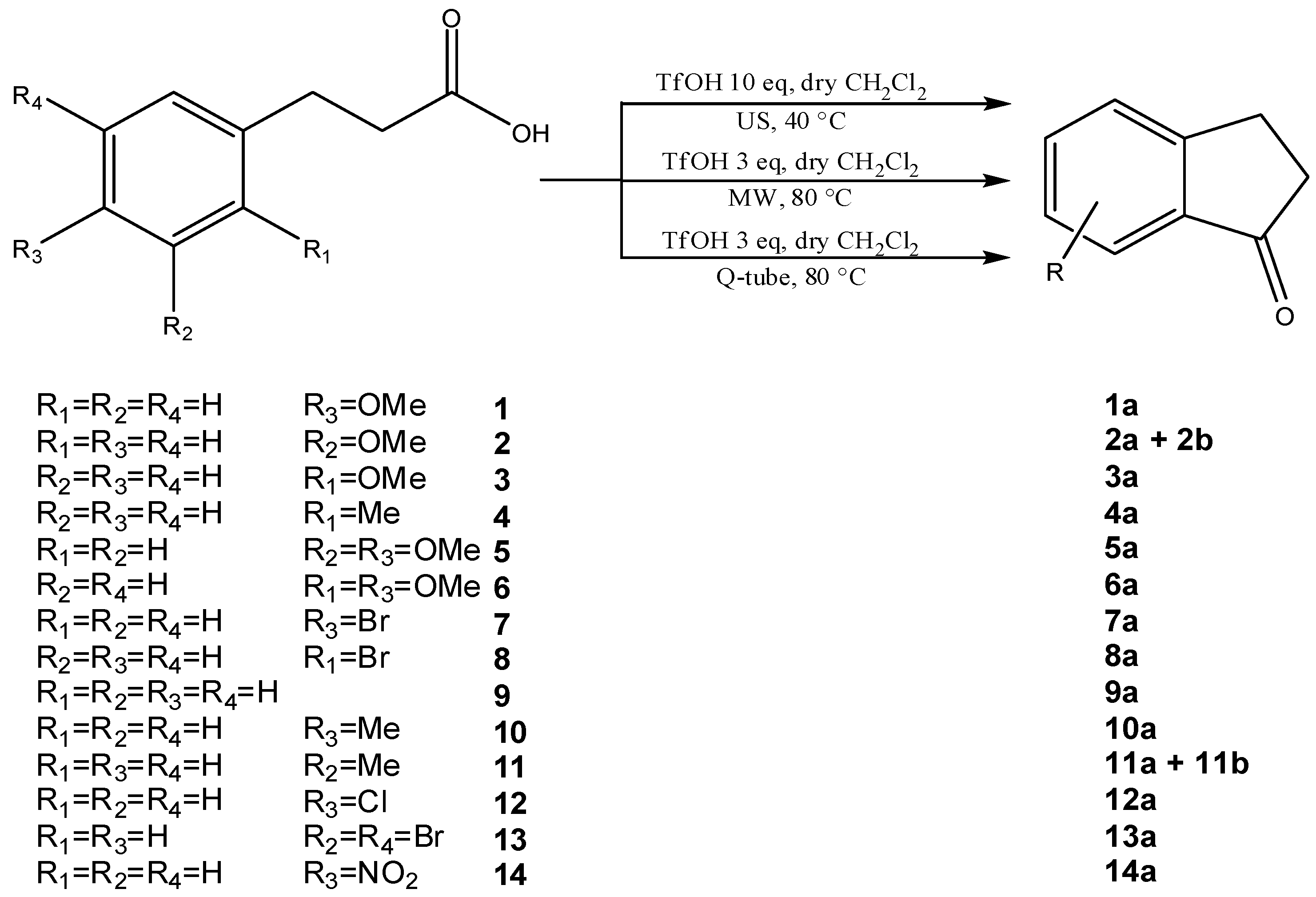{kind=link}
| Entry | Catalyst (%mol) | Solvent | T (°C) | Method | Time (min) | Conv. (%) | Yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | Tb(OTf)3 (20) | PEG a | 180 | MW | 30 | - | - |
| 2 | Tb(OTf)3 (20) | n-BuOH a | 180 | MW | 30 | - | - |
| 3 | Tb(OTf)3 (20) | Ethyl lactate a | 180 | MW | 30 | - | - |
| 4 | Tb(OTf)3 (20) | H2O | 180 | MW | 30 | - | - |
| 5 | Tb(OTf)3 (20) | Toluene b | 180 | MW c | 30 | 10 | 10 |
| 6 | Tb(OTf)3 (20) | Xylene b | 180 | MW c | 30 | - | - |
| 7 | Tb(OTf)3 (20) | Isooctane c | 250 | MW c | 10 | - | - |
| 8 | Tb(OTf)3 (20) | Cl-benzene | 250 | MW | 60 | 85 | 33 d |
| 9 | TfOH (10 eq.) | CH2Cl2 (dry) | 25 | r.t. | 1440 | 85 | 61 d |
| 10 | TfOH (3 eq.) | CH2Cl2 (dry) | 80 | MW | 60 | 100 | 100 |
| 11 | TfOH (1 eq.) | CH2Cl2 (dry) | 80 | MW | 120 | 50 | 20 d |
| 12 | TfOH (2 eq.) | CH2Cl2 (dry) | 80 | MW | 60 | 75 | 53 d |
| 13 | TfOH (3 eq.) | CH2Cl2 (dry) | 110 | MW | 30 | 100 | 100 |
| 14 | TfOH (3 eq.) | CH2Cl2 (dry) | 40 | US | 120 | - | - |
| 15 | TfOH (5 eq.) | CH2Cl2 (dry) | 40 | US | 1260 | 80 | 80 |
| 16 | TfOH (10 eq.) | CH2Cl2 (dry) | 40 | US | 150 | 100 | 100 |
| 17 | TfOH-SiO2 (30) | CH2Cl2 (dry) | 110 | MW | 60 | - | - |
| 18 | TfOH-SiO2 (30) | CH2Cl2 (dry) | 40 | US | 60 | - | - |
| Entry | Catalyst (%mol) | Solvent | T (°C) | Time (min) | Conv. (%) | Yield (%) a |
|---|---|---|---|---|---|---|
| 1 | TfOH (3 eq.) | CH2Cl2 (dry) | 80 | 60 | 100 | 100 |
| 2 | TfOH (3 eq.) | CH2Cl2(dry) | 110 | 30 | 100 | 96 |
| 3 | TfOH (3 eq.) | CH2Cl2(dry) | 150 | 10 | 100 | trace b |
| 4 | Tb(OTf)3 (10) | C6H5Cl | 180 | 180 | 100 | 40 |
| 5 | Tb(OTf)3 (10) | toluene | 150 | 180 | 100 | 86 c |
| 6 | Tb(OTf)3 (10) | n-C7H14 | 180 | 240 | - | - |
| 7 | Tb(OTf)3 (20) | n-C7H14 | 250 | 120 | 18 | 45 |
| 8 | Tb(OTf)3 (10) | isooctane | 250 | 240 | 32 | 20 |
| 9 | TfOH-SiO2(30 ) | CH2Cl2 (dry) | 25 | 120 | - | - |
| 10 | TfOH-SiO2 (30) | CH2Cl2 (dry) | 180 | 180 | - | - |

| Entry | Product | US-Assisted Reaction a | MW-Assisted Reaction a | Q-Tube-Assisted Reaction a | ||||||
| Time (min) | Conv (%) | Yield (%) | Time (min) | Conv (%) | Yield(%) | Time (min) | Conv (%) | Yield(%) | ||
| 1 |  | 150 | 100 | 100 | 60 | 100 | 100 | 60 | 100 | 100 |
| 210 b | 100 | 100 | 90 b | 100 | 100 | 90 b | 100 | 100 | ||
| 2 |  | 60 | 100 | 88/12 | 60 | 100 | 90/10 | 60 | 100 | 90/10 |
| (2b)/(2a) | (2b)/(2a) | (2b)/(2a) | ||||||||
| 3 |  | 120 | 100 | - c | 180 | 100 | - c | 180 | 100 | - c |
| 4 |  | 60 | 100 | 100 | 60 | 100 | 100 | 60 | 100 | 100 |
| 5 |  | 60 | 100 | 100 | 60 | 100 | 100 | 60 | 100 | 100 |
| 6 |  | 360 | - | - | 180 | - | - | 180 | - | - |
| 7 |  | 360 | 100 | 100 | 180 d | 42 | 33 | 180 d | 54 | 36 |
| 60 e | 100 | 100 | 60 e | 100 | 100 | |||||
| 8 |  | 1200 | 90 | 90 | 180 d | 58 | 48 | 180 d | 44 | 43 |
| 60 e | 100 | 100 | 60 e | 100 | 100 | |||||
| 9 |  | 60 | 100 | 100 | 180 | 100 | 100 | 180 | 100 | 100 |
| 10 |  | 60 | 100 | 100 | 60 | 100 | 100 | 60 | 100 | 100 |
| 11 |  | 60 | 100 | 85/15 | 60 | 100 | 88/12 | 60 | 100 | 88/12 |
| (11b)/(11a) | (11b)/(11a) | (11b)/(11a) | ||||||||
| 12 |  | 400 | 100 | 100 | 180 d | 38 | 30 | 180 d | 45 | 33 |
| 60 e | 100 | 100 | 60 e | 100 | 100 | |||||
| 13 |  | 460 | 100 | 100 | 180 d | 32 | 28 | 180 d | 40 | 36 |
| 120 e | 100 | 100 | 120 e | 100 | 100 | |||||
| 14 |  | 1200 d | - | - | 360 e | - | - | 360 e | - | - |

3. Experimental
3.1. General Information
3.2. General US-Assisted Procedure
3.3. General MW-Assisted Procedure
3.4. General Q-tubetm-Assisted Procedure
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Gawande, M.B.; Bonifácio, V.D.B.; Luque, R.; Branco, P.S.; Varma, R.S. Benign by design: Catalyst-free in-water, on-water green chemical methodologies in organic synthesis. Chem. Soc. Rev. 2013, 42, 5522–5551. [Google Scholar] [CrossRef]
- Wu, C.; Nakamura, H.; Murai, A.; Inouye, S. Chemical studies on the chiral indanone derivatives as the inhibitor of Renilla luciferase. Tetrahedron 2001, 57, 9575–9583. [Google Scholar] [CrossRef]
- Kerr, D.J.; Hamel, E.; Jung, M.K.; Flynn, B.L. The concise synthesis of chalcone, indanone and indenone analogues of combretastatin A4. Bioorg. Med. Chem. 2007, 15, 3290–3298. [Google Scholar] [CrossRef]
- Bansal, R.; Narang, G.; Zimmer, C.; Hartmann, R.W. Synthesis of some imidazolyl-substituted 2-benzylidene indanone derivatives as potent aromatase inhibitors for breast cancer therapy. Med. Chem. Res. 2011, 20, 661–669. [Google Scholar] [CrossRef]
- Byrne, A.J.; Barlow, J.W.; Walsh, J.J. Synthesis and pharmacological evaluation of the individual stereoisomers of 3-[methyl(1,2,3,4-tetrahydro-2-naphthalenyl)amino]-1-indanone, a potent mast cell stabilising agent. Bioorg. Med. Chem. Lett. 2011, 21, 1191–1194. [Google Scholar] [CrossRef]
- Saxena, H.O.; Faridi, U.; Srivastava, S.; Kumar, J.K.; Darokar, M.P.; Luqman, S.; Chanotiya, C.S.; Krishna, V.; Negi, A.S.; Khanuja, S.P.S. Gallic acid-based indanone derivatives as anticancer agents. Biorg. Med. Chem. Lett. 2008, 18, 3914–3918. [Google Scholar] [CrossRef]
- Schumann, H.; Stenzel, O.; Girgsdies, F. (–)-2-Menthylindenyl and (–)-2-menthyl-4,7-dimethylindenyl complexes of rhodium, iridium, cobalt, and molybdenum. Organometallics 2001, 20, 1743–1751. [Google Scholar] [CrossRef]
- Herzog, M.N.; Chien, J.C.W.; Rausch, M.D. Novel 2-indenyl ansa-zirconocenes for the polymerization of α-olefins. J. Organomet. Chem. 2002, 654, 29–35. [Google Scholar] [CrossRef]
- Leoni, L.M.; Hamel, E.; Genini, D.; Shih, H.; Carrera, C.J.; Cottam, H.M.; Carson, D.A. Indanocine, a microtubule-binding indanone and a selective inducer of apoptosis in multidrug-resistantm cancer cells. J. Natl. Cancer Inst. 2000, 92, 217–224. [Google Scholar] [CrossRef]
- Beukes, D.R.; Davies-Coleman, M.T.; Kelly-Borges, M.; Harper, M.K.; Faulkner, D.J.; Dilemmaones, A.-C. Dilemmaones A−C, unusual indole alkaloids from a mixed collection of South African Sponges. J. Nat. Prod. 1998, 61, 699–701. [Google Scholar] [CrossRef]
- Pellissier, H. Recent developments in the Nazarov process. Tetrahedron 2005, 61, 6479–6517. [Google Scholar] [CrossRef]
- Frontier, A.J.; Collinson, C. The Nazarov cyclization in organic synthesis. Recent advances. Tetrahedron 2005, 61, 7577–7606. [Google Scholar] [CrossRef]
- Coyanis, E.M.; Panayides, J.L.; Fernandes, M.A.; de Koning, C.B.; van Ottoerlo, W.A.L. Ring-closing metathesis for the synthesis of substituted indenols, indenones, indanones and indenes: Tandem RCM-dehydrogenative oxidation and RCM-formal redox isomerization. J. Organomet. Chem. 2006, 691, 5222–5224. [Google Scholar] [CrossRef]
- Clark, W.M.; Tickner-Eldridge, A.M.; Huang, G.K.; Pridgen, L.N.; Olsen, M.A.; Mills, R.J.; Lantos, I.; Baine, N.H. A catalytic enantioselective synthesis of the endothelin receptor antagonists SB-209670 and SB-217242. A base-catalyzed stereospecific formal 1,3-hydrogen transfer of a chiral 3-arylindenol. J. Am. Chem. Soc. 1998, 120, 4550–4551. [Google Scholar] [CrossRef]
- Wang, G.; Zheng, C.; Zhao, G. Asymmetric reduction of substituted indanones and tetralones catalyzed by chiral dendrimer and its application to the synthesis of (+)-sertraline. Tetrahedron: Asymmetry 2006, 17, 2074–2081. [Google Scholar] [CrossRef]
- Kajiro, H.; Mitamura, S.; Mori, A.; Hiyama, T. Enantioselective synthesis of 2-hydroxy-1-indanone, a key precursor of enantiomerically pure 1-amino-2-indanol. Tetrahedron: Asymmetry 1998, 9, 907–910. [Google Scholar] [CrossRef]
- Torisawa, Y.; Aki, S.; Minamikawa, J. Conversion of indanone oximes into isocarbostyrils. Bioorg. Med. Chem. Lett. 2007, 17, 453–455. [Google Scholar] [CrossRef]
- Yadav, J.S.; Reddy, G.S.K.K.; Sabitha, G.; Krishna, A.D.; Prasad, A.R.; Rahaman, H.R.R.; Rao, K.V.; Rao, A.B. Daucus carota and baker’s yeast mediated bio-reduction of prochiral ketones. Tetrahedron: Asymmetry 2007, 18, 717–723. [Google Scholar] [CrossRef]
- Sharma, A.K.; Subramani, A.V.; Gorman, C.B. Efficient synthesis of halo indanones via chlorosulfonic acid mediated Friedel-Crafts cyclization of aryl propionic acids and their use in alkylation reactions. Tetrahedron 2007, 63, 389–395. [Google Scholar] [CrossRef]
- Fukuoka, M.; Yoshihira, K.; Natori, S.; Mihashi, K.; Nishi, M. Carbon-13 nuclear magnetic resonance spectra of pterosin-sesquiterpenes and related indan-1-one derivatives. Chem. Pharm. Bull. 1983, 31, 3113–3128. [Google Scholar] [CrossRef]
- Sugimoto, H. Structure-activity relationships of acetylcholinesterase inhibitors: Donepezil hydrochloride for the treatment of Alzheimer’s Disease. Pure Appl. Chem. 1999, 71, 2031–2037. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Segmuller, B.; Ybarra, A. Preparation of acrylophenones and 2-alkyl indanones utilizing hexamethylenetetramine as an inexpensive mannich reagent. Synth. Commun. 1996, 26, 1775–1784. [Google Scholar] [CrossRef]
- De Castro, C.; Primo, J.; Corma, A. Heteropolyacids and large-pore zeolites as catalysts in acylation reactions using α,β-unsaturated organic acids as acylating agents. J. Mol. Catal. A-Chem. 1998, 134, 215–222. [Google Scholar] [CrossRef]
- Gevorgyan, V.; Quan, L.G.; Yamamoto, Y. Synthesis of indenols and indanones via catalytic cyclic vinylpalladation of aromatic aldehydes. Tetrahedron Lett. 1999, 40, 4089–4092. [Google Scholar] [CrossRef]
- Rudler, H.; Denise, B. Copper(II)-catalyzed aerobic oxidation of indane in the presence of aldehydes: Intermediate formation of hydroperoxides. J. Mol. Catal. A-Chem. 2000, 154, 277–279. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Yan, P.; Torok, B.; Olah, G.A. Gallium(III) trifluoro-methanesulfonate: A Water tolerant, reusable lewis acid catalyst for Friedel-Crafts reactions. Catal. Lett. 2003, 87, 109–112. [Google Scholar] [CrossRef]
- Gagnier, S.V.; Larock, R.C. Palladium-catalyzed carbonylative cyclization of unsaturated aryl iodides and dienyl triflates, iodides, and bromides to indanones and 2-cyclopentenones. J. Am. Chem. Soc. 2003, 125, 4804–4807. [Google Scholar] [CrossRef]
- Fillion, E.; Fishlock, D. Convenient access to polysubstituted 1-indanones by Sc(OTf)3-Catalyzed intramolecular Friedel-Crafts acylation of benzyl meldrum’s acid derivatives. Org. Lett. 2003, 5, 4653–4656. [Google Scholar] [CrossRef]
- Johnson, W.S. The formation of cyclic ketones by intramolecular acylation. Org. React. 1949, 2, 114–177. [Google Scholar]
- Popp, F.D.; McEwen, W.E. Polyphosphoric acids as a reagent in organic chemistry. Chem. Rev. 1958, 58, 321–401. [Google Scholar] [CrossRef]
- Miller, R.B.; Frincke, J.M. Stereospecific total synthesis of (.+-.)-vetiselinenol. J. Org. Chem. 1981, 46, 2972–2974. [Google Scholar] [CrossRef]
- Eaton, P.E.; Carlson, G.R.; Lee, J.T. Phosphorus pentoxide-methanesulfonic acid. Convenient alternative to polyphosphoric acid. J. Org. Chem. 1973, 38, 4071–4073. [Google Scholar] [CrossRef]
- Cui, D.M.; Zhang, C.; Kawamura, M.; Shimada, S. Synthesis of 1-indanones by intramolecular Friedel-Crafts reaction of 3-arylpropionic acids catalyzed by Tb(OTf)3. Tetrahedron Lett. 2004, 45, 1741–1745. [Google Scholar] [CrossRef]
- Mason, T.J. Ultrasound in synthetic organic chemistry. Chem. Soc. Rev. 1997, 26, 443–451. [Google Scholar] [CrossRef]
- Cravotto, G.; Nano, G.M.; Palmisano, G.; Tagliapietra, S.; Demetri, A.; Penoni, A. The aldol reaction under high-intensity ultrasound: A novel approach to an old reaction. Eur. J. Org. Chem. 2003, 22, 4438–4444. [Google Scholar]
- Kappe, C.O.; Dallinger, D.; Murphree, S.S. Practical Microwave Synthesis for Organic Chemists, 1st ed.; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Kappe, C.O. Controlled microwave heating in modern organic synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6284. [Google Scholar] [CrossRef]
- Procopio, A.; Gaspari, M.; Nardi, M.; Oliverio, M.; Tagarelli, A.; Sindona, G. Simple and efficient MW-assisted cleavage of acetals and ketals in pure water. Tetrahedron. Lett. 2007, 48, 8623–8627. [Google Scholar]
- Procopio, A.; de Luca, G.; Nardi, M.; Oliverio, M.; Paonessa, R. General MW-assisted grafting of MCM-41: Study of the dependence on time dielectric heating and solvent. Green Chem. 2009, 11, 770–773. [Google Scholar] [CrossRef]
- Procopio, A.; Costanzo, P.; Dalpozzo, R.; Maiuolo, L.; Nardi, M.; Oliverio, M. Efficient ring opening of epoxides with trimethylsilyl azide and cyanide catalyzed by erbium (III) triflate. Tetrahedron Lett. 2010, 51, 5150–5153. [Google Scholar] [CrossRef]
- Procopio, A.; Cravotto, G.; Oliverio, M.; Costanzo, P.; Nardi, M.; Paonessa, R. An eco-sustainable erbium(III)-catalysed method for formation/cleavage of O-tert-butoxy carbonates. Green Chem. 2011, 13, 436–443. [Google Scholar] [CrossRef]
- Nardi, M.; Cozza, A.; Maiuolo, L.; Oliverio, M.; Procopio, A. 1,5-Benzoheteroazepines through eco-friendly general condensation reactions. Tetrahedron Lett. 2011, 52, 4827–4834. [Google Scholar] [CrossRef]
- Cravotto, G.; Procopio, A.; Oliverio, M.; Orio, L.; Carnaroglio, D. Simple sonochemical protocols for fast and repeatable Grignard reactions. Green Chem. 2011, 13, 2806–2809. [Google Scholar] [CrossRef]
- Liu, P.N.; Xia, F.; Wang, Q.W.; Ren, Y.J.; Chen, J.Q. Triflic acid adsorbed on silica gel as an efficient and recyclable catalyst for the addition of β-dicarbonyl compounds to alcohols and alkenes. Green Chem. 2010, 12, 1049–1055. [Google Scholar]
- Duddeck, H.; Toth, G.; Simon, A. Chemical Shifts for Oxygen-Springer; Verlag Berlin: Heidelberg, Germany, 2002. [Google Scholar]
- Gomez-Lor, B.; Frutos, O.D.; Ceballos, P.A.; Granier, T.; Echavarren, A.M. Synthesis of new C3h and C3v Truxene derivatives. Eur. J. Org. Chem. 2001, 11, 2107–2114. [Google Scholar]
- Takeuchi, R.; Yasue, H. Rhodium complex-catalyzed desilylative cyclocarbonylation of 1-aryl-2-(trimethylsilyl)acetylenes: A new route to 2,3-dihydro-1H-inden-1-ones. J. Org. Chem. 1993, 58, 5386–5392. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Oliverio, M.; Nardi, M.; Costanzo, P.; Cariati, L.; Cravotto, G.; Giofrè, S.V.; Procopio, A. Non-Conventional Methodologies in the Synthesis of 1-Indanones. Molecules 2014, 19, 5599-5610. https://doi.org/10.3390/molecules19055599
Oliverio M, Nardi M, Costanzo P, Cariati L, Cravotto G, Giofrè SV, Procopio A. Non-Conventional Methodologies in the Synthesis of 1-Indanones. Molecules. 2014; 19(5):5599-5610. https://doi.org/10.3390/molecules19055599
Chicago/Turabian StyleOliverio, Manuela, Monica Nardi, Paola Costanzo, Luca Cariati, Giancarlo Cravotto, Salvatore Vincenzo Giofrè, and Antonio Procopio. 2014. "Non-Conventional Methodologies in the Synthesis of 1-Indanones" Molecules 19, no. 5: 5599-5610. https://doi.org/10.3390/molecules19055599