Molecular Docking and Fluorescence Characterization of Benzothieno[3,2-d]pyrimidin-4-one Sulphonamide Thio-Derivatives, a Novel Class of Selective Cyclooxygenase-2 Inhibitors

Abstract
:1. Introduction
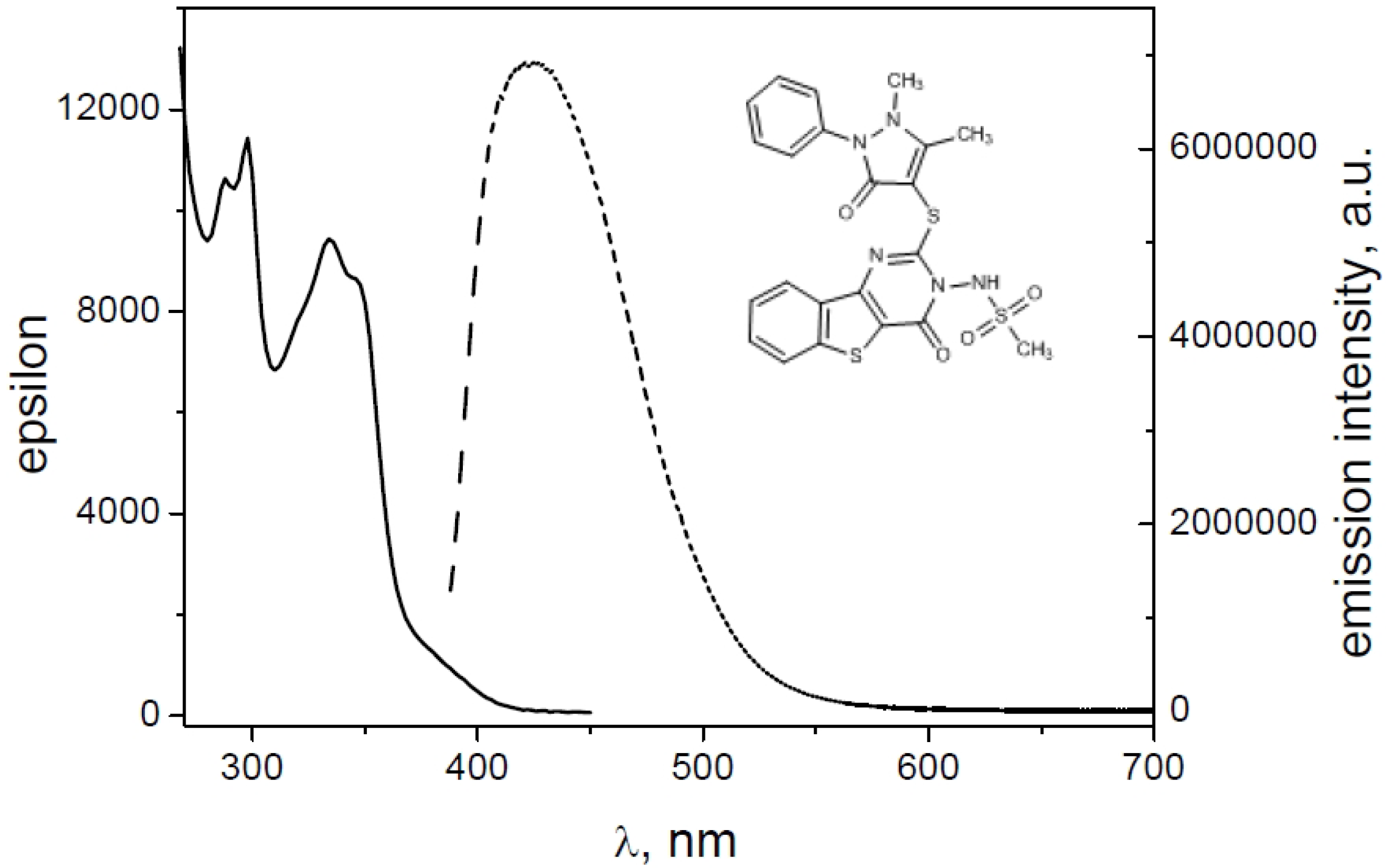
- N-[2-[(1,3-dimethyl-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)thio]-4-oxo[1]benzothieno[3,2-d]pyrimidin-3(4H)yl]methanesulfonamide (1);
- N-[2-[(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)thio]-4oxo[1]benzothieno [3,2-d]pyrimidin-3(4H)yl]methanesulfonamide (2);
- N-[2-[(2,4-nitrophenyl)thio]-4-oxo[1]benzothieno[3,2-d]pyrimidin-3(4H)yl]methanesulfonamide (4);
- N-[2-(cyclohexylthio)-4-oxo[1]benzothieno[3,2-d]pyrimidin-3(4H)yl]methanesulfonamide (8);
- N-[2-[(2,4-difluorophenyl)thio]-4-oxo[1]benzothieno[3,2-d]pyrimidin-(4H)yl]methane-sulfonamide (9);
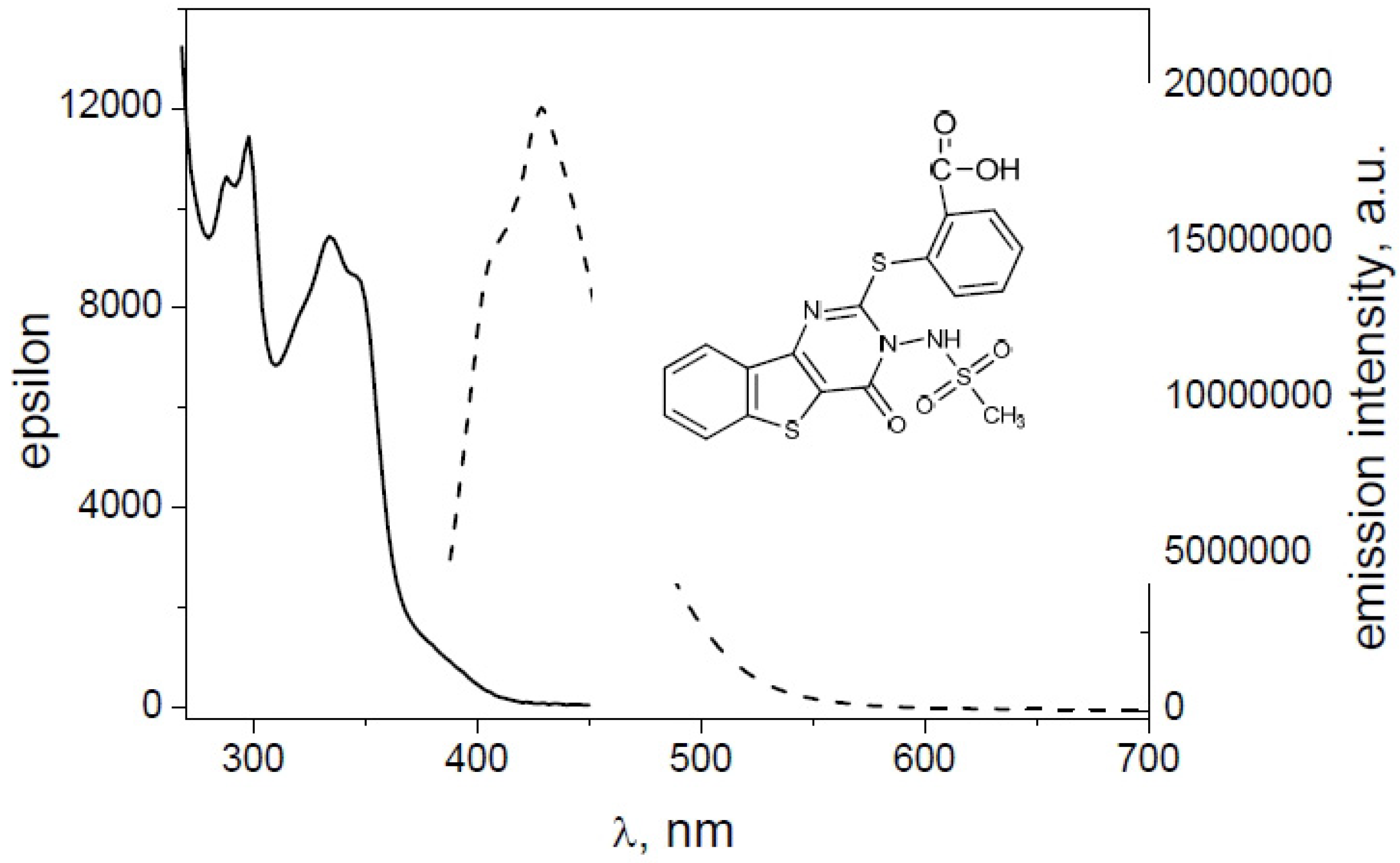
- 2-({3-[(methylsulfonyl)amino]-4-oxo-3,4-dihydro[1]benzothieno[3,2-d]pyrimidin-2-yl}thio)benzoic acid (10).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | NCTC2544 | J774 | ||
|---|---|---|---|---|
| iNOS | COX-2 | iNOS | COX-2 | |
| 1 | 7.0 ± 0.2 | 6.8 ± 0.5 | 7.3 ± 0.3 | 6.9 ± 0.9 |
| 2 | 7.0 ± 0.9 | 6.7 ± 0.3 | 6.5 ± 0.5 | 6.9 ± 0.5 |
| 4 | 5.0 ± 0.2 | 4.5 ± 0.5 | 4.8 ± 0.3 | 5.0 ± 0.1 |
| 8 | 5.8 ± 0.3 | 6.1 ± 1.5 | 6.5 ± 0.9 | 6.2 ± 0.6 |
| 9 | 8.0 ± 0.3 | 8.0 ± 1.0 | 7.8 ± 0.9 | 8.2 ± 0.4 |
| 10 | 6.5 ± 1.3 | 6.2 ± 0.8 | 6.5 ± 1.5 | 6.2 ± 0.5 |
2. Results and Discussion
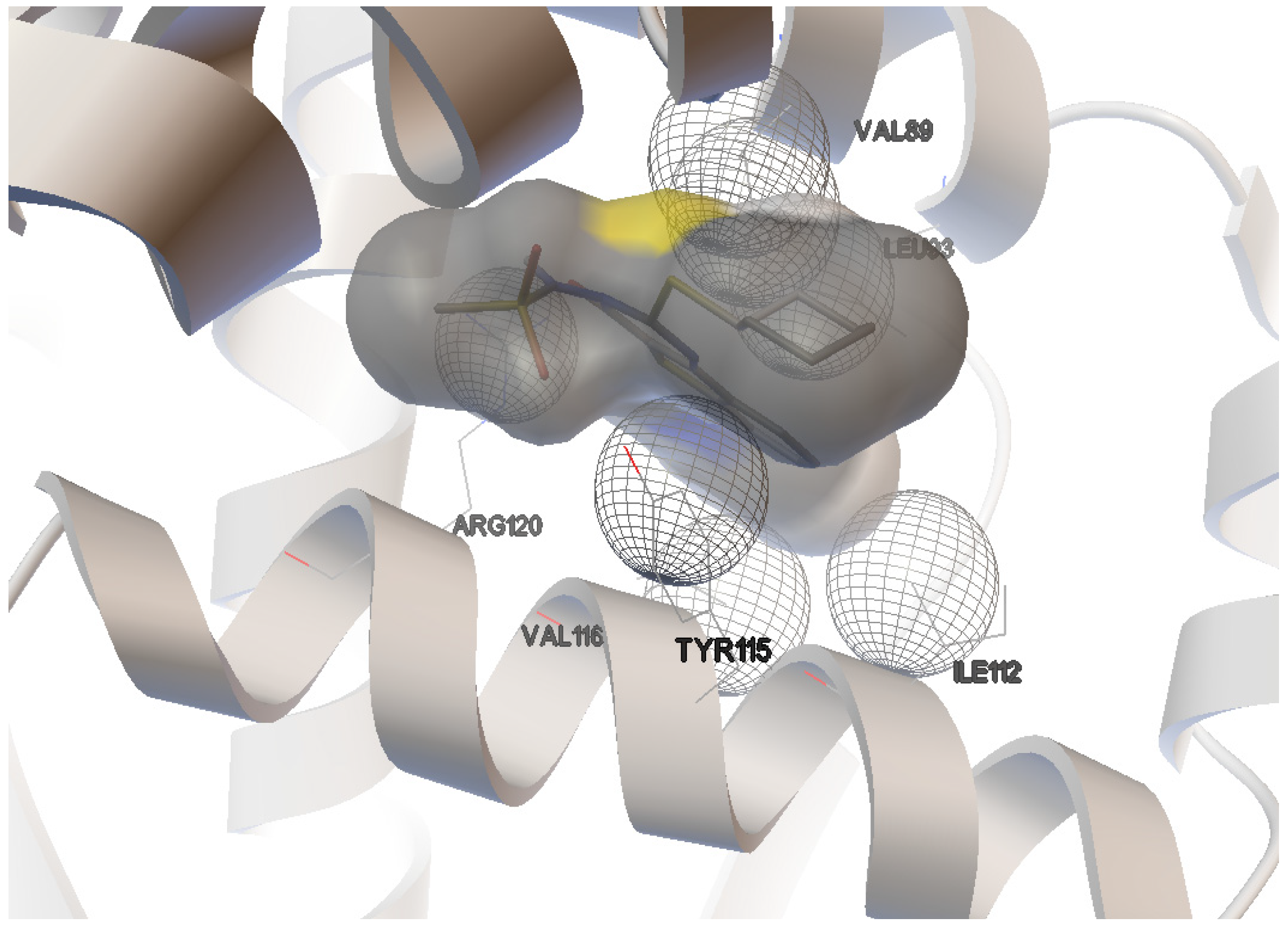
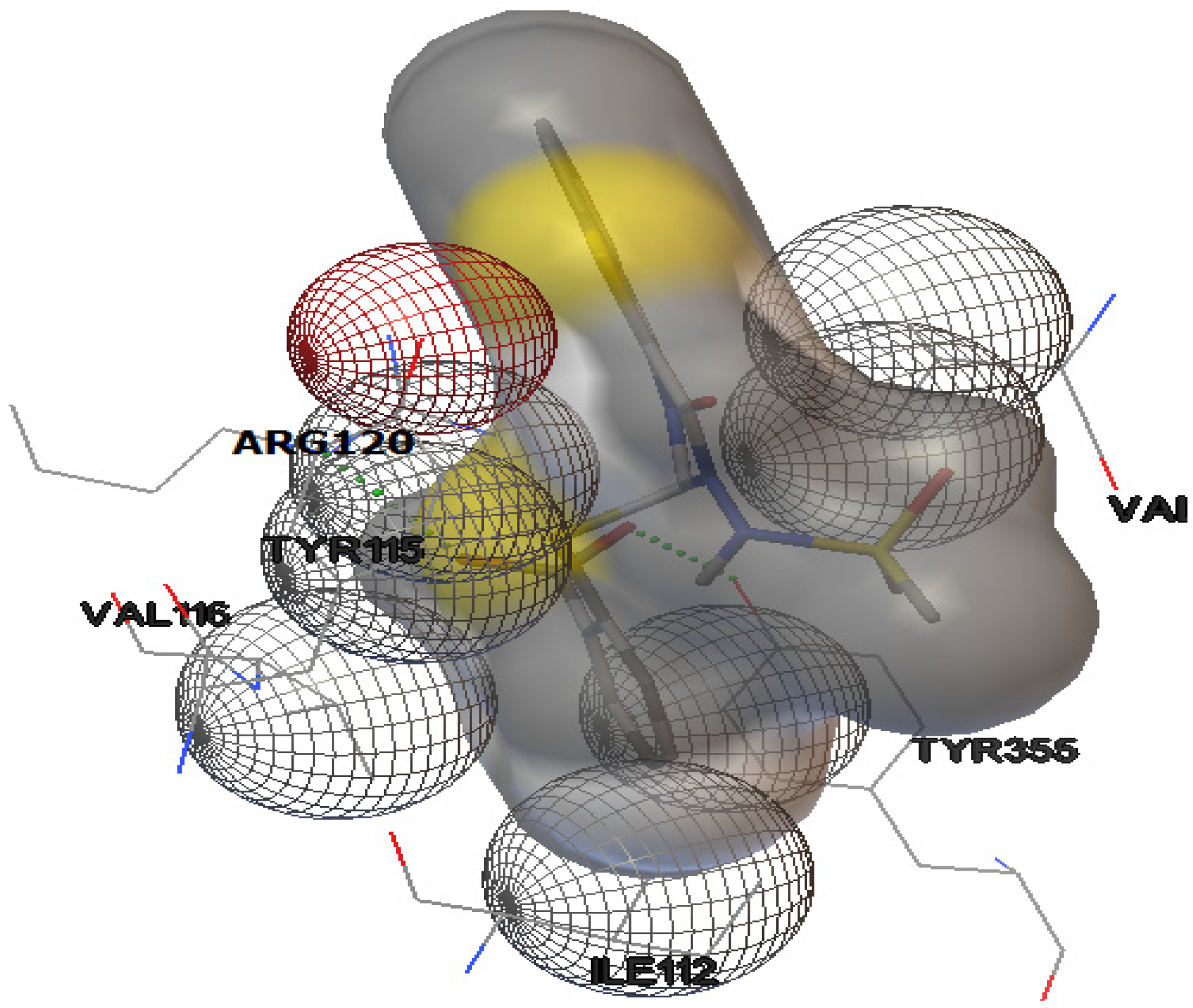
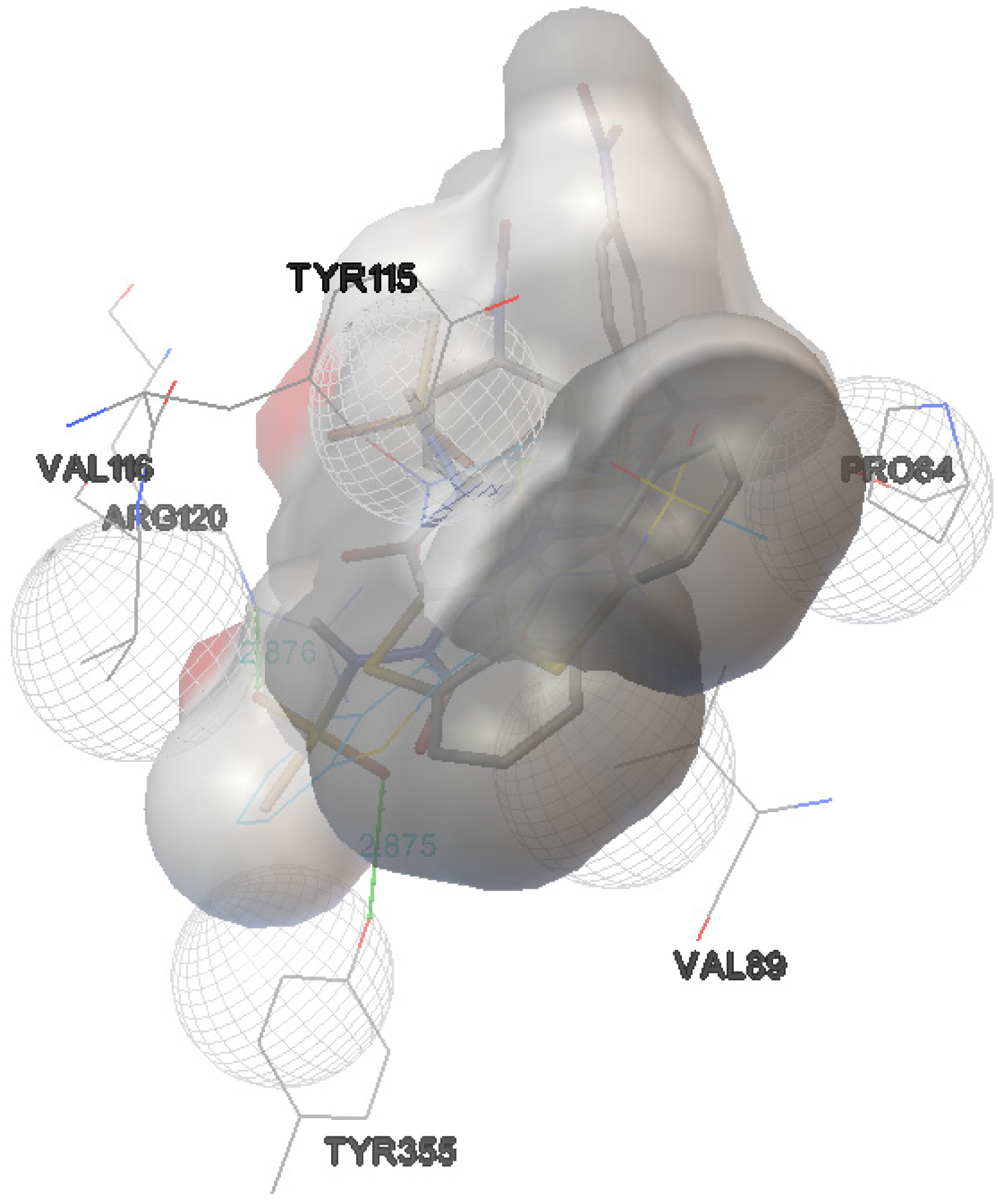
2.1. Docking Study
| Compound | Docking energies (ΔG) (kcal/mol) | Number of H-bonds | Donor/Acceptor Hydrogen bond | H-bond distance (Å) |
|---|---|---|---|---|
| Naproxen | −9.0 | 2 | ARG-120:NH1 | 1.863 |
| TYR-355:OH 1 | 2.165 | |||
| Nimesulide | −7.3 | 2 | ARG-120:NH1 | 3.088 |
| TYR-355:OH 1 | 2.737 | |||
| Derivative 1 | −7.9 | 2 | ARG-120:NH1 | 3.149 |
| TYR-112:OH 1 | 2.806 | |||
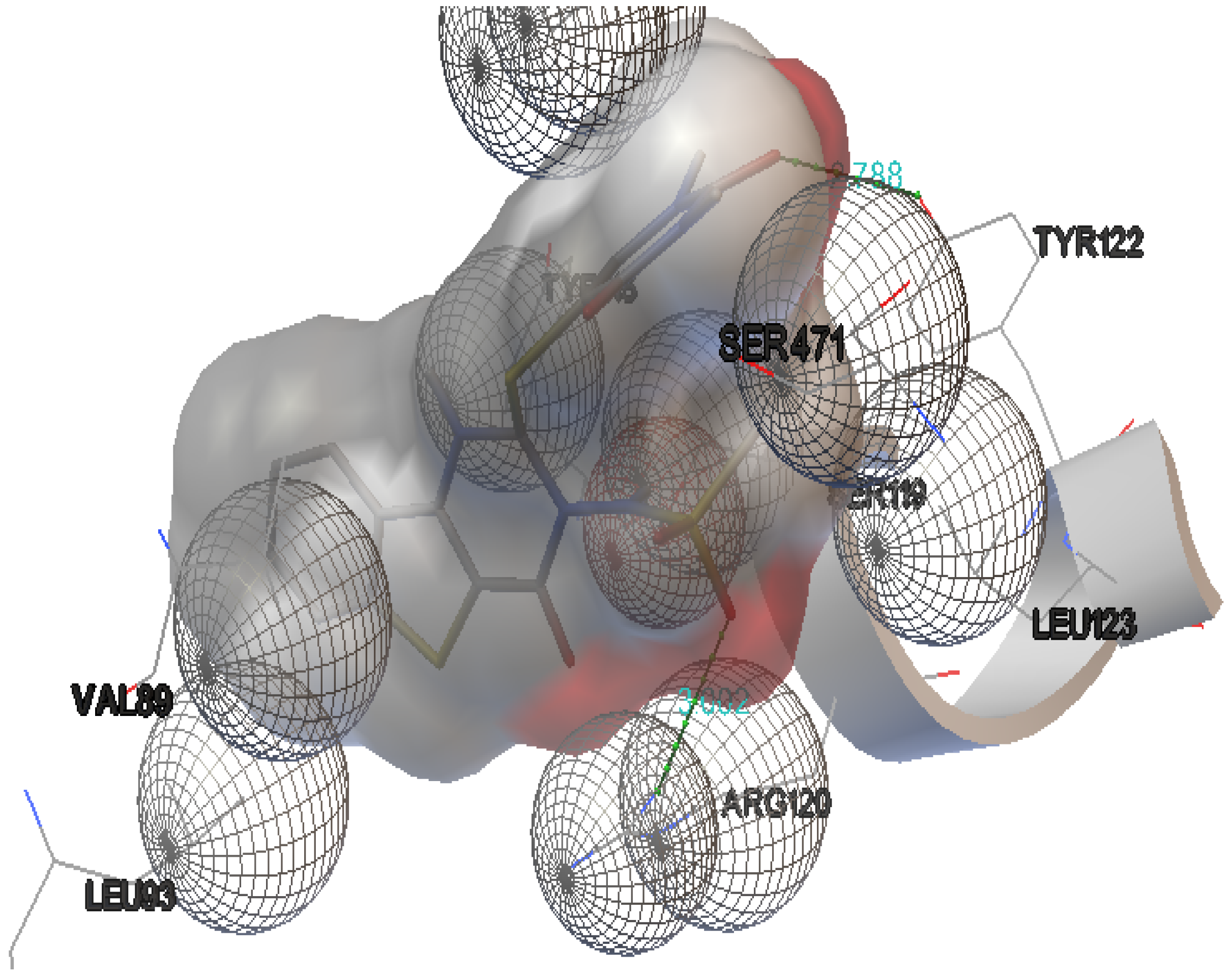
| Derivative 2 | −8.1 | 2 | ARG-120:NH1 | 3.002 |
| TYR-112:OH 1 | 2.788 | |||
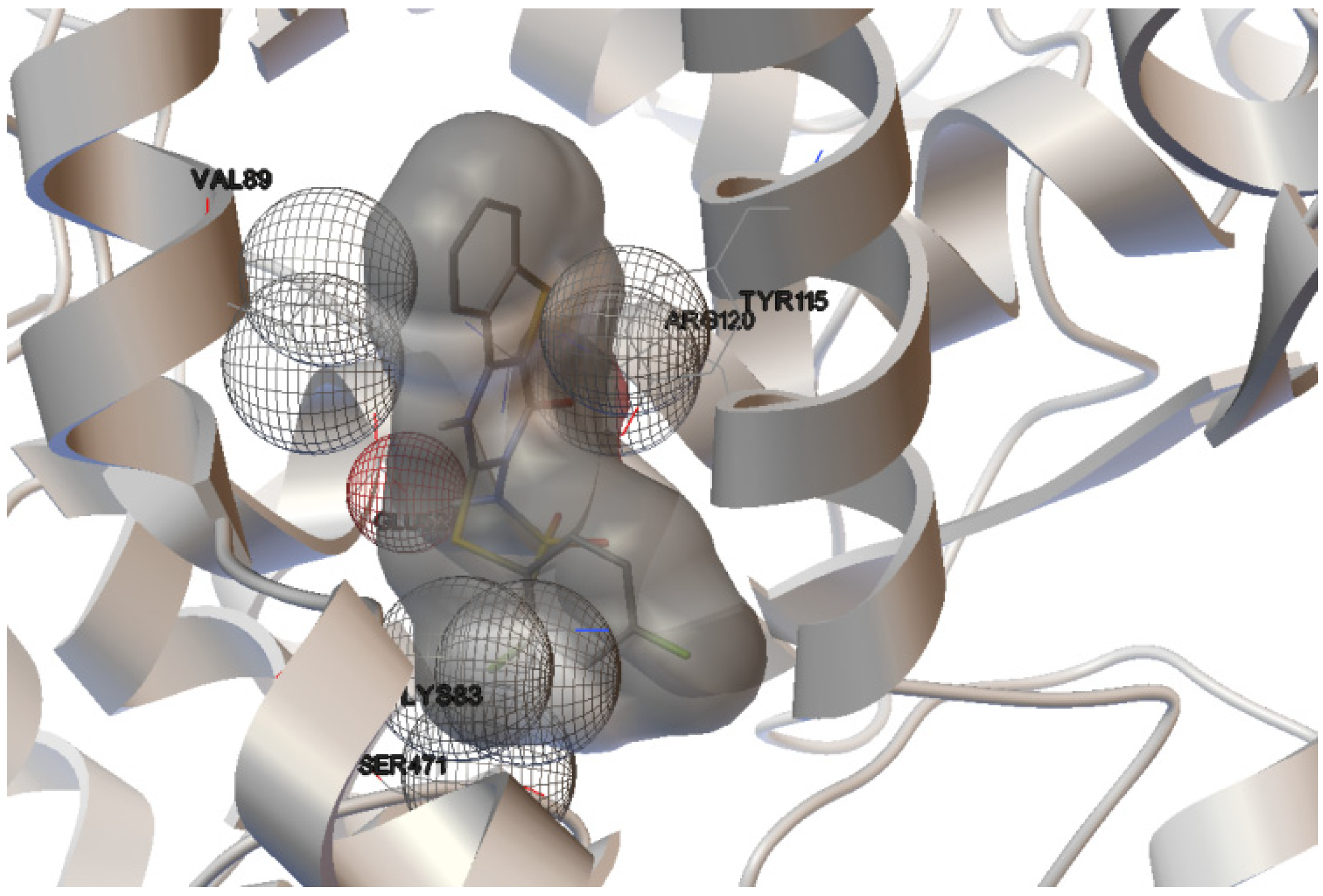
| Derivative 4 | −9.4 | 3 | ASN-382:NH2 | 2.867 |
| HIS-386:NH2 | 2.945 | |||
| THR-212:OH1 | 2.692 | |||
| Derivative 8 | −8.9 | 1 | ARG-120:NH1 | 2.882 |
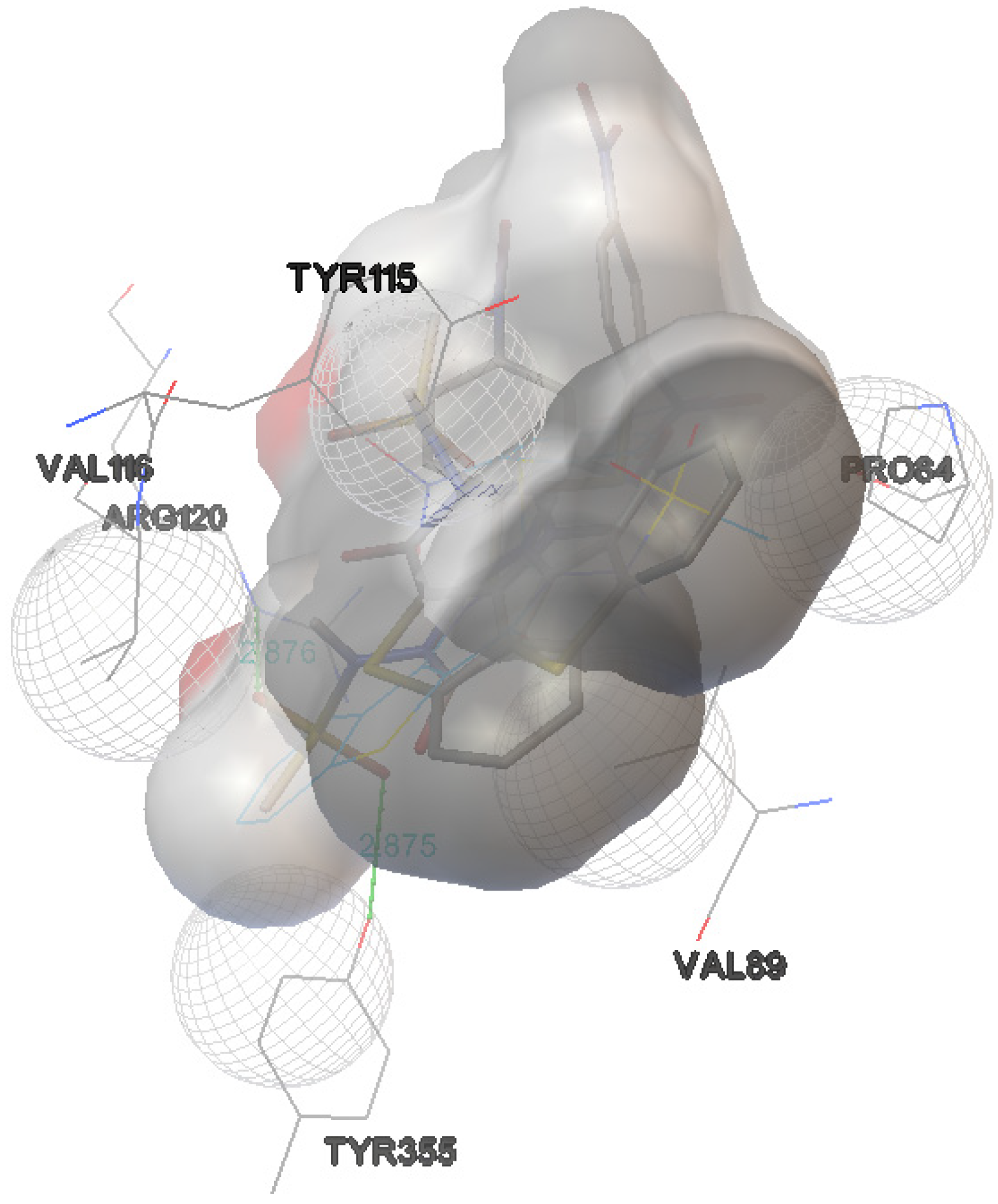
| Derivative 9 | −9.1 | 2 | TYR-355:OH | 2.875 |
| ARG-120:NH | 2.876 | |||
| Derivative 10 | −8.2 | 2 | ARG-120:NH | 2.970 |
| TYR-355:OH | 3.120 |





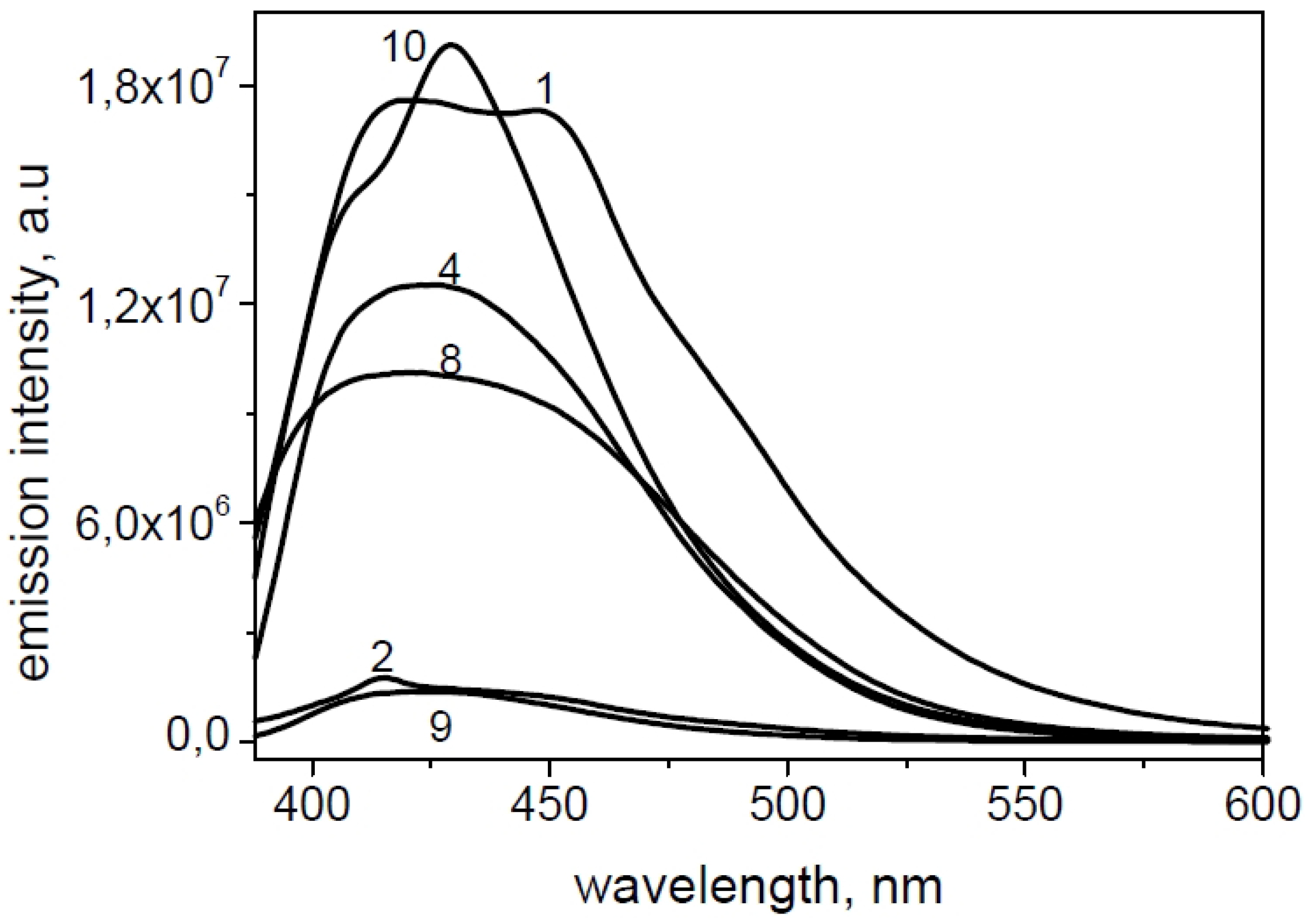
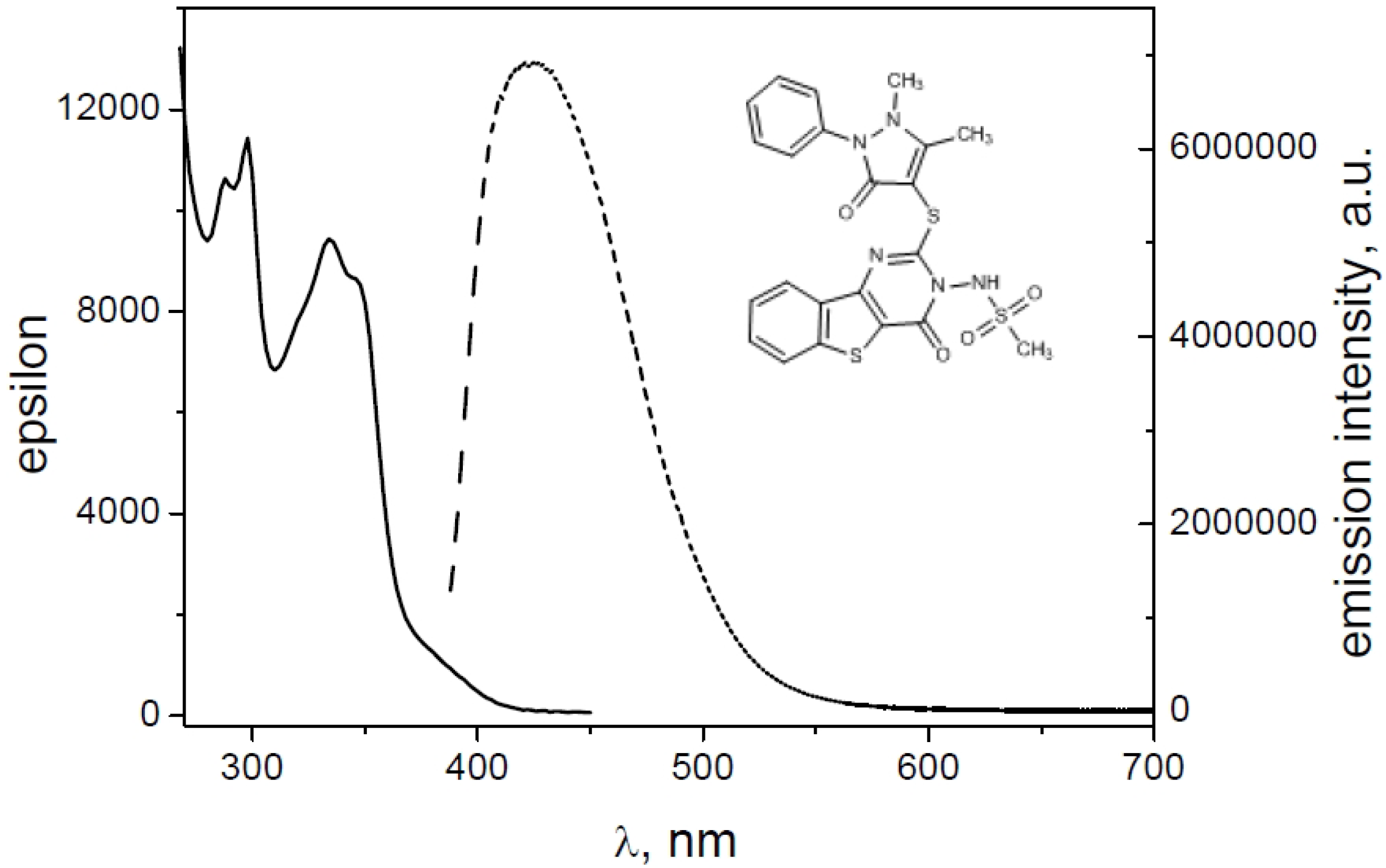
2.2. Spectroscopic Characterization
| Derivative | ε (λmax) | ε (λmax) | Φfl |
|---|---|---|---|
| 1 | 10,991 (340) | 9916 (354) | 0.080 |
| 2 | 8304 (338) | 7500 (352) | 0.013 |
| 4 | 9784 (338) | 8905 (352) | 0.032 |
| 8 | 9662 (338) | 8199 (350) | 0.055 |
| 9 | 10,773 (338) | 9855 (352) | 0.007 |
| 10 | 9435 (334) | 8643 (346) | 0.087 |

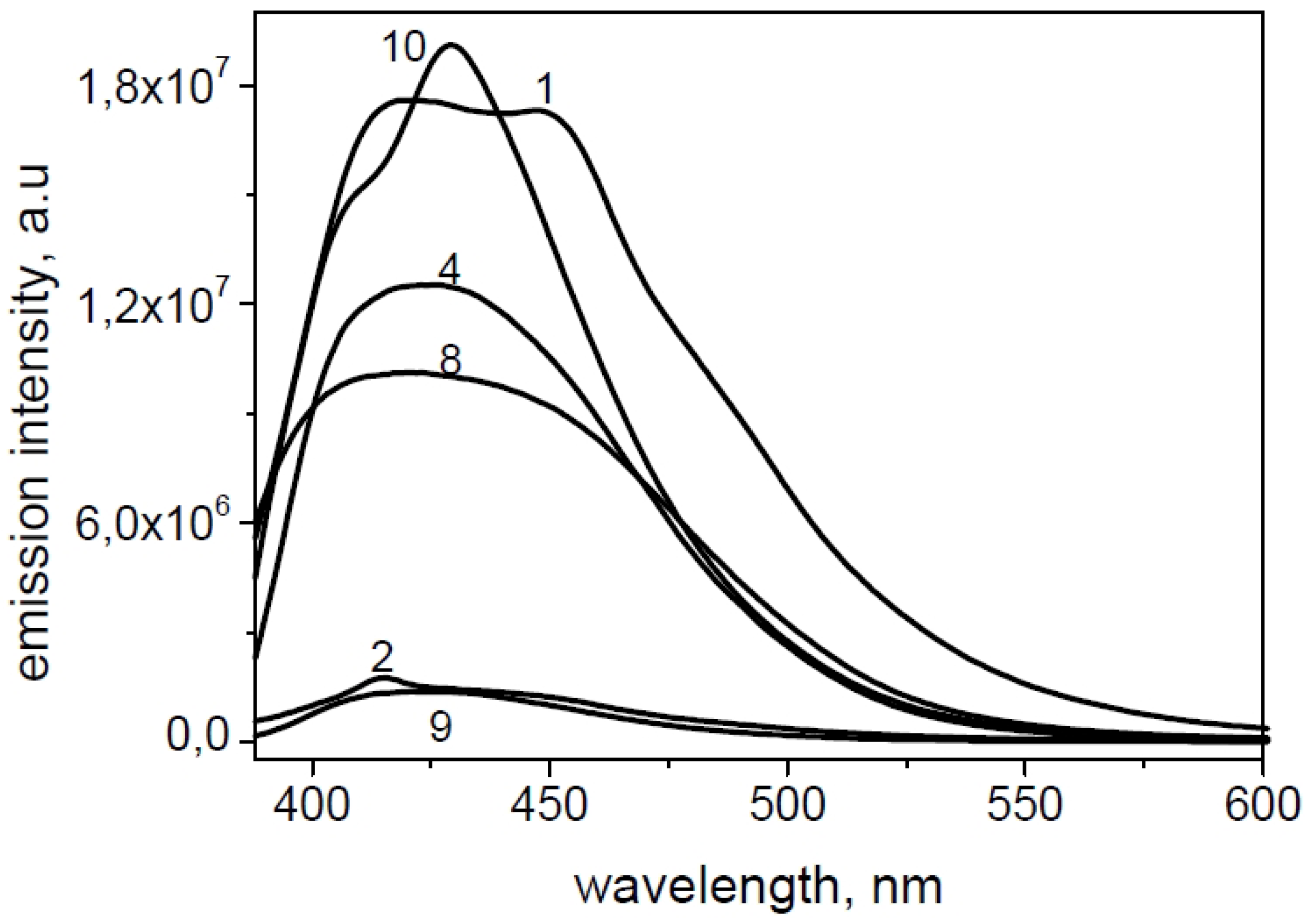
2.3. Fluorescence Microscopy

3. Experimental
3.1. General Information
3.2. Computational Method
3.3. Absorption and Emission Spectroscopy
3.4. Fluorescence Microscopy
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Santagati, A.; Modica, M.; Santagati, M. New synthetic approaches to bridgehead nitrogen heterocycles. Derived from 3-amino-2,3-dihydro-5,6-dimethyl-2-thioxothieno[2,3-d]pyrimidin-4(1H)-one and its salts. J. Heterocyclic. Chem. 2000, 37, 1161–1164. [Google Scholar] [CrossRef]
- Barone, M.; Graziano, A.C.E.; Marrazzo, A.; Gemmellaro, P.; Santagati, A.; Cardile, V. Synthesis and biological evaluation of new benzo-thieno[3,2-d]pyrimidin-4-one sulphonamide thio-derivatives as potential selective cyclooxygenase-2 inhibitors. Mol. Divers. 2013, 17, 445–458. [Google Scholar] [CrossRef]
- Sporn, M.B.; Roberts, A.B. Peptide growth factors and inflammation, tissue repair, and cancer. J. Clin. Invest. 1986, 78, 329–332. [Google Scholar] [CrossRef]
- Moneada, S.; Palmer, R.M.J.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar]
- Anggard, E. Nitric oxide: Mediator, murderer and medicine. Lancet 1994, 343, 1199–1206. [Google Scholar] [CrossRef]
- Nathan, C.F.; Xie, Q.W. Nitric oxide synthases: Roles, tolls, and controls. Cell 1994, 78, 915–918. [Google Scholar] [CrossRef]
- Seibert, K.; Masferrer, J. Role of inducible cyclooxygenase (COX-2) in inflammation. Receptor 1994, 94, 17–23. [Google Scholar]
- Sano, H.; Kawahito, Y.; Wilder, R.L.; Hashiramoto, A.; Mukai, S.; Asai, K.; Kimura, S.; Kalo, H.; Kondo, M.; Hla, T. Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Res. 1995, 55, 3785–3789. [Google Scholar]
- Eberhart, C.E.; DuBois, R.N. Eicosanoids and the gastrointestinal tract. Gastroenterology 1995, 109, 285–301. [Google Scholar]
- Misko, T.P.; Trotter, J.L.; Cross, A.H. Mediation of inflammation by encephalitogenic cells: Interferon gamma induction of nitric oxide synthase and cyclooxygenase 2. J. Neuroimmunol. 1995, 61, 195–204. [Google Scholar] [CrossRef]
- Thun, M.J.; Henkey, S.J.; Patrono, C.J. Nonsteroidal anti-inflammatory drugs as anticancer agents: Mechanistic, pharmacologic, and clinical issues. Natl. Cancer Inst. 2001, 94, 252–266. [Google Scholar] [CrossRef]
- Boland, G.P.; Butt, I.S.; Prasad, R.; Knox, W.F.; Bundred, N.J. COX-2 expression is associated with an aggressive phenotype in ductal carcinoma in situ. Br. J. Cancer 2001, 90, 423–429. [Google Scholar]
- Marnett, L.J.; DuBois, R.N. COX-2: A target for colon cancer prevention. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 55–80. [Google Scholar] [CrossRef]
- Wang, J.Y.; Chen, B.K.; Wang, Y.S.; Tsai, Y.T.; Chen, W.C.; Chang, W.C.; Hou, M.F.; Wu, Y.C.; Chang, W.C. Involvement of store-operated calcium signaling in EGF-mediated COX-2 gene activation in cancer cells. Cell. Signal. 2012, 24, 169–169. [Google Scholar]
- Guo, Y.C.; Chang, C.M.; Hsu, W.L.; Chiu, S.J.; Tsai, Y.T.; Chou, Y.H.; Hou, M.F.; Wang, J.Y.; Lee, M.H.; Tsai, K-L.; et al. Indomethacin inhibits cancer cell migration via attenuation of cellular calcium mobilization. Molecules 2013, 18, 6584–6596. [Google Scholar] [CrossRef]
- Wu, T.Y.; Yang, I.H.; Tsai, Y.T.; Wang, J.W.; Shiurba, R.; Hsieh, T.J.; Chang, F.R.; Chang, W.C. Isodesacetyluvaricin, an annonaceous acetogenin, specifically inhibits gene expression of cyclooxygenase-2. J. Nat. Prod. 2012, 75, 572–576. [Google Scholar] [CrossRef]
- Hsu, W.L.; Chiu, S.J.; Tsai, Y.T.; Chang, C.M.; Wang, J.Y.; Wang, E.T.; Hou, M.F.; Huang, C.Y.; Sheu, J.H.; Chang, W.C. A soft coral natural product, 11-episinulariolide acetate, inhibits gene expression of cyclooxygenase-2 and interleukin-8 through attenuation of calcium signaling. Molecules 2013, 18, 7023–7034. [Google Scholar] [CrossRef]
- Silverstein, F.E.; Faich, G.; Goldstein, J.L.; Simon, L.S.; Pincus, T.; Whelton, A.; Makuch, R.; Eisen, G.; Agrawal, N.M.; Stenson, W.F.; et al. Gastrointestinal toxicity with celecoxib vs. nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: The CLASS study: A randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. JAMA 2000, 284, 1247–1255. [Google Scholar] [CrossRef]
- Tsujii, M.; Kawano, S.; Tsuji, S.; Sawaoka, H.; Hori, M.; DuBois, R.N. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 1998, 93, 705–716. [Google Scholar] [CrossRef]
- Bhaedwaj, A.; Kaur, J.; Sharma, S.K.; Huang, Z.; Wuest, F.; Knaus, E.E. Hybrid fluorescent conjugates of COX-2 inhibitors: Search for a COX-2 isozyme imaging cancer biomarker. Biorg. Med. Chem. Lett. 2013, 23, 163–168. [Google Scholar]
- Bhardwaj, A.; Kaur, J.; Wuest, F.; Knaus, E.E. Fluorophore-labeled cyclooxygenase-2 inhibitors for the imaging of cyclooxygenase-2 overexpression in cancer: Synthesis and biological studies. Chem. Med. Chem. 2014, 9, 109–116. [Google Scholar] [CrossRef]
- Laneuville, O.; Breuer, D.K.; Dewitt, D.L.; Hla, T.; Funk, C.D.; Smith, W.L. Differential inhibition of human prostaglandin endoperoxide H synthases-1 and -2 by nonsteroidal anti-inflammatory drugs. J. Pharm. Exp. Ther. 1994, 271, 927–934. [Google Scholar]
- Picot, D.; Loll, P.J.; Garavito, R.M. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature 1994, 367, 243–249. [Google Scholar] [CrossRef]
- Kurumbail, R.G.; Stevens, A.M.; Gierse, J.K.; McDonald, J.J.; Stegeman, R.A.; Pak, J.Y.; Gildehaus, D.; Miyashiro, J.M.; Penning, T.D.; Seibert, K.; et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996, 384, 644–648. [Google Scholar]
- Kaur, J.; Bhardwaj, A.; Sharma, S.K.; Wuest, F. 1,4-Diaryl-substituted triazoles as cyclooxygenase-2 inhibitors: Synthesis, biological evaluation and molecular modeling studies. Bioorg. Med. Chem. 2013, 21, 4288–4295. [Google Scholar]
- Singh, P.; Sharma, P.; Bisetty, K.; Mahaian, M.P. Synthesis and docking studies of thiophene scaffolds in COX-2. Arkivoc 2011, 11, 55–70. [Google Scholar]
- Yoshikawa, K.; Koso, K.; Shimomura, M.; Tanaka, M.; Yamamoto, H.; Imagawa, H.; Arihara, S.; Hashimoto, T. Yellow pigments, fomitellanols A and B, and drimane sesquiterpenoids, cryptoporic acids p and q, from Fomitella fraxinea and their inhibitory activity against COX and 5-LO. Molecules 2013, 18, 4181–4191. [Google Scholar] [CrossRef]
- Schuttelkopf, A.W.; van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar]
- Oostenbrink, C.; Soares, T.A.; van der Vegt, N.F.; van Gunsteren, W.F. Validation of the 53A6 GROMOS force field. Eur. Biophys. J. 2005, 34, 273–284. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. AutomatedDocking Using a Lamarckian Genetic Algorithm and Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Duggan, K.C.; Walters, M.J.; Musee, J.; Harp, J.M.; Kiefer, J.R.; Oates, J.A.; Marnett, L.J. Molecular basis for cyclooxygenase inhibition by the non-steroidal anti-inflammatory drug naproxen. J. Biol. Chem. 2005, 285, 34950–34959. [Google Scholar]
- D’Mello, P.; Gadhwal, M.K.; Joshi, U.; Shetgiri, P. Modelling of COX-2 Inhibitory Activity of Flavonoids. Int. J. Pharm. Pharm. Sci. 2011, 4, 33–40. [Google Scholar]
- Bikadi, K.; Hazai, Z. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J. Chem. Inf. 2009, 1, 15–21. [Google Scholar]
- Meech, S.R.; Phillips, D. Photophysics of some common fluorescence standard. J. Photochem. 1983, 23, 193–217. [Google Scholar] [CrossRef]
- Demas, J.N.; Crosby, G.A. The measurement of photoluminescence quantum yields. J. Phys. Chem. 1971, 75, 991–1024. [Google Scholar] [CrossRef]
- Sample Availability: Samples of all compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Barone, M.; Pannuzzo, G.; Santagati, A.; Catalfo, A.; De Guidi, G.; Cardile, V. Molecular Docking and Fluorescence Characterization of Benzothieno[3,2-d]pyrimidin-4-one Sulphonamide Thio-Derivatives, a Novel Class of Selective Cyclooxygenase-2 Inhibitors. Molecules 2014, 19, 6106-6122. https://doi.org/10.3390/molecules19056106
Barone M, Pannuzzo G, Santagati A, Catalfo A, De Guidi G, Cardile V. Molecular Docking and Fluorescence Characterization of Benzothieno[3,2-d]pyrimidin-4-one Sulphonamide Thio-Derivatives, a Novel Class of Selective Cyclooxygenase-2 Inhibitors. Molecules. 2014; 19(5):6106-6122. https://doi.org/10.3390/molecules19056106
Chicago/Turabian StyleBarone, Mariarita, Giovanna Pannuzzo, Andrea Santagati, Alfio Catalfo, Guido De Guidi, and Venera Cardile. 2014. "Molecular Docking and Fluorescence Characterization of Benzothieno[3,2-d]pyrimidin-4-one Sulphonamide Thio-Derivatives, a Novel Class of Selective Cyclooxygenase-2 Inhibitors" Molecules 19, no. 5: 6106-6122. https://doi.org/10.3390/molecules19056106