Synthesis, Cytotoxicity and Mechanistic Evaluation of 4-Oxoquinoline-3-carboxamide Derivatives: Finding New Potential Anticancer Drugs

Abstract
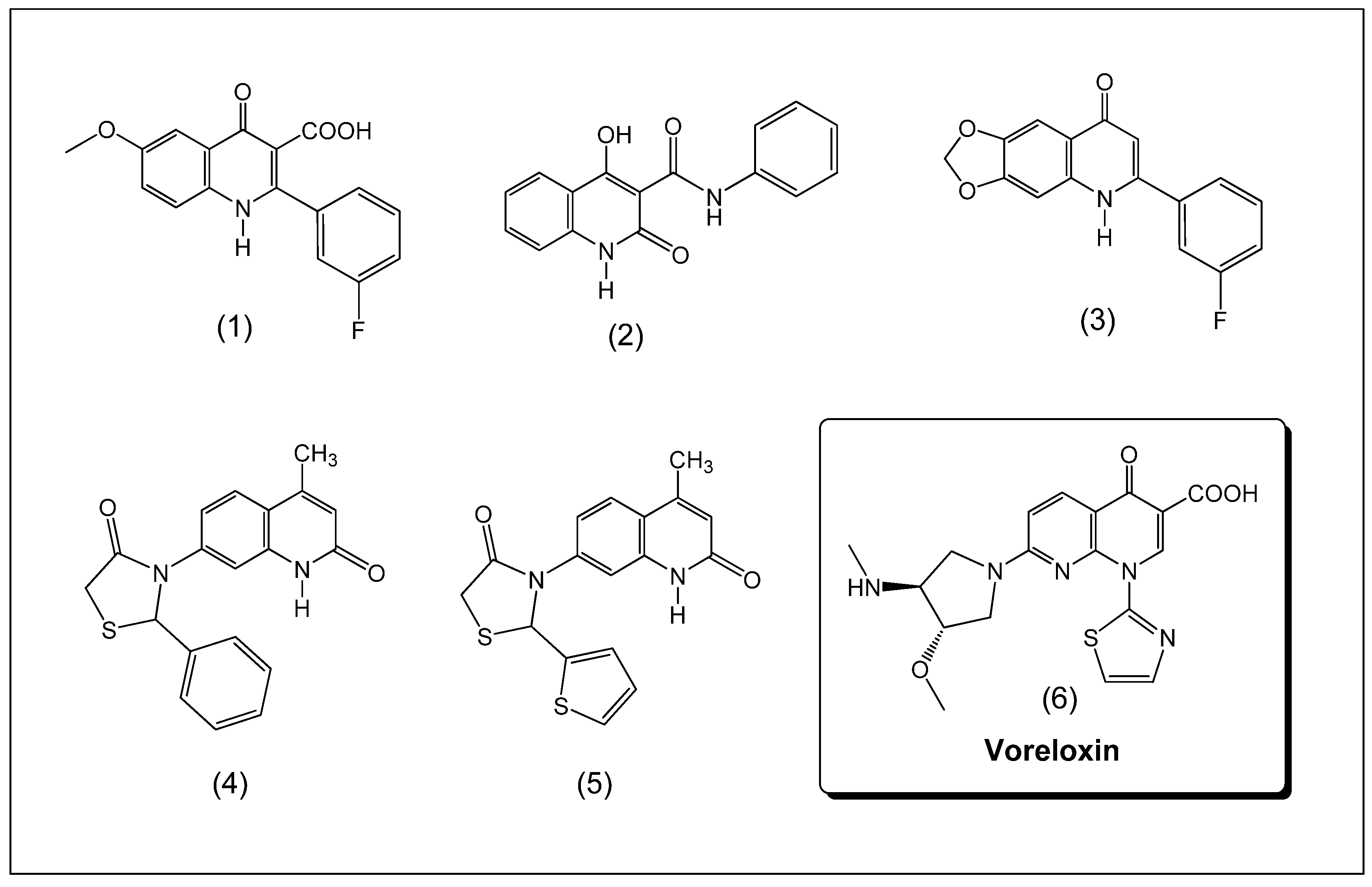
:1. Introduction
2. Results and Discussion
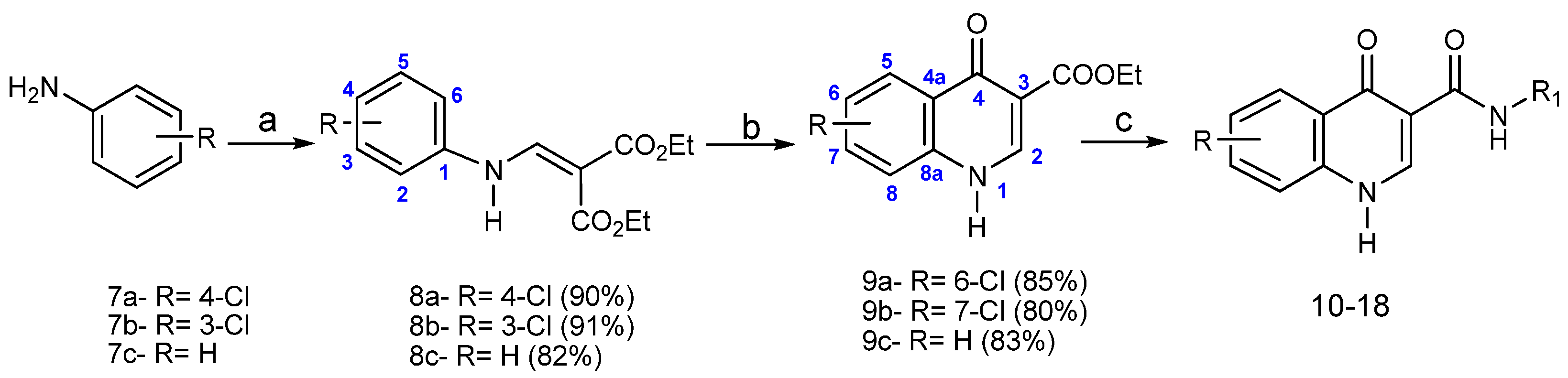
2.1. Chemistry


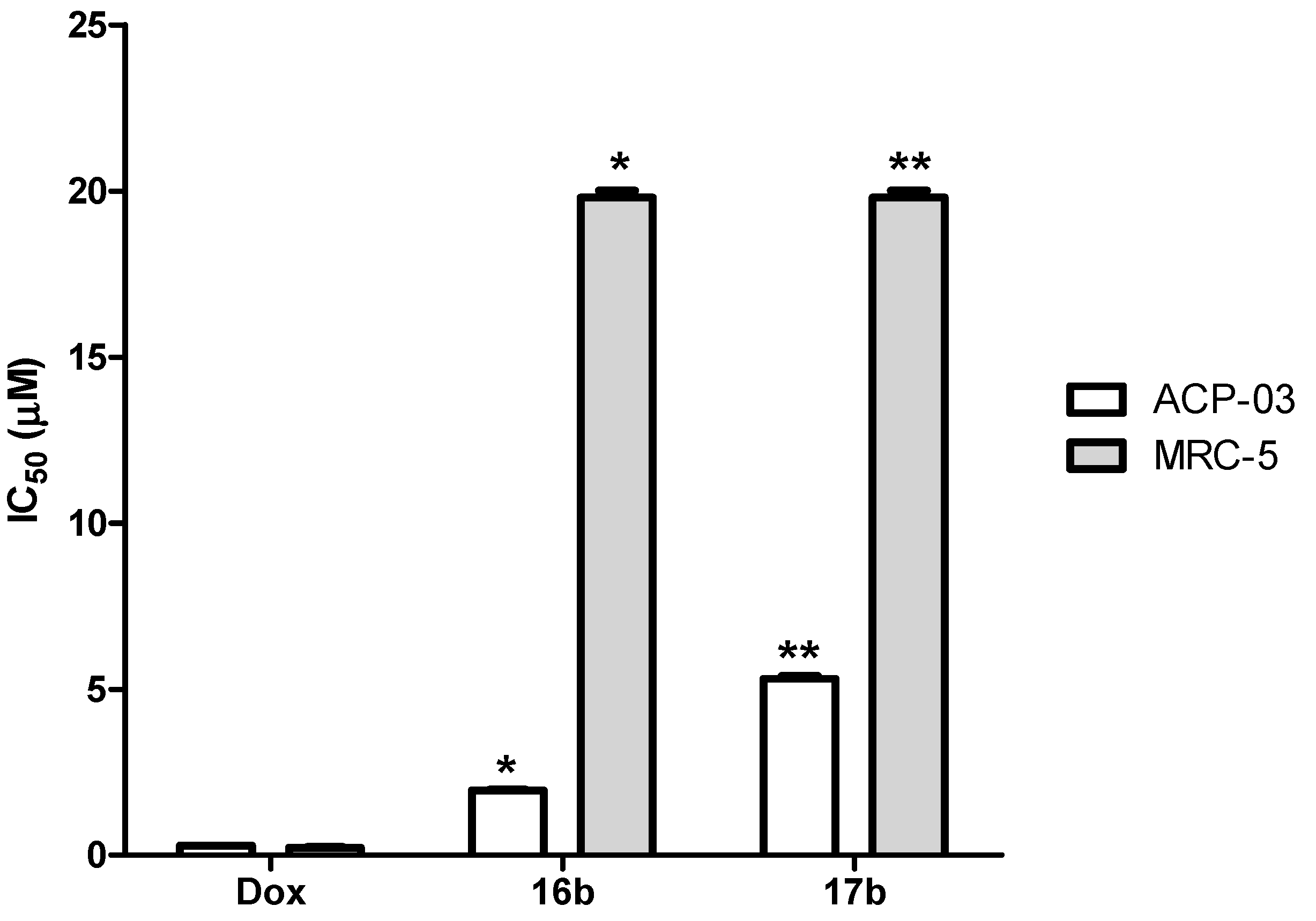
2.2. Evaluation of Anticancer Activity in Vitro

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Derivative | R | R1 | Yield (%) | MP (°C) |
|---|---|---|---|---|
| 10a | H | 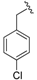 | 96 | 244–246 |
| 10b | 6-Cl | 85 | 262–263 | |
| 10c | 7-Cl | 79 | 220–221 | |
| 11a | H | 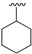 | 30 | 209–212 |
| 11b | 6-Cl | 58 | 140–142 | |
| 11c | 7-Cl | 75 | 140–141 | |
| 12a | H | 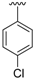 | 96 | >300 |
| 12b | 6-Cl | 98 | >300 | |
| 13a | H |  | 94 | >300 |
| 13b | 7-Cl | 80 | >300 | |
| 14a | H | 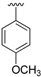 | 86 | >300 |
| 14b | 7-Cl | 87 | 270–273 | |
| 15a | H |  | 79 | >300 |
| 15b | 6-Cl | 85 | >300 | |
| 15c | 7-Cl | 83 | >300 | |
| 16a | H | 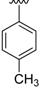 | 94 | >300 |
| 16b | 6-Cl | 96 | >300 | |
| 16c | 7-Cl | 86 | >300 | |
| 17a | H |  | 66 | >300 |
| 17b | 6-Cl | 98 | >300 | |
| 17c | 7-Cl | 63 | >300 | |
| 18a | H | 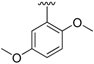 | 57 | 250–251 |
| 18b | 6-Cl | 44 | 256–258 | |
| 18c | 7-Cl | 45 | 261–262 |
| Derivatives | IC50 µM | Hemolysis (μg/mL) | |||
|---|---|---|---|---|---|
| ACP-03 | HCT-116 | MDAMB231 | MRC5 | ||
| 10a | >10 | >10 | >10 | ND | >200 |
| 10b | >10 | >10 | >10 | ND | >200 |
| 10c | >10 | >10 | >10 | ND | >200 |
| 11a | >10 | >10 | >10 | ND | >200 |
| 11b | >10 | >10 | >10 | ND | >200 |
| 11c | >10 | >10 | >10 | ND | >200 |
| 12a | >10 | >10 | >10 | ND | >200 |
| 12b | >10 | >10 | >10 | ND | >200 |
| 13a | >10 | >10 | >10 | ND | >200 |
| 13b | >10 | >10 | >10 | ND | >200 |
| 14a | >10 | >10 | >10 | ND | >200 |
| 14b | >10 | >10 | >10 | ND | >200 |
| 15a | >10 | >10 | >10 | ND | >200 |
| 15b | >10 | >10 | >10 | ND | >200 |
| 15c | >10 | >10 | >10 | ND | >200 |
| 16a | >10 | >10 | >10 | ND | >200 |
| 16b | 1.92 (1.39–2.66) | >10 | >10 | >20 | >200 |
| 16c | >10 | >10 | >10 | ND | >200 |
| 17a | >10 | >10 | >10 | ND | >200 |
| 17b | 5.18 (3.61–7.45) | >10 | >10 | >20 | >200 |
| 17c | >10 | >10 | >10 | ND | >200 |
| 18a | >10 | >10 | >10 | ND | >200 |
| 18b | >10 | >10 | >10 | ND | >200 |
| 18c | >10 | >10 | >10 | ND | >200 |
| DOXORUBICIN | 0.274 (0.22–0.33) | 0.1 (0.047–0.28) | 0.43 (0.36–0.52) | 0.2 (0.16–0.25) | >200 |
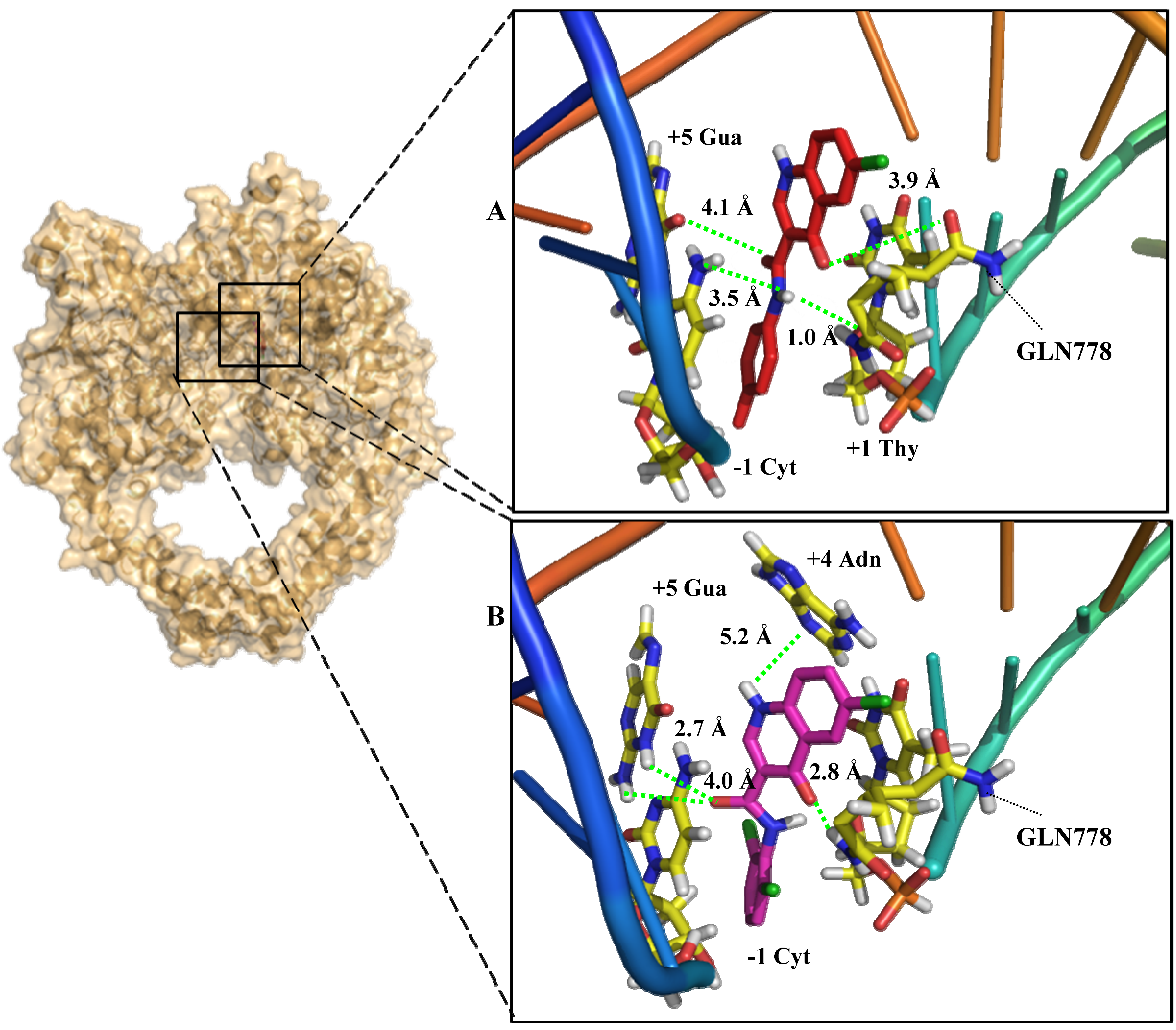
2.3. In Silico Mechanism Analysis: Topoisomerase II as a Feasible Target


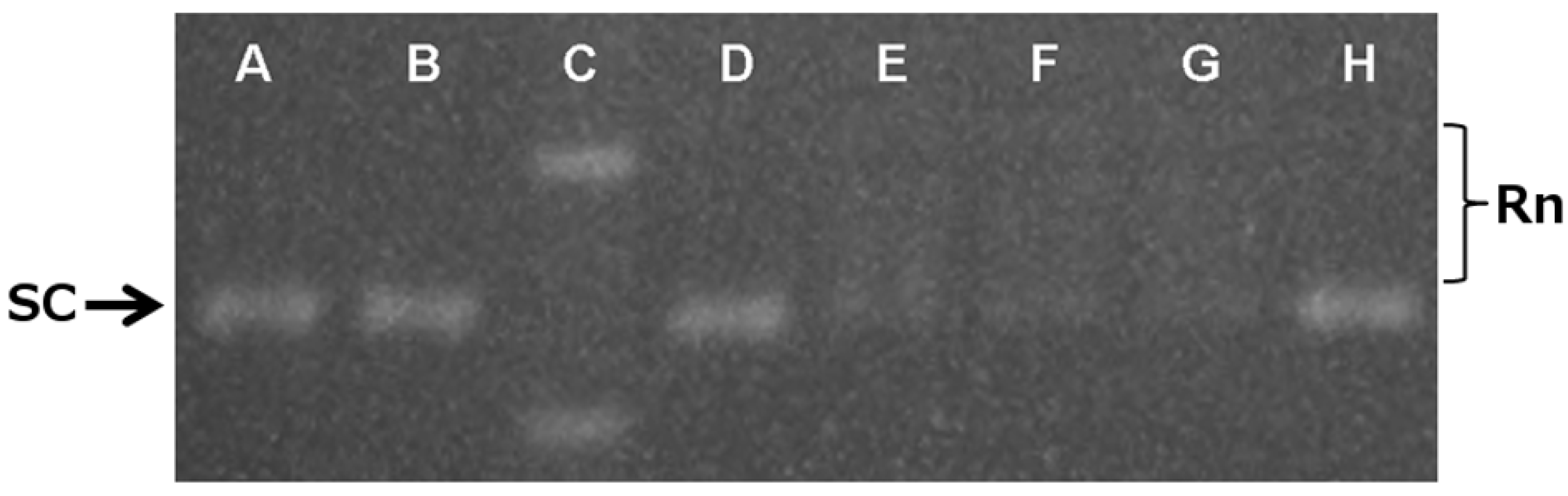
2.4. In Vitro Mechanistic Evaluation: Topoisomerase II as a Target


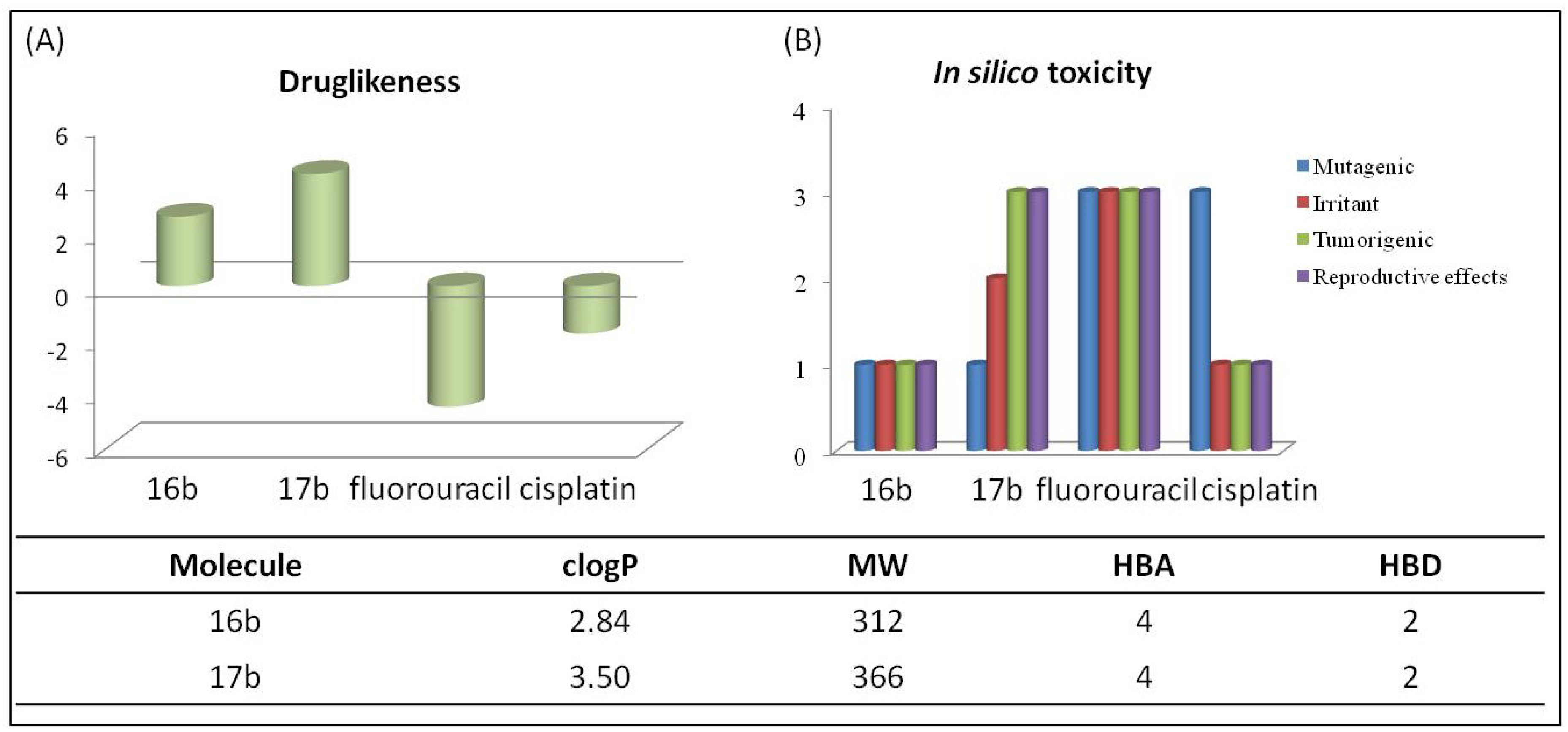
2.5. In Silico Pharmacokinetic Analysis
3. Experimental
3.1. General Information
3.2. Synthesis
3.2.1. General Procedure for the Synthesis of Anilinomethylenemalonates 8a–c
3.2.2. General Procedure for the Synthesis of Oxoquinolines 9a–c
3.2.3. General Procedure for the Synthesis of Oxoquinolines 10–18
3.3. Instrumental Parameters for HPLC
3.4. Molecular Docking Studies
3.5. DNA Relaxation Assay
3.6. In Silico Pharmacokinetics and Toxicity Analysis
3.7. Anticancer Assays
3.7.1. Cytotoxicity against Cancer Cell Lines
3.7.2. Cell Membrane Disruption
3.7.3. Analysis of the Results
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and notes
- Suthar, S.K.; Jaiswal, W.; Lohan, S.; Bansal, S.; Chaudhary, A.; Tiwari, A.; Alex, A.T.; Joesph, A. Novel quinolone substituted thiazolidin-4-ones as anti-inflammatory, anticancer agents: Design, synthesis and biological screening. Eur. J. Med. Chem. 2013, 63, 589–602. [Google Scholar] [CrossRef]
- Mitscher, L.A. Bacterial topoisomerase inhibitors: Quinolone and pyridone antibacterial agents. Chem. Rev. 2005, 105, 559–592. [Google Scholar] [CrossRef]
- Ahmed, A.; Daneshtalab, M. Nonclassical Biological Activities of Quinolone Derivatives. J. Pharm. Pharm. Sci. 2012, 15, 52–72. [Google Scholar]
- Mugnaini, C.; Pasquini, S.; Corelli, F. The 4-quinolone-3-carboxylic acid motif as a multivalent Scaffold in Medicinal Chemistry. Curr. Med. Chem. 2009, 16, 1746–1767. [Google Scholar] [CrossRef]
- Audisio, D.; Messaoudi, S.; Peyrat, J.F.; Brion, J.D.; Alami, M.J. A general copper powder-catalyzed ullmann-type reaction of 3-halo-4(1H)-quinolones with variousnitrogen-containingnucleophiles. J. Org. Chem. 2011, 76, 4995–5005. [Google Scholar] [CrossRef]
- Xia, Y.; Yang, Z.; Xia, P.; Hackl, T.; Hamel, E.; Mauger, A.; Wu, J.; Lee, K. Antitumor agents. 211. Fluorinated 2-phenyl-4-quinolone derivatives as antimitotic antitumor agents. J. Med. Chem. 2001, 44, 3932–3936. [Google Scholar] [CrossRef]
- Abbas, J.A.; Stuart, R.K. Vosaroxin: A novel antineoplasic quinolone. Expert Opin. Investig. Drugs 2012, 21, 1223–1233. [Google Scholar] [CrossRef]
- Advani, R.H.; Hurwitz, H.I.; Gordon, M.S.; Ebbinghaus, S.W.; Mendelson, D.S.; Wakelee, H.A. Voreloxin, a first-in-class anticancer quinolone derivative, in relapsed/refractory solid tumors: A report on two dosing schedules. Clin. Cancer Res. 2010, 16, 2167–2175. [Google Scholar] [CrossRef]
- Lai, Y.; Huang, L.; Lee, K.; Xiao, Z.; Bastow, K.F.; Yamiri, T.; Kuo, S. Synthesis and biological relationships of 3',6-substituted 2-phenyl-4-quinolone-3-carboxylic acid derivatives as antimitotic agents. Bioorg. Med. Chem. 2005, 13, 265–275. [Google Scholar] [CrossRef]
- Rajabalian, S.; Foroumadi, A.; Shafiee, A.; Emami, A. Functionalized N-(2-oxyiminoethyl) piperazinyl quinolones as new cytotoxic agents. J. Pharm. Pharm. Sci. 2007, 10, 153–158. [Google Scholar]
- Abe, H.; Kawada, M.; Inoue, H.; Ohba, S.; Nomoto, A.; Watanabe, T.; Shibasaki, M. Synthesis of intervenolin, an antitumor natural quinolone with unusual substituents. Org. Lett. 2013, 15, 2124–2127. [Google Scholar]
- Hawtin, R.E.; Stockett, D.E.; Byl, J.A.W.; Mcdowell, R.S.; Tan, N.; Arkin, M.R. Structural biology of human H3K9 methyltransferases. PLoS One 2010, 5, e8570. [Google Scholar]
- Foroumadi, A.; Emami, S.; Rajabalian, S.; Badinloo, M.; Mohammdhosseini, N.; Shafiee, A. N-Substituted piperazinyl quinolones as potential cytotoxic agents: Structure-activity relationships study. Biomed. Pharmacother. 2009, 63, 216–220. [Google Scholar] [CrossRef]
- Chen, Y.; Chung, C.; Chen, I.; Chen, P.; Jeng, H. Synthesis and cytotoxic activity evaluation of indolo-, pyrrolo-, and benzofuro-quinolin-2(1H)-ones and 6-anilinoindoloquinoline derivatives. Bioorg. Med. Chem. 2002, 10, 2705–2712. [Google Scholar] [CrossRef]
- Sabbah, D.; Simms, N.; Wang, W.; Dong, Y.; Ezell, E.L.; Brattain, M.G.; Vennerstrom, J.L.; Zhong, H.A. N-Phenyl-4-hydroxy-2-quinolone-3-carboxamides as selective inhibitors of mutant H1047R phosphoinositide-3-kinase (PI3Kα). Bioorg. Med. Chem. 2012, 20, 7175–7183. [Google Scholar] [CrossRef]
- Voreloxin is not in the market (but in clinical trial) and literature still ask about its ability of replacing the anthracycline drugs, requesting future investigation to confirm its efficacy. Altogether these data point doxorubicin as the best control drug to compare our active experimental profile against cancer cell lines see: Freeman, C.; Keane, N.; Swords, R.; Giles, F. Vosaroxin: A new valuable tool with the potential to replace anthracyclines in the treatment of AML? Expert Opi. Pharmacother. 2013, 14, 1417–1427. [Google Scholar] [CrossRef]
- Gould, R.G.; Jacobs, W.A. The Synthesis of Certain Substituted Quinolines and 5,6-Benzoquinolines. J. Am. Chem. Soc. 1939, 61, 2890–2895. [Google Scholar] [CrossRef]
- Snyder, H.R.; Freier, H.E.; Kovacic, P.; Heyningen, E.M.V. Synthesis of 4-hydroxyquinolines. VIII. Some halogen containing 4-aminoquinoline derivatives. J. Am. Chem. Soc. 1947, 69, 371–373. [Google Scholar] [CrossRef]
- Stern, E.; Muccioli, G.G.; Millet, R.; Goossens, J.-F.; Farce, A.; Chavatte, P.; Poupaert, J.H.; Lambert, D.M.; Depreux, P.; Hénichart, J.-P. Novel 4-oxo-1,4-dihydroquinoline-3-carboxamide derivatives as new CB2 cannabinoid receptors agonists: Synthesis, pharmacological properties, and molecular modeling. J. Med. Chem. 2006, 49, 70–79. [Google Scholar] [CrossRef]
- Riegel, B.; Lappin, G.R.; Adelson, B.H.; Jackson, R.I.; Albisetti C.J., Jr.; Dodson, R.M.; Baker, R.H. The synthesis of some 4-quinolinols and 4-chloroquinolines by the ethoxymethylenemalonic ester method. J. Am. Chem. Soc. 1946, 68, 1264–1266. [Google Scholar] [CrossRef]
- Matta, A.D.; Bernardino, A.M.R.; Romeiro, G.A.; Oliveira, M.R.P.; Souza, M.C.B.V.; Ferreira, V.F. Nucleosides having quinolone derivatives as nitrogenated base: Regiospecific and stereospecific ribosylation of 3-carbethoxy-1,4-dihydro-4-oxoquinolines. Nucleos. Nucleot. Nucleic Acids 1996, 15, 889–898. [Google Scholar] [CrossRef]
- Doxorubicin is one of the most important anticancer chemotherapeutic drug currently in use. Importantly, despite of its side effects, it is still in use and new formulations options are still being investigated due to its significant therapeutic profile, see: Deepa, K.; Singha, S.; Panda, T.J. doxorubicin nanoconjugates. Nanosci. Nanotechnol. 2014, 14, 892–904. [Google Scholar] [CrossRef]
- Wu, C.C.; Li, T.K.; Farh, L.; Lin, L.Y.; Lin, T.S.; Yu, Y.J.; Yen, T.J.; Chiang, C.W.; Chan, N.L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef]
- Esteves-Souza, A.; Rodrigues-Santos, C.E.; del Cistia, C.N.; Silva, D.R.; Sant’anna, C.M.R.; Echevarria, A. Solvent-free synthesis, DNA-topoisomerase II activity and molecular docking study of new asymmetrically N,N'-substituted ureas. Molecules 2012, 17, 12882–12894. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 4, 337–341. [Google Scholar]
- Gajbhiye, A.; Chaturvedi, L. Synthesis of 4-quinolones derivatives for their antihistaminic activity. Int. J. Pharm. Pharm. Sci. 2013, 5, 223–227. [Google Scholar]
- Shah, K.J.; Coats, E.A. Design, synthesis, and correlation analysis of 7-Substituted4-hydroxyquinoline-3-carboxylic acids as inhibitors of cellular respiration. J. Med. Chem. 1977, 20, 1001–1006. [Google Scholar] [CrossRef]
- Wathen, L.K.; Wathen, L.M. Method of Preventing or Treating Atherosclerosis or Restenosis. U.S. Patent WO2004019932 (A1), 11 March 2004. [Google Scholar]
- Sheth, U.; Fanning, T.T.D.; Numa, M.; Binch, H.; Hurley, D.J.; Zhou, J.; Hadida, R.S.S.; Hazlewood, A.R.; Silina, A.; Vairagoundar, R.; et al. 4-Oxo-1H-Quinoline-3-Carboxamides as Modulators of ATP-Binding Cassette Transporters. U.S. Patent WO2011072241 (A9), 16 June 2011. [Google Scholar]
- Singh, A.; van Goor, F.; Worley, L.J.F.; Jennings, F.; Knapp, T. Compounds Useful in Cftr Assays and Methods Therewith. U.S. Patent WO 2007075946 (A1), 5 July 2007. [Google Scholar]
- Verdonk, M.L.; Berdini, V.; Hartshorn, M.L.; Mooij, W.T.; Murray, C.W.; Taylor, R.D.; Watson, P. Virtual screening using protein-ligand docking: Avoiding artificial enrichment. J. Chem. Inf. Comput. Sci. 2004, 44, 793–806. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein–ligand docking using GOLD. Proteins: Struct. Funct. Genet. 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Sander, T. Actelion Pharmaceuticals Ltd. Available online: http://www.organic-chemistry.org/ (accessed on 13 May 2014).
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 10–18 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Forezi, L.D.S.M.; Tolentino, N.M.C.; De Souza, A.M.T.; Castro, H.C.; Montenegro, R.C.; Dantas, R.F.; Oliveira, M.E.I.M.; Silva, Jr., F.P.; Barreto, L.H.; Burbano, R.M.R.; et al. Synthesis, Cytotoxicity and Mechanistic Evaluation of 4-Oxoquinoline-3-carboxamide Derivatives: Finding New Potential Anticancer Drugs. Molecules 2014, 19, 6651-6670. https://doi.org/10.3390/molecules19056651
Forezi LDSM, Tolentino NMC, De Souza AMT, Castro HC, Montenegro RC, Dantas RF, Oliveira MEIM, Silva, Jr. FP, Barreto LH, Burbano RMR, et al. Synthesis, Cytotoxicity and Mechanistic Evaluation of 4-Oxoquinoline-3-carboxamide Derivatives: Finding New Potential Anticancer Drugs. Molecules. 2014; 19(5):6651-6670. https://doi.org/10.3390/molecules19056651
Chicago/Turabian StyleForezi, Luana Da S. M., Nathalia M. C. Tolentino, Alessandra M. T. De Souza, Helena C. Castro, Raquel C. Montenegro, Rafael F. Dantas, Maria E. I. M. Oliveira, Floriano P. Silva, Jr., Leilane H. Barreto, Rommel M. R. Burbano, and et al. 2014. "Synthesis, Cytotoxicity and Mechanistic Evaluation of 4-Oxoquinoline-3-carboxamide Derivatives: Finding New Potential Anticancer Drugs" Molecules 19, no. 5: 6651-6670. https://doi.org/10.3390/molecules19056651