Direct 2,3-O-Isopropylidenation of α-D-Mannopyranosides and the Preparation of 3,6-Branched Mannose Trisaccharides

Abstract
:
1. Introduction
2. Results and Discussion



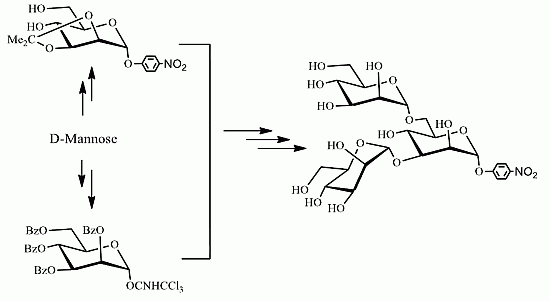{kind=link}
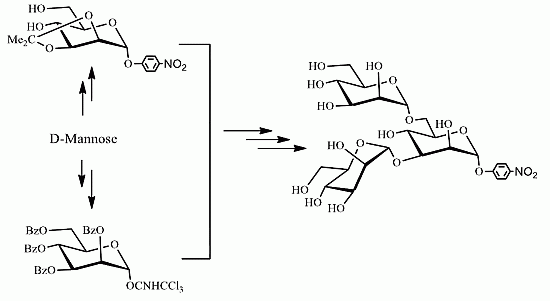{kind=link}
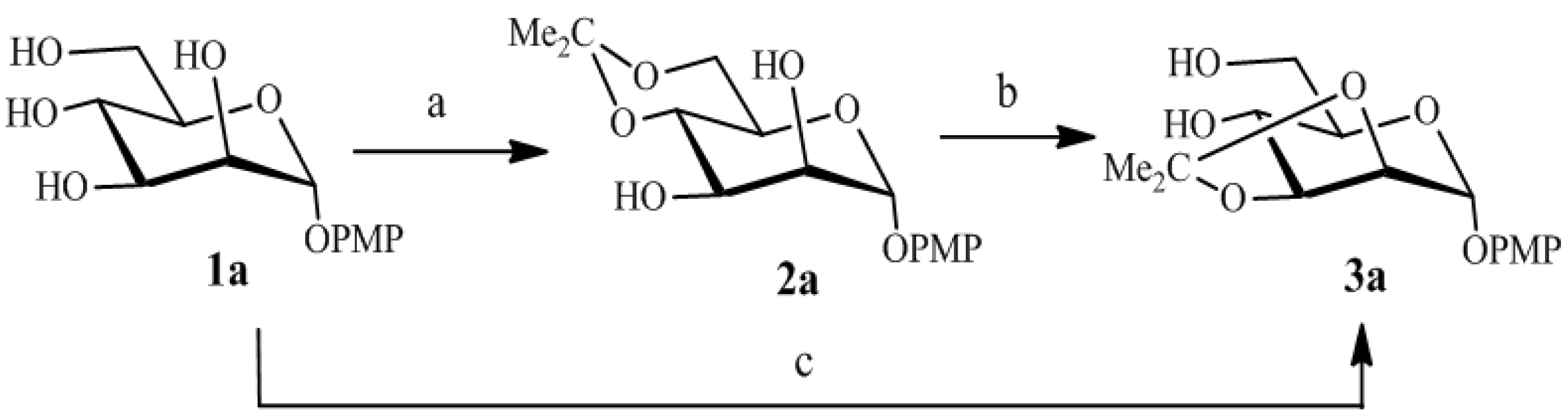{kind=link}
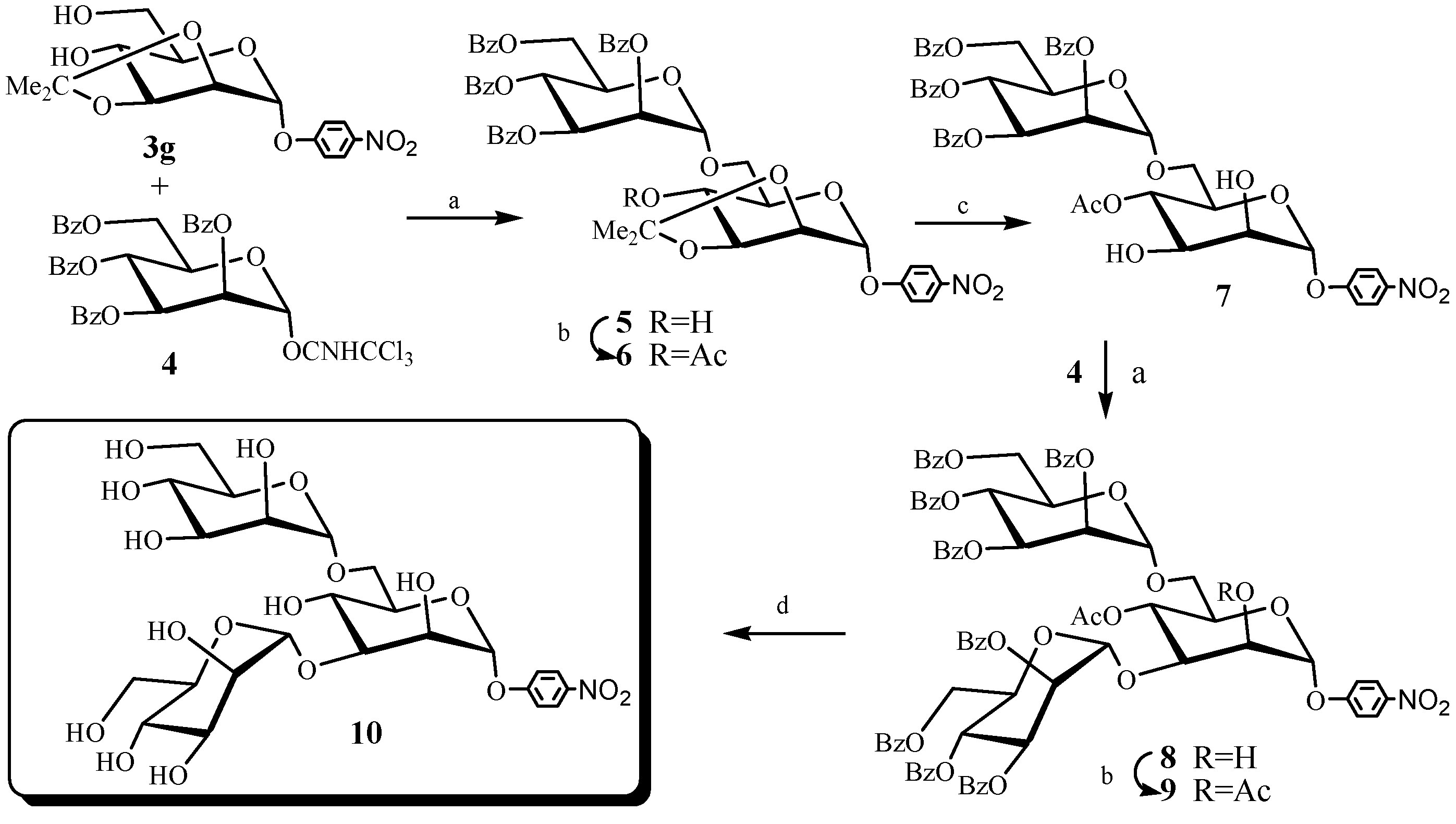{kind=link}
| Entry | Reaction | R | TsOH (equiv) | T(°C) | Time(h) | Yield (%) | Ref. * |
|---|---|---|---|---|---|---|---|
| 1 | 1a  3a 3a |  | 0.1 | 70 | 4 | 93 | [5] |
| 2 | 1b 3b |  | 0.3 | 50 | 1.5 | 89 | - |
| 3 | 1c 3c |  | 0.1 | 70 | 2 | 91 | [6] |
| 4 | 1d 3d |  | 0.3 | 70 | 3.5 | 92 | [7] |
| 5 | 1e 3e |  | 0.5 | 50 | 4.5 | 85 | [21] |
| 6 | 1f 3f |  | 0.1 | 70 | 2 | 88 | [22] |
| 7 | 1g 3g |  | 0.3 | 70 | 2 | 90 | [23] |

3. Experimental Section
3.1. General Methods
3.2. Chemical Synthesis: Representative Procedure for the Synthesis of 2,3-O-Isopropylidene-α-d-mannopyranosides 3a~3f
 +69.7° (c 1.0 CHCl3). 1H-NMR (300 MHz, CDCl3) δ 7.00–6.82 (2m, 4H, Ar-H ), 5.67 (s, 1H, H-1), 4.38 (d, J2,3 = 5.7 Hz, 1H, H-2), 4.32 (m, 1H, H-3), 3.84–3.78 (m, 7H, H-4, H-5, H-6, OCH3), 2.82 (d, J = 3.9 Hz, 1H, 4-OH), 2.04 (m, 1H, 6-OH), 1.56, 1.41 (2s, 6H, Me2C). 13C-NMR (75 MHz, CDCl3): δ 155.1, 149.7, 117.8, 114.6, 109.8, 96.4, 78.5, 75.6, 70.2, 69.0, 61.8, 55.5, 27.9, 26.1. HRMS for C16H26NO7 (M+NH4)+ 344.17038. Found: 344.17035. +128.7° (c 1.0 CHCl3). 1H-NMR (300 MHz, CDCl3) δ 5.63 (s, 1H, H-1), 4.18 (d, J2,3 = 5.5 Hz, 1H, H-2), 4.11 (m, 1H, H-3), 4.01 (m, 1H, H-5), 3.88–3.77 (m, 3H, H-4, H-6), 3.10 (m, 1H, SCH(CH3)2), 2.89 (d, J = 4.0 Hz, 1H, OH), 2.16 (m, 1H, OH), 1.54, 1.36 (2s, 6H, Me2C). 1.35 (d, J = 4.8 Hz, 3H, CH(CH3)2), 1.30 (d, J = 6.9 Hz, 3H, CH(CH3)2). HRMS for C12H26SNO5 (M+NH4)+ 296.15262. Found: 296.15259. +68.2° (c 1.0 CHCl3). 1H-NMR (300 MHz, CDCl3) δ 5.96–5.85 (m, 1H, OCH2CHCH2), 5.33–5.19 (m, 2H, CH2CHCH2O), 4.89 (d, J1,2 = 1.2 Hz, 1H, H-1), 4.21–3.68 (m, 8H, H-2, H-3, H-4, H-5, H-6, CH2CHCH2O), 2.63–2.57 (m, 2H, 2OH), 1.53,1.43 (2s, 6H, Me2C). HRMS for C12H21O6 (M+H)+ 261.13326. Found: 261.13321. +77.0° (c 1.0 CHCl3). 1H-NMR (300 MHz, CDCl3) δ 7.40–7.26 (m, 5H, ArH), 5.12 (s, 1H, H-1), 4.74 (d, J = 11.7 Hz, 1H, OCH2Ph), 4.54 (d, J = 11.7 Hz, 1H, OCH2Ph), 4.20–4.16 (m, 2H), 3.85–3.83 (m, 2H), 3.78–3.66 (m, 2H), 3.30–2.88 (s, 1H, OH), 2.35 (s, 1H, OH), 1.52, 1.34 (2s, 6H, Me2C). HRMS for C16H26NO6 (M+NH4)+ 328.17546. Found: 328.17548. +136.8° (c 1.0 CHCl3). 1H-NMR (300 MHz, CDCl3) δ 5.58 (s, 1H, H-1), 4.19 (m, 1H, H-2), 4.14 (m, 1H, H-3), 3.99–3.93 (m, 1H, H-5), 3.88–3.77 (m, 3H, H-4, H-6), 2.74–2.49 (m, 3H, SCH2, OH), 2.07 (t, J = 6.5 Hz, 1H, OH), 1.54 (s, 3H, Me2C), 1.40–1.25 (m, 6H, SCH2CH3, Me2C). HRMS for C11H24SNO5 (M+NH4)+ 282.13697. Found: 282.13696. +197.8° (c 1.0 CHCl3). 1H-NMR (300 MHz, CDCl3) δ 7.50–7.26 (m, 5H, ArH), 5.81 (s, 1H, H-1), 4.35 (d, J2,3 = 5.5 Hz, 1H, H-2), 4.18 (dd, J2,3 = 5.5 Hz, J3,4 = 7.5 Hz, 1H, H-3), 4.08–4.02 (m, 1H, H-5), 3.83–3.70 (m, H-4, H-6), 2.92 (d, J = 4.0 Hz, OH), 1.95 (t, 1H, J = 6.3 Hz, OH), 1.54,1.38 (2s, 6H, Me2C). HRMS for C15H24SNO5 (M+NH4)+ 330.13697. Found: 330.13699. +94.8° (c 1.0 CHCl3). 1H-NMR (300 MHz, CDCl3) δ ppm 8.25–8.20 (m, 2H, Ar-H), 7.18–7.13 (m, 2H, Ar-H), 5.90 (s, 1H, H-1), 4.38 (d, J2,3 = 5.7 Hz, 1H, H-2), 4.32 (dd, J2,3 = 5.7 Hz, J3,4 = 7.3 Hz, 1H, H-3), 3.88–3.78 (m, 3H, H-4, 2 × H-6), 3.67–3.62 (m, 1H, H-5), 2.96 (d, 1H, J = 4.0 Hz, OH), 2.05 (d, 1H, J = 3.6 Hz, OH), 1.58, 1.42 (2s, 6H, 2 × C-CH3). HRMS for C15H23NO8 (M+NH4)+ 359.1449. Found: 359.14481. −62.6° (c 0.5 CHCl3). 1H-NMR (300 MHz, CDCl3): δ 8.31–7.23 (m, 24H, Bz-H, Ar-H), 6.07 (t, J3,4 = J4,5 = 10.0 Hz, 1H, H-4'), 5.94 (s, 1H, H-1'), 5.74 (dd, J2,3 = 3.3 Hz, J3,4 = 10.0 Hz, 1H, H-3'), 5.56 (dd, J1,2 = 1.7 Hz, J2,3 = 3.3 Hz, 1H, H-2'), 5.10 (d, 1H, J1,2 = 1.7 Hz, H-1), 4.74–4.68 (m, 1H), 4.50–4.41 (m, 3H), 4.34 (m, 1H, H-4), 4.01 (m, 1H), 3.91–3.82 (m, 3H), 2.68 (s, 1H, OH), 1.60, 1.43 (2s, 6H, 2 × C-CH3). 13C-NMR (75 MHz, CDCl3): δ 166.2, 165.6, 165.2(2) (4 × COPh), 160.6 (CNO2), 142.9, 133.4, 133.1, 133.0, 129.8, 129.7, 129.6, 129.2, 129.0, 128.9, 128.5, 128.4, 128.3, 128.2, 126.0, 116.2, 110.3, 97.6, 95.7 (2 × C-1), 78.5, 75.3, 70.3, 70.0, 69.6, 69.2, 68.9, 67.1, 66.7, 62.9, 28.0, 26.3 (2 × C-CH3). HRMS for C49H49N2O17 (M+NH4)+ 937.30257. Found: 937.30133. −53.3° (c 0.3 CHCl3). 1H-NMR (300 MHz, CDCl3): δ 8.29 (d, J = 9.1 Hz, 2H, Bz-H), 8.10, 7.80 (2d, J = 7.4 Hz, 4H, C6H4NO2 ), 7.58–7.24 (m, 14H, Bz-H ), 6.05 (t, J3,4 = J4,5 = 10.0 Hz, 1H, H-4'), 5.95 (s, 1H, H-1'), 5.65 (dd, J2,3 = 3.3 Hz, J3,4 = 10.0 Hz, 1H, H-3'), 5.46 (m, 1H, H-2'), 5.10 (t, J3,4 = J4,5 = 10.3 Hz, 1H, H-4), 5.01 (s, 1H, H-1), 4.73 (d, J3,4 = 10.3 Hz, 1H, H-3), 4.49–4.40 (m, 4H), 4.05–3.88 (m, 2H), 3.53 (m, 1H), 2.21 (s, 3H, CH3CO), 1.60, 1.42 (2s, 6H, 2 × C-CH3). 13C-NMR (75 MHz, CDCl3): δ 169.7 (COCH3), 166.1, 165.6, 165.2, 165.1 (4 × COPh), 160.4(CNO2), 143.1, 133.4, 133.1, 133.0, 129.8, 129.7, 129.6, 129.1, 129.0, 128.9, 128.5, 128.4, 128.2, 126.0, 116.1, 110.6, 97.3, 95.4 (2 × C-1), 77.1, 75.4, 75.1, 70.2, 69.5, 69.3, 69.0, 68.3, 66.9, 66.8, 62.9, 27.5, 26.3(2 × C-CH3), 20.8 (COCH3). HRMS for C51H51N2O18 (M+NH4)+ 979.31314. Found: 979.31201. –14.4° (c 1.0 CHCl3). 1H-NMR (300 MHz, CDCl3): δ 8.31-7.23 (m, 24H, Bz-H, Ar-H), 6.05 (t, J3,4 = J4,5 = 10.1Hz, 1H, H-4'), 5.81 (d, J2,3= 3.2 Hz, J3,4 = 10.1 Hz, 1H, H-3'), 5.77 (d, J1,2 = 1.6 Hz, 1H, H-1'), 5.65 (dd, J1,2 = 1.6 Hz, J2,3 = 3.2 Hz, 1H, H-2'), 5.32 (t, J3,4 = J4,5 =9.6 Hz, 1H, H-4), 5.13 (d, J1,2 = 1.6 Hz, 1H, H-1'), 4.73–4.69 (m, 1H, H-3), 4.50–4.43 (m, 2H), 4.27–4.20 (m, 2H), 3.99–3.91 (m, 2H), 3.64 (d, 1H, J 9.5 Hz, OH), 3.36–3.30 (m, 2H, H-6, OH), 2.17 (s, 3H, COCH3). 13C-NMR (75 MHz, CDCl3): δ 171.3 (COCH3), 166.1, 165.6, 165.5, 165.4 (4 × COPh), 160.6(CNO2), 142.9, 133.5, 133.4, 133.2, 133.1, 129.8, 129.7, 129.6, 129.1, 129.0, 128.8, 128.6, 128.5, 128.4, 128.2, 126.0, 116.2, 97.73, 97.70 (2 × C-1), 77.2, 70.4, 70.3, 70.1, 69.9, 69.8, 69.3, 69.1, 66.8, 66.6, 62.8, 20.8 (COCH3). HRMS for C48H47N2O18 (M+NH4)+ 939.28184. Found: 939.28088. –27.2° (c 0.3 CHCl3). 1H-NMR (300 MHz, CDCl3): δ 8.30–7.16 (m, 44H, Bz-H, Ar-H), 6.09 (t, J3,4 = J4,5 = 10.1 Hz, 2H, H-4', H-4''), 5.93 (dd, J2,3 = 3.3 Hz, J3,4 = 10.1 Hz, 1H, H-3'), 5.80 (dd, J2,3 = 3.3 Hz, J3,4 = 10.1 Hz, 1H, H-3''), 5.63–5.51 (m, 4H, H-2', H-2'', H-1', H-4), 5.42 (s, 1H, H-1''), 5.10 (d, J1,2 = 1.6 Hz, 1H, H-1), 4.77–4.63 (m, 4H), 4.52–4.44 (m, 3H), 4.35 (dd, J2,3 = 3.1 Hz, J3,4 = 9.5 Hz, 1H, H-3), 4.0 (d, J = 8.8 Hz, 2H, 2H-6), 3.61 (d, J = 8.8 Hz, 1H, H-6), 3.06 (d, J = 4.9 Hz, 1H, OH), 2.27 (s, 3H, COCH3). 13C NMR (75 MHz, CDCl3): δ 170.3 (COCH3), 166.2, 166.1, 165.6(2), 165.4, 165.3(2), 165.0 (8 × COPh), 160.3 (CNO2), 143.0, 133.6, 133.4, 133.3, 133.0, 129.8, 129.75, 129.7, 129.6, 129.56, 129.50, 129.1, 129.0, 128.99, 128.91, 128.8, 128.7, 128.6, 128.5, 128.4, 128.3, 128.28, 128.22, 128.1, 125.9, 116.2, 99.6, 97.6, 97.5 (3 × C-1), 79.0, 77.2, 70.8, 70.6, 70.3, 69.8, 69.7, 69.6, 69.4, 69.0, 67.1, 67.0, 66.7, 63.4, 62.8, 20.7 (COCH3). HRMS for C82H69NO27Na (M+Na)+ 1522.39508. Found: 1522.39463. +54.2° (c 0.1 DMSO). 1H-NMR (300 MHz, D2O): δ 8.20 (d, J = 9.2 Hz, 2H, Ar-H), 7.20 (d, J = 9.2 Hz, 2H, Ar-H), 5.69 (s, 1H, H-1), 5.13 (s, 1H, H-1'), 4.30 (m, 1H, H-2), 4.11 (dd, J2,3 = 3.2 Hz, J3,4 = 9.3 Hz, 1H, H-3), 4.04 (m, 1H, H-2'), 3.90–3.80 (m, 5H), 3.78–3.74 (m, 3H), 3.68 (m, 2H), 3.64–3.61 (m, 2H), 3.57–3.54 (m, 2H). 13C-NMR (75 MHz, D2O): δ 160.6 (CNO2), 142.4, 126.1(2), 116.9(2) (4 × Ar-C), 102.4, 98.9, 97.6 (3 × C-1), 77.9, 73.2, 72.6, 71.9, 70.6, 70.4, 70.1, 69.9, 69.2, 66.8, 66.7, 65.8. 65.2, 61.0, 60.9. HRMS for C24H35NO18Na (M+Na)+ 648.1752. Found: 648.1754.
+69.7° (c 1.0 CHCl3). 1H-NMR (300 MHz, CDCl3) δ 7.00–6.82 (2m, 4H, Ar-H ), 5.67 (s, 1H, H-1), 4.38 (d, J2,3 = 5.7 Hz, 1H, H-2), 4.32 (m, 1H, H-3), 3.84–3.78 (m, 7H, H-4, H-5, H-6, OCH3), 2.82 (d, J = 3.9 Hz, 1H, 4-OH), 2.04 (m, 1H, 6-OH), 1.56, 1.41 (2s, 6H, Me2C). 13C-NMR (75 MHz, CDCl3): δ 155.1, 149.7, 117.8, 114.6, 109.8, 96.4, 78.5, 75.6, 70.2, 69.0, 61.8, 55.5, 27.9, 26.1. HRMS for C16H26NO7 (M+NH4)+ 344.17038. Found: 344.17035. +128.7° (c 1.0 CHCl3). 1H-NMR (300 MHz, CDCl3) δ 5.63 (s, 1H, H-1), 4.18 (d, J2,3 = 5.5 Hz, 1H, H-2), 4.11 (m, 1H, H-3), 4.01 (m, 1H, H-5), 3.88–3.77 (m, 3H, H-4, H-6), 3.10 (m, 1H, SCH(CH3)2), 2.89 (d, J = 4.0 Hz, 1H, OH), 2.16 (m, 1H, OH), 1.54, 1.36 (2s, 6H, Me2C). 1.35 (d, J = 4.8 Hz, 3H, CH(CH3)2), 1.30 (d, J = 6.9 Hz, 3H, CH(CH3)2). HRMS for C12H26SNO5 (M+NH4)+ 296.15262. Found: 296.15259. +68.2° (c 1.0 CHCl3). 1H-NMR (300 MHz, CDCl3) δ 5.96–5.85 (m, 1H, OCH2CHCH2), 5.33–5.19 (m, 2H, CH2CHCH2O), 4.89 (d, J1,2 = 1.2 Hz, 1H, H-1), 4.21–3.68 (m, 8H, H-2, H-3, H-4, H-5, H-6, CH2CHCH2O), 2.63–2.57 (m, 2H, 2OH), 1.53,1.43 (2s, 6H, Me2C). HRMS for C12H21O6 (M+H)+ 261.13326. Found: 261.13321. +77.0° (c 1.0 CHCl3). 1H-NMR (300 MHz, CDCl3) δ 7.40–7.26 (m, 5H, ArH), 5.12 (s, 1H, H-1), 4.74 (d, J = 11.7 Hz, 1H, OCH2Ph), 4.54 (d, J = 11.7 Hz, 1H, OCH2Ph), 4.20–4.16 (m, 2H), 3.85–3.83 (m, 2H), 3.78–3.66 (m, 2H), 3.30–2.88 (s, 1H, OH), 2.35 (s, 1H, OH), 1.52, 1.34 (2s, 6H, Me2C). HRMS for C16H26NO6 (M+NH4)+ 328.17546. Found: 328.17548. +136.8° (c 1.0 CHCl3). 1H-NMR (300 MHz, CDCl3) δ 5.58 (s, 1H, H-1), 4.19 (m, 1H, H-2), 4.14 (m, 1H, H-3), 3.99–3.93 (m, 1H, H-5), 3.88–3.77 (m, 3H, H-4, H-6), 2.74–2.49 (m, 3H, SCH2, OH), 2.07 (t, J = 6.5 Hz, 1H, OH), 1.54 (s, 3H, Me2C), 1.40–1.25 (m, 6H, SCH2CH3, Me2C). HRMS for C11H24SNO5 (M+NH4)+ 282.13697. Found: 282.13696. +197.8° (c 1.0 CHCl3). 1H-NMR (300 MHz, CDCl3) δ 7.50–7.26 (m, 5H, ArH), 5.81 (s, 1H, H-1), 4.35 (d, J2,3 = 5.5 Hz, 1H, H-2), 4.18 (dd, J2,3 = 5.5 Hz, J3,4 = 7.5 Hz, 1H, H-3), 4.08–4.02 (m, 1H, H-5), 3.83–3.70 (m, H-4, H-6), 2.92 (d, J = 4.0 Hz, OH), 1.95 (t, 1H, J = 6.3 Hz, OH), 1.54,1.38 (2s, 6H, Me2C). HRMS for C15H24SNO5 (M+NH4)+ 330.13697. Found: 330.13699. +94.8° (c 1.0 CHCl3). 1H-NMR (300 MHz, CDCl3) δ ppm 8.25–8.20 (m, 2H, Ar-H), 7.18–7.13 (m, 2H, Ar-H), 5.90 (s, 1H, H-1), 4.38 (d, J2,3 = 5.7 Hz, 1H, H-2), 4.32 (dd, J2,3 = 5.7 Hz, J3,4 = 7.3 Hz, 1H, H-3), 3.88–3.78 (m, 3H, H-4, 2 × H-6), 3.67–3.62 (m, 1H, H-5), 2.96 (d, 1H, J = 4.0 Hz, OH), 2.05 (d, 1H, J = 3.6 Hz, OH), 1.58, 1.42 (2s, 6H, 2 × C-CH3). HRMS for C15H23NO8 (M+NH4)+ 359.1449. Found: 359.14481. −62.6° (c 0.5 CHCl3). 1H-NMR (300 MHz, CDCl3): δ 8.31–7.23 (m, 24H, Bz-H, Ar-H), 6.07 (t, J3,4 = J4,5 = 10.0 Hz, 1H, H-4'), 5.94 (s, 1H, H-1'), 5.74 (dd, J2,3 = 3.3 Hz, J3,4 = 10.0 Hz, 1H, H-3'), 5.56 (dd, J1,2 = 1.7 Hz, J2,3 = 3.3 Hz, 1H, H-2'), 5.10 (d, 1H, J1,2 = 1.7 Hz, H-1), 4.74–4.68 (m, 1H), 4.50–4.41 (m, 3H), 4.34 (m, 1H, H-4), 4.01 (m, 1H), 3.91–3.82 (m, 3H), 2.68 (s, 1H, OH), 1.60, 1.43 (2s, 6H, 2 × C-CH3). 13C-NMR (75 MHz, CDCl3): δ 166.2, 165.6, 165.2(2) (4 × COPh), 160.6 (CNO2), 142.9, 133.4, 133.1, 133.0, 129.8, 129.7, 129.6, 129.2, 129.0, 128.9, 128.5, 128.4, 128.3, 128.2, 126.0, 116.2, 110.3, 97.6, 95.7 (2 × C-1), 78.5, 75.3, 70.3, 70.0, 69.6, 69.2, 68.9, 67.1, 66.7, 62.9, 28.0, 26.3 (2 × C-CH3). HRMS for C49H49N2O17 (M+NH4)+ 937.30257. Found: 937.30133. −53.3° (c 0.3 CHCl3). 1H-NMR (300 MHz, CDCl3): δ 8.29 (d, J = 9.1 Hz, 2H, Bz-H), 8.10, 7.80 (2d, J = 7.4 Hz, 4H, C6H4NO2 ), 7.58–7.24 (m, 14H, Bz-H ), 6.05 (t, J3,4 = J4,5 = 10.0 Hz, 1H, H-4'), 5.95 (s, 1H, H-1'), 5.65 (dd, J2,3 = 3.3 Hz, J3,4 = 10.0 Hz, 1H, H-3'), 5.46 (m, 1H, H-2'), 5.10 (t, J3,4 = J4,5 = 10.3 Hz, 1H, H-4), 5.01 (s, 1H, H-1), 4.73 (d, J3,4 = 10.3 Hz, 1H, H-3), 4.49–4.40 (m, 4H), 4.05–3.88 (m, 2H), 3.53 (m, 1H), 2.21 (s, 3H, CH3CO), 1.60, 1.42 (2s, 6H, 2 × C-CH3). 13C-NMR (75 MHz, CDCl3): δ 169.7 (COCH3), 166.1, 165.6, 165.2, 165.1 (4 × COPh), 160.4(CNO2), 143.1, 133.4, 133.1, 133.0, 129.8, 129.7, 129.6, 129.1, 129.0, 128.9, 128.5, 128.4, 128.2, 126.0, 116.1, 110.6, 97.3, 95.4 (2 × C-1), 77.1, 75.4, 75.1, 70.2, 69.5, 69.3, 69.0, 68.3, 66.9, 66.8, 62.9, 27.5, 26.3(2 × C-CH3), 20.8 (COCH3). HRMS for C51H51N2O18 (M+NH4)+ 979.31314. Found: 979.31201. –14.4° (c 1.0 CHCl3). 1H-NMR (300 MHz, CDCl3): δ 8.31-7.23 (m, 24H, Bz-H, Ar-H), 6.05 (t, J3,4 = J4,5 = 10.1Hz, 1H, H-4'), 5.81 (d, J2,3= 3.2 Hz, J3,4 = 10.1 Hz, 1H, H-3'), 5.77 (d, J1,2 = 1.6 Hz, 1H, H-1'), 5.65 (dd, J1,2 = 1.6 Hz, J2,3 = 3.2 Hz, 1H, H-2'), 5.32 (t, J3,4 = J4,5 =9.6 Hz, 1H, H-4), 5.13 (d, J1,2 = 1.6 Hz, 1H, H-1'), 4.73–4.69 (m, 1H, H-3), 4.50–4.43 (m, 2H), 4.27–4.20 (m, 2H), 3.99–3.91 (m, 2H), 3.64 (d, 1H, J 9.5 Hz, OH), 3.36–3.30 (m, 2H, H-6, OH), 2.17 (s, 3H, COCH3). 13C-NMR (75 MHz, CDCl3): δ 171.3 (COCH3), 166.1, 165.6, 165.5, 165.4 (4 × COPh), 160.6(CNO2), 142.9, 133.5, 133.4, 133.2, 133.1, 129.8, 129.7, 129.6, 129.1, 129.0, 128.8, 128.6, 128.5, 128.4, 128.2, 126.0, 116.2, 97.73, 97.70 (2 × C-1), 77.2, 70.4, 70.3, 70.1, 69.9, 69.8, 69.3, 69.1, 66.8, 66.6, 62.8, 20.8 (COCH3). HRMS for C48H47N2O18 (M+NH4)+ 939.28184. Found: 939.28088. –27.2° (c 0.3 CHCl3). 1H-NMR (300 MHz, CDCl3): δ 8.30–7.16 (m, 44H, Bz-H, Ar-H), 6.09 (t, J3,4 = J4,5 = 10.1 Hz, 2H, H-4', H-4''), 5.93 (dd, J2,3 = 3.3 Hz, J3,4 = 10.1 Hz, 1H, H-3'), 5.80 (dd, J2,3 = 3.3 Hz, J3,4 = 10.1 Hz, 1H, H-3''), 5.63–5.51 (m, 4H, H-2', H-2'', H-1', H-4), 5.42 (s, 1H, H-1''), 5.10 (d, J1,2 = 1.6 Hz, 1H, H-1), 4.77–4.63 (m, 4H), 4.52–4.44 (m, 3H), 4.35 (dd, J2,3 = 3.1 Hz, J3,4 = 9.5 Hz, 1H, H-3), 4.0 (d, J = 8.8 Hz, 2H, 2H-6), 3.61 (d, J = 8.8 Hz, 1H, H-6), 3.06 (d, J = 4.9 Hz, 1H, OH), 2.27 (s, 3H, COCH3). 13C NMR (75 MHz, CDCl3): δ 170.3 (COCH3), 166.2, 166.1, 165.6(2), 165.4, 165.3(2), 165.0 (8 × COPh), 160.3 (CNO2), 143.0, 133.6, 133.4, 133.3, 133.0, 129.8, 129.75, 129.7, 129.6, 129.56, 129.50, 129.1, 129.0, 128.99, 128.91, 128.8, 128.7, 128.6, 128.5, 128.4, 128.3, 128.28, 128.22, 128.1, 125.9, 116.2, 99.6, 97.6, 97.5 (3 × C-1), 79.0, 77.2, 70.8, 70.6, 70.3, 69.8, 69.7, 69.6, 69.4, 69.0, 67.1, 67.0, 66.7, 63.4, 62.8, 20.7 (COCH3). HRMS for C82H69NO27Na (M+Na)+ 1522.39508. Found: 1522.39463. +54.2° (c 0.1 DMSO). 1H-NMR (300 MHz, D2O): δ 8.20 (d, J = 9.2 Hz, 2H, Ar-H), 7.20 (d, J = 9.2 Hz, 2H, Ar-H), 5.69 (s, 1H, H-1), 5.13 (s, 1H, H-1'), 4.30 (m, 1H, H-2), 4.11 (dd, J2,3 = 3.2 Hz, J3,4 = 9.3 Hz, 1H, H-3), 4.04 (m, 1H, H-2'), 3.90–3.80 (m, 5H), 3.78–3.74 (m, 3H), 3.68 (m, 2H), 3.64–3.61 (m, 2H), 3.57–3.54 (m, 2H). 13C-NMR (75 MHz, D2O): δ 160.6 (CNO2), 142.4, 126.1(2), 116.9(2) (4 × Ar-C), 102.4, 98.9, 97.6 (3 × C-1), 77.9, 73.2, 72.6, 71.9, 70.6, 70.4, 70.1, 69.9, 69.2, 66.8, 66.7, 65.8. 65.2, 61.0, 60.9. HRMS for C24H35NO18Na (M+Na)+ 648.1752. Found: 648.1754.4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, C.C.; Lee, J.C.; Luo, S.Y.; Kulkarni, S.S.; Huang, Y.W.; Lee, C.C.; Chang, K.L.; Hung, S.C. Regioselective one-pot protection of carbohydrates. Nature 2007, 446, 896–899. [Google Scholar]
- Seeberger, P.H.; Werz, D.B. Synthesis and medical applications of oligosaccharides. Nature 2007, 446, 1046–1051. [Google Scholar]
- Guo, J.; Ye, X.S. Protecting groups in carbohydrate chemistry: Influence on stereoselectivity of glycosylations. Molecules 2010, 15, 7235–7265. [Google Scholar]
- Bertozzi, C.R.; Kiessling, L.L. Chemical glycobiology. Science 2001, 291, 2357–2364. [Google Scholar]
- Liu, C.; Skogman, F.; Cai, Y.; Lowary, T.L. Synthesis of the ‘primer–adaptor’ trisaccharide moiety of Escherichia coli O8, O9, and O9a lipopolysaccharide. Carbohydr. Res. 2007, 342, 2818–2825. [Google Scholar]
- Gigg, J.; Gigg, R.; Payne, S.; Conant, R. Synthesis of propyl 4-O-(3,6-di-O-methyl-β-d-glucopyranosyl)-2,3-di-O-methyl-α-d-rhamnopyranoside. Carbohydr.Res 1985, 141, 91–97. [Google Scholar]
- Chung, S.K.; Moon, S.H. Synthesis and biological activities of (4,6-di-O-phosphonato-β-d-mannopyranosyl)-methylphosphonate as an analogue of 1l-myo-inositol 1,4,5-trisphosphate. Carbohydr. Res. 1994, 260, 39–50. [Google Scholar]
- Evans, M.E.; Parrish, F.W. Monomolar acetalations of methyl α-d-mannosides-synthesis of methyl α-d-talopyranoside. Carbohydr. Res. 1977, 54, 105–114. [Google Scholar]
- Fleet, G.W.; Smith, P.W. Enantiospecific syntheses of deoxymannojirimycin, fagomine and 2r, 5r-dihydroxymethyl-3r, 4r-dihydroxypyrrolidine from d-glucose. Tetrahedron Lett. 1985, 26, 1469–1472. [Google Scholar]
- Yadav, J.; Chander, M.C.; Reddy, K.K. Stereoselective synthesis of 10(S), 11(R), 12(R)-trihydroxyeicosa-5(Z), 8(Z), 14(Z)-trienoic acid from d-mannose. Tetrahedron Lett. 1992, 33, 135–138. [Google Scholar]
- Manna, S.; Viala, J.; Yadagiri, P.; Falck, J. Synthesis of 12(S), 20-, 12(S), 19(R)-, and 12(S), 19(S)-dihydroxyeicosa-cis-5,8,14-trans-10-tetraenoic acids, metabolites of 12(S)-hete. Tetrahedron Lett 1986, 27, 2679–2682. [Google Scholar]
- Vijayasaradhi, S.; Singh, J.; Aidhen, I.S. An Efficient, Selective Hydrolysis of Terminal Isopropylidene Acetal Protection by Zn (NO3)2 6H2O in Acetonitrile. Synlett 2000, 1, 110–112. [Google Scholar]
- Swamy, N.R.; Venkateswarlu, Y. A mild and efficient method for chemoselective deprotection of acetonides by bismuth (III) trichloride. Tetrahedron Lett. 2002, 43, 7549–7552. [Google Scholar]
- Ki, H.P.; Yong, J.Y.; Sang, G.L. Efficient cleavage of terminal acetonide group: Chirospecific synthesis of 2, 5-dideoxy-2, 5-imino-d-mannitol. Tetrahedron Lett. 1994, 35, 9737–9740. [Google Scholar]
- Kim, K.S.; Song, Y.H.; Lee, B.H.; Hahn, C.S. Efficient and selective cleavage of acetals and ketals using ferric chloride adsorbed on silica gel. J. Org. Chem. 1986, 51, 404–407. [Google Scholar]
- Mahender, G.; Ramu, R.; Ramesh, C.; Das, B. A simple and facile chemo-and regioselective deprotection of acetonides using silica supported sodium hydrogen sulfate as a heterogeneous catalyst. Chem. Lett. 2003, 8, 734–735. [Google Scholar]
- Agarwal, A.; Vankar, Y.D. Selective deprotection of terminal isopropylidene acetals and trityl ethers using HClO4 supported on silica gel. Carbohydr. Res. 2005, 340, 1661–1667. [Google Scholar]
- Fauré, R.; Shiao, T.C.; Damerval, S.; Roy, R. Practical synthesis of valuable d-rhamnoside building blocks for oligosaccharide synthesis. Tetrahedron Lett. 2007, 48, 2385–2388. [Google Scholar]
- Zong, G.; Yu, N.; Xu, Y.; Zhang, J.; Wang, D.; Liang, X. Synthesis of a mannose hexasaccharide related to the cell wall mannan of candida dubliniensis and trychophyton mentagrophytes. Synthesis 2010, 10, 1666–1672. [Google Scholar]
- Cheng, L.; Chen, Q.; Liu, J.; Du, Y. Synthesis of a fluorescence-labeled K30 antigen repeating unit using click chemistry. Carbohydr. Res. 2007, 342, 975–981. [Google Scholar]
- Zegelaar-Jaarsveld, K.; Duynstee, H.I.; van der Marel, G.A.; van Boom, J.H. Iodonium ion-assisted synthesis of tetrameric fragments corresponding to the cell wall phenolic glycolipids of Mycobacterium kansasii serovars II and IV. Tetrahedron 1996, 52, 3575–3592. [Google Scholar]
- Lemanski, G.; Ziegler, T. Intramolecular mannosylations of glucose derivatives via prearranged glycosides. Helv. Chim. Acta 2000, 83, 2655–2675. [Google Scholar]
- Rana, S.S.; Barlow, J.J.; Matta, K.L. Synthetic studies in carbohydrates. Part XVIII. Synthesis of p-nitrophenyl 6-O-(2-acetamido-2-deoxy-β-d-glucopyranosyl)-α-d-mannopyranoside. Carbohydr. Res. 1981, 96, 79–85. [Google Scholar]
- Sparrow, L.G.; Lawrence, M.C.; Gorman, J.J.; Strike, P.M.; Robinson, C.P.; McKern, N.M.; Ward, C.W. N-linked glycans of the human insulin receptor and their distribution over the crystal structure. Proteins: Struct. Funct. Bioinform. 2008, 71, 426–439. [Google Scholar]
- De Leoz, M.L.A.; Young, L.J.T.; An, H.J.; Kronewitter, S.R.; Kim, J.; Miyamoto, S.; Borowsky, A.D.; Chew, H.K.; Lebrilla, C.B. High-mannose glycans are elevated during breast cancer progression. Mol. Cell. Proteomics 2011, 10, 1–9. [Google Scholar]
- Moremen, K.W.; Tiemeyer, M.; Nairn, A.V. Vertebrate protein glycosylation: Diversity, synthesis and function. Nat. Rev. Mol. Cell Bio. 2012, 13, 448–462. [Google Scholar]
- Nairn, A.V.; Aoki, K.; dela Rosa, M.; Porterfield, M.; Lim, J.M.; Kulik, M.; Pierce, J.M.; Wells, L.; Dalton, S.; Tiemeyer, M.; et al. Regulation of glycan structures in murine embryonic stem cells. combined transcript profiling of glycan-related genes and glycan structural analysis. J. Biol. Chem. 2012, 287, 37835–37856. [Google Scholar]
- Zhang, J.J.; Kong, F.Z. Efficient and practical syntheses of mannose tri-, tetra-, penta-, hexa-, hepta-, and octasaccharides existing in N-glycans. Tetrahedron: Asymmetry 2002, 13, 243–252. [Google Scholar]
- Zhang, J.J.; Kong, F.Z. A facile large scale synthesis of the core mannose pentasaccharide of N-linked glycoprotein and its isomer. Acta Chim. Sinica 2002, 1, 150–156. [Google Scholar]
- Mikkelsen, L.M.; Krintel, S.L.; Jiménez-Barbero, J.; Skrydstrup, T. Application of the anomeric samarium route for the convergent synthesis of the C-linked trisaccharide α-d-Man-(1→ 3)-[α-d-man-(1→ 6)]-d-man and the disaccharides α-d-man-(1→ 3)-d-man and α-d-man-(1→ 6)-d-man. J. Org. Chem. 2002, 67, 6297–6308. [Google Scholar]
- Liu, Y.; Chen, G. Chemical synthesis of N-linked glycans carrying both mannose-6-phosphate and glcnac-mannose-6-phosphate motifs. J. Org. Chem. 2011, 76, 8682–8689. [Google Scholar]
- Ogawa, T.; Katano, K.; Matsui, M. Regio- and stereo-controlled synthesis of core oligosaccharides of glycopeptides. Carbohydr. Res. 1978, 64, C3–C9. [Google Scholar]
- Winnik, F.M.; Brisson, J.R.; Carver, J.P.; Krepinsky, J.J. Syntheses of model oligosaccharides of biological significance. Synthesis of methyl 3,6-di-O-(α-d-mannopyranosyl)-α-d-mannopyranoside and the corresponding mannobiosides. Carbohydr. Res. 1982, 103, 15–28. [Google Scholar]
- Kaur, K.J.; Alton, G.; Hindsgaul, O. Use of N-acetylglucosaminyltransferases I and II in the preparative synthesis of oligosaccharides. Carbohydr. Res. 1991, 210, 145–153. [Google Scholar]
- Kaur, K.J.; Hindsgaul, O. A simple synthesis of octyl 3,6-di-O-(α-d-mannopyranosyl)-β-d-mannopyranoside and its use as an acceptor for the assay of N-acetylglucosaminyltransferase-I activity. Glycoconjugate J. 1991, 8, 90–94. [Google Scholar]
- Oscarson, S.; Tiden, A.K. Synthesis of the octyl and tetradecyl glycosides of 3,6-di-O-α-d-mannopyranose and of 3,4-di-O-α-d-mannopyranosyl-α-d-mannopyranose. A new way for 2,4-di-O-protection of mannopyranosides. Carbohydr. Res. 1993, 247, 323–328. [Google Scholar]
- Figueroa-Perez, S.; Verez-Bencomo, V.J. Synthesis of neoglycolipids containing oligosaccharides based on 3,6-branched-α-d-mannopyranosides as the carbohydrate moieties. Carbohydr. Chem. 1998, 17, 851–868. [Google Scholar]
- Tanaka, H.; Nishida, Y.; Kobayashi, K. A facile synthesis of a glycoconjugate cationic polymer carrying the 3,6-branched α-d-mannosyl trisaccharide cluster. J. Carbohydr. Chem. 2000, 19, 413–418. [Google Scholar]
- Ratner, D.M.; Plante, O.J.; Seeberger, P.H. A linear synthesis of branched high-mannose oligosaccharides from the HIV-1 viral surface envelope glycoprotein gp120. Eur. J. Org. Chem. 2002, 5, 826–833. [Google Scholar]
- Abronina, P.I.; Backinowsky, L.V.; Grachev, A.A.; Sedinkin, S.L.; Malysheva, N.N. An easy access to a 3,6-branched mannopentaoside bearing one terminal [1-13C]-labeled d-mannopyranose residue. Russ. Chem. Bull. 2005, 54, 1287–1293. [Google Scholar]
- Mukhopadhyay, B.; Maurer, S.V.; Rudolph, N.; van Well, R.M.; Russell, D.A.; Field, R.A. From solution phase to “on-column” chemistry: Trichloroacetimidate-based glycosylation promoted by perchloric acid-silica. J. Org. Chem. 2005, 70, 9059–9062. [Google Scholar]
- Arnarp, J.; Lonngren, J. Synthesis of 3,6-di-O-(α-d-mannopyranosyl)-d-mannose. Acta Chem. Scand. Ser. B 1978, B32, 696. [Google Scholar]
- Sample Availability: Samples of the compounds 1a, 2a, 3a–g, 4–8, 10 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jiang, R.; Zong, G.; Liang, X.; Jin, S.; Zhang, J.; Wang, D. Direct 2,3-O-Isopropylidenation of α-D-Mannopyranosides and the Preparation of 3,6-Branched Mannose Trisaccharides. Molecules 2014, 19, 6683-6693. https://doi.org/10.3390/molecules19056683
Jiang R, Zong G, Liang X, Jin S, Zhang J, Wang D. Direct 2,3-O-Isopropylidenation of α-D-Mannopyranosides and the Preparation of 3,6-Branched Mannose Trisaccharides. Molecules. 2014; 19(5):6683-6693. https://doi.org/10.3390/molecules19056683
Chicago/Turabian StyleJiang, Rui, Guanghui Zong, Xiaomei Liang, Shuhui Jin, Jianjun Zhang, and Daoquan Wang. 2014. "Direct 2,3-O-Isopropylidenation of α-D-Mannopyranosides and the Preparation of 3,6-Branched Mannose Trisaccharides" Molecules 19, no. 5: 6683-6693. https://doi.org/10.3390/molecules19056683