Applications of Liquid-Phase Microextraction in the Sample Preparation of Environmental Solid Samples

Abstract
:1. Introduction
2. Modes and Variations of Liquid-Phase Microextraction


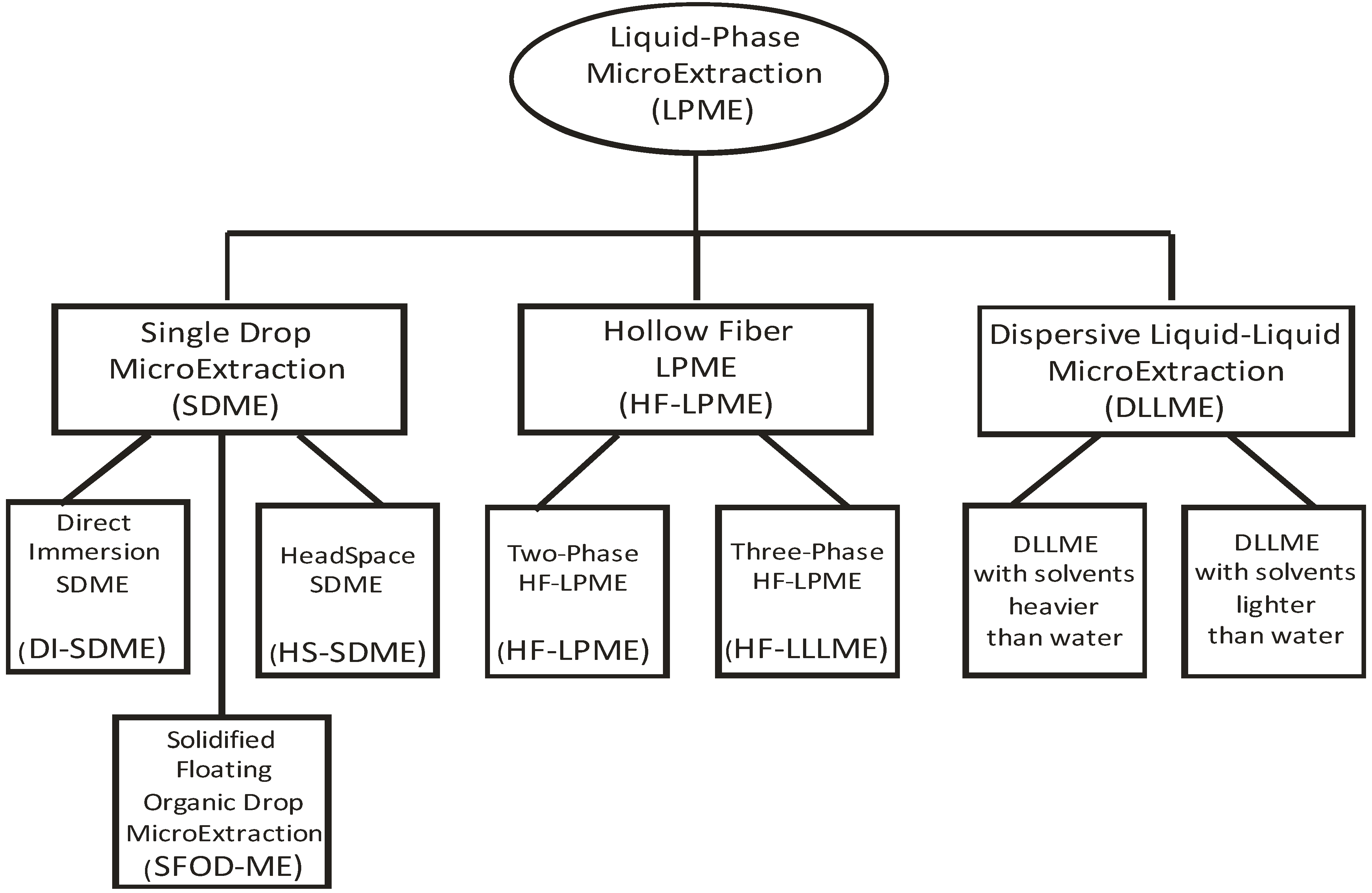{kind=link}
| Technique | Solvent properties | Solvent volume | Sample preparation; other equipment | Mixing/ stirring | Extraction time | Typical analytes | Automation | Other considerations & modifications |
|---|---|---|---|---|---|---|---|---|
| SDME | immiscible with water; usually GC-compatible HS-SDME: low vapor pressure, also water; recent: ionic liquids | 1–8 µL | GC syringe sample: filtration in DI-SDME; adjustment of ionic strength, T | DI-SDME: up to 600 rpm HS-SDME: higher rates | min. 1–15 min, usually longer | non-polar, semi-volatile or volatile (HS-SDME) | semi-automatic in dynamic mode; continuous flow ME (CF-ME) [34] | simple; ready-to-analyze extracts; modifications: dynamic mode possible in-needle or in-syringe; LLLME with back-extraction into droplet of 2nd immiscible solvent; exhaustive extraction by multiple HS-SDME [35] |
| HF-LPME | immiscible with water; compatible with HF material; low volatility & viscosity, e.g., toluene, n-octanol, also di-n-hexyl ether HF-LLLME: above valid for solvent in the HF wall; in the HF lumen: aqueous acceptor phase or ionic liquid or immiscible organic solvent | 4–20 µL | small-diameter porous tube (fiber), usually polypropylene, one end sealed, other attached to syringe sample: adjustment of pH and ionic strength | vigorous stirring or vibration, microwaves | 20–60 min (except for dynamic HF-LPME or EME) | non-polar; ionizable (in HF-LLLME) | yes, with autosampler; dynamic HF-LPME [36]; still each fiber manually prepared | applicable to »dirty« samples; modifications: dynamic HF-LPME [36]; solvent-bar microextraction (SBE) [37]; air in HF wall with aqueous solvent in lumen for volatile analytes [38] |
| DLLME | disperser solvent: miscible with water, e.g., acetone, methanol, ethanol, acetonitrile, THF; extraction solvent: ρsolv. > ρaq, e.g., C2Cl4, Cl-benzene, CH2Cl2, CHCl3, CCl4, ionic liquid; OR ρsolv. < ρaq, e.g., 1- or 2-dodecanol, 1-undecanol, hexadecane (Tmp ≈ room T), also cyclohexane, n-hexanol, tri-n-butyl-phosphate | Disp.s.: 0, 1–2 mL Extr.s.: 10–150 µL | centrifuge (ρsolv. > ρaq); ice bath or special extracting vessel (ρsolv. < ρaq); sample: filtration, adjustment of pH and ionic strength | not needed; ultrasound, vortex [9] | equilibration in few seconds; phase separation 1–20 min | non-polar | barely possible, although attempts [39] | modifications: temperature-controlled DLLME with ionic liquids, mixing and separation of phases at high/low T [40] |
| LPME | Analytical techn. | Sample | Analytes | Extraction procedure for solid sample | Optimized extraction conditions for s.s. | LPME procedure | Method performance | Ref. |
|---|---|---|---|---|---|---|---|---|
| DLLME | GC-ECD | soil | 5 PCBs | 1 g s.s. + 10 mL AC; mech. shaking 30 min; upper layer | extraction solvent | 1.0 mL AC extract (DS) + 30 µL Cl-benzene (ES) inj. into 5.0 mL w.; centrif.; sedim. phase evaporated, rediss. in 20 µL n-hexane | η 82.3%–113.6% (3 levels); RSD < 6.4%; LOD 0.20–0.50 ng/g | [60] |
| US-DLLME | GC-MS (SIM) | soil | endosulfan & 5 metab. | 0.5 g s.s. + 1.25 mL AC; US (10 min); centrif. | not given | AC extract (DS) + 58 μL TCE (ES) inj. into 5.0 mL w. + 7% Na2SO4; US (2 min); centrif.; direct injection | experimental design for US-DLLME optimization; η 89.0%–99.7%; RSD < 6.3%; LOD 0.316–2.494 ng/g; no interference from sample matrix observed | [61] |
| DLLME | HPLC-FLD | sediment | PAHs | 0.2 g s.s. + 2 mL ACN, VA (2 min), centrif. | extraction solvent, vortex time | 1.0 mL ACN extract (DS) + 80 μL CH2Cl2 (ES) inj. into 5.0 mL w.; centrif.; sedim. phase evaporated, rediss. in 40 μL ACN | η 72.9%–97.8% (3 levels); RSD < 8.0%; LOD 2.3–6.8 ng/g; no interference from sample matrix observed | [62] |
| DLLME | LC-FLD | soil | carbaryl, triazophos | 1 g s.s. + 10 mL MeOH, mech. shaking (30 min); filtered | extraction solvent | 1.0 mL MeOH extract (DS) + 50 µL TtCE (ES) inj. into 5.0 mL w.; centrif.; sedim. phase evaporated, rediss. in 25 μL MeOH | η 80.8%–111.1% (3 levels); RSD < 4.3%; LOD 0.014–0.110 ng/g; some matrix interferences present in chromatograms | [63] |
| US-IL-DLLME | HPLC-UV | soil | 3 pesticides | 10 g s.s. + 30 mL sol. (60% MeOH, 5 mg NaCl); US, centrif. + filtr.; repeat; evaporated to dryness, rediss. in 10 mL MeOH sol., pH adjust to 4.0 | not given | 1.0 mL sample sol., inj. 0.3 mL MeOH (DS) + 70 μL [BMIM]TFSI (ES); shake, US (2 min); centrif.; sedim. phase dissolved in 0.5 mL MeOH | not given for overall method (just for DLLME) | [64] |
| IL-DLLME | HPLC-FLD | soil | 5 pesticides 2 metabol. | 3 g s.s. + 20 mL MeOH + 2.5% NaCl; manual shaking + US; centrif. + filtr.; repeat; evaporated to dryness, rediss. in 10.0 mL w., pH adjust to 5.2 | extraction solvent, Vsolv, NaCl add., US time, amount of sample | add. NaCl to 30%; MeOH (DS) + IL ([HMIm][PF6], ES); centrif.; 80 µL sedim. phase dissolved in 1120 µL ACN-phosphate buffer | central composite experimental design for optimization of IL-DLLME conditions; η 88%–119%; LOD 0.02–27.1 ng/g | [65] |
| IL-DLLME | HPLC-FLD | soil | 7 pesticides and metabol. | 3 g s.s. + 20 mL MeOH + 2.5% NaCl; manual shaking + US (10 min), centrif. + filtr.; repeat; evaporated to dryness, rediss. in 10.0 mL w., pH adjust to 5.2 | not given | comparison of 2 IL as ES: [PPIm][PF6] and [HMIm][PF6]; add. NaCl (2.5 g); MeOH (DS) 418 μL + IL 117.5 mg (ES); VA 1 min; centrif.; 80 µL sedim. phase dissolved in 1120 µL ACN-phosphate buffer | η 93%–118%; RSD < 20%; LOD 0.02–60.5 ng/g | [66] |
| DLLME | sweeping MEKC-DAD | soil | 5 sulfonylurea herbicides | 10 g s.s. + 10 mL ACN (5% HCOOH, pH 3.0), shaking; added 4 g MgSO4 + 1 g NaCl, shaking; centrif.; 5 mL supernatant + 250 mg C18 + 1.5 g MgSO4, shaking; centrif., filtered | extraction solvent, pH of sample solution for DSPE, DSPE sorbent | 1.0 mL extract (DS) + 50 µL ClBz inj. into 5.0 mL w. (pH 2.0, HCl), VA (5 s); centrif.; sedim. phase evaporated, rediss. in 20.0 µL phosphate buffer (pH 10.0) | η 76.0%–93.5% (3 levels); RSD < 6.8%; LOD 0.5–1.0 ng/g | [67] |
| DLLME | HPLC-DAD | soil | 4 sulfonylurea herbicides | 10 g s.s. + 20 mL AC/0.15 M NaHCO3 (2:8), shaking 30 min; filtered; 10 mL filtrate + 0.15 g C18, shaking 5 min; filtered; pH adj. to 2.0 and dil. to 25 mL with AC/w. (2:8, pH 2.0) | organic solvent, DSPE sorbent | 5.0 mL solution (with AC 20% as DS) + 60 µL ClBz (ES), VA 5 s; centrif.; sedim. phase evaporated, rediss. in 15 µL ACN | η 78.0%–92.5% (3 levels); RSD < 7.2%; LOD 0.5–1.2 ng/g; some interferences present in chromatograms | [68] |
| DLLME | GC-MS/MS | sediment | 4 PBDEs | 0.25 g s.s. + 1.5 mL AC; US (35 °C) 6 × 5 min; centrif.; 1.2 mL leachate + 100 mg SiO2, VA (30 s); centrif. | leaching solvent, Vsolv, DSPE sorbent, US time & mode, US-transmitting liq., leaching T | 1.0 mL AC extract (DS) + 60 µL CCl4 (ES) inj. into 5.0 mL w.; shaking, 5 min in bath (35 °C); centrif.; direct injection | η 80%–112% (2 levels); RSD < 9.8%; LOD 0.02–0.06 ng/g; extraction method comparable efficiency with Soxhlet's | [69] |
| DLLME | GC-MS/MS | sediment | 4 PBDEs | 1 g s.s. + 1.2 mL MeOH; US (40 °C) 2 × 9.2 min; centrif. | leaching solvent type (also a DS) & V, T, US time & cycles | 0.1 mL MeOH (DS) + 22 mg 1-dodecanol (ES) inj. into 0.4 mL leachate + 1.0 mL 6.15 M NaCl + 4.4 mL w. at 40 °C; SFOD form. at 10 min in ice bath; collected, melt, add. 3 µL i-octane; direct injection | factorial (2k) screening & central composite design for optimization; η 71%–104% (2 levels); RSD < 9.2%; LOD 0.5–1.8 pg/g | [70] |
| HLLE | GC-ECD | soil | 3 organo-phosph. & pyrethroid pesticides | 4.0 g s.s. + 10 mL AC; mech. shaking (30 min); supernatant decanted | extraction solvent, Vsolv | 1.0 mL AC extract (CS) + 40 µL CCl4 (ES) inj. into 5.0 mL w.; phase separation by 0.3 g NaCl; centrif.; direct injection | η 79.2%–113.1%; RSD < 9.6%; LOD 0.01–0.04 ng/g | [71] |
| USA-EME | GC-FID | soil | diazinon, chlorpyrifos | 2 g s.s. + 2.5 mL MeOH; US (2 min pulse on/off); centrif., filtered | not given | 1.5 mL extract + 10.5 mL w.; 14 µL toluene slowly injected during US, US (30 s); centrif.; upper phase collection, direct injection | η 90.0%–105%; RSD < 9.2%; LOD not given for soil; several interferences in the chromatogram | [72] |
| HF-LPME | GC-ICP-MS | soil, dust | 4 PBDEs | 0.5 g soil/0.05 g dust + 3 mL MeOH; US (30 min); centrif.; supernatant diluted to 10 mL with w. | not given | 3.0 mL extract, 4 µL decane (ES) in 1.5-cm HF; stirring 20 min at 40 °C & 1000 rpm; direct injection | η 86.7%–110.9%; RSD < 10.4%; LOD not given for soil & dust; several interferences present in the chromatogram (brominated compounds?) | [73] |
| HF-LPME | GC-ECD | sediment | vinclozoline | 5 g s.s. + 10 mL ACN-MeOH (9:1); US 30 min; centrif., evapor. to 0.05 mL | extraction solvent | extract + 5 mL w., 3 µL toluene (ES) in 1.3-cm HF; stirring 20 min at 800 rpm; direct injection | η 94%–96% (2 levels); RSD 6.1%; LOD 0.5 ng/g | [74] |
| HF-LPME | HPLC-FLD | soil | 7 pesticides and metabol. | 3 g s.s. + 20 mL MeOH + 2.5% NaCl; manual shaking + US (10 min); centrif. + filtr.; repeat; evaporated to dryness, rediss. in 10.0 mL w., filtered | not given | extract (pH adj. to 9.0, NaCl to 20%), 20 µL 1-octanol (ES) in 2.0-cm HF; stirring 30 min at 1440 rpm; ES evaporated, rediss. in 50 µL mobile phase for HPLC | η 85%–117%; RSD variable, up to 71% at low levels; LOD 0.001–6.94 ng/g | [75] |
| HF-LLLME | GC-ECD | soil | chlorophenols | 2 g s.s. + 3 mL MeOH; US (2 min pulse on/off); centrif., filtered | not given except for MeOH effect on HF-LLLME | 2 mL extract dil. to 20 mL with w., dodecane in 8-cm HF wall (ES) & 25 µL ACN (AS) + IS in HF lumen; stirring 30 min at 400 rpm; direct injection | η 86.3%–110%; RSD < 9.3%; LOD not given for soil | [58] |
| HF-LLLME, dynamic | GC-FID | soil | PAHs | 2 g s.s. + 3 mL MeOH; US (2 min pulse on/off); centrif., filtered | not given except for MeOH effect on HF-LLLME | 2 mL extract dil. to 20 mL with w. + dodecane in 8-cm HF wall (ES) & 25 µL ACN (AS) in HF lumen; stirring 20 min at 1000 rpm; dynamic extr. (syringe plunger); direct injection | η 84.4%–110%; RSD < 9.3%; LOD not given for soil | [36] |
3. Liquid-Phase Microextraction Applied to (Semi)Solid Environmental Samples as a Step for Preconcentration and Clean-up of Extracts
3.1. LPME Combined with Conventional Solvent Extraction
| LPME | Analytical techn. | Sample | Analytes | Extraction procedure for solid sample | Optimized extraction conditions for s.s. | LPME procedure | Method performance | Ref. |
|---|---|---|---|---|---|---|---|---|
| US-DLLME | GC-ECD | soil | 3 pyrethroids | MSPD: 0.1 g s.s. + 0.3 g SiO2 (d 38 µm) blended in mortar; transf. to cartridge with 0.1 g Na2SO4(anhyd); eluted with 3 mL AC; evaporated to 0.5 mL | sorbent, sample/sorb. ratio, eluting solv. type and V, | AC extract (DS) + 50 µL TtCEt (ES) inj. into 5 mL w.; US 2 min; centrif.; sedim. phase evaporated, rediss. in 20 µL n-hexane | η 83.6%–98.5%; RSD < 7.3%; LOD 0.45–1.13 ng/g | [77] |
| DLLME | HPLC-FLD | soil | carbendazime thiabendazole | 20 g s.s. + 40 mL 0.1 mol/L HCl; mech. shaking 30 min; filtered, pH adj. to 7.0 | not given | 0.75 mL THF (DS) + 80 µL CHCl3 (ES) inj. into 5.0 mL solution + 0.5 g NaCl; centrif.; sedim. phase evaporated, rediss. in 15 µL MeOH | η 82.0%–93.4% (2 levels); RSD < 7.3%; LOD 1.0–1.6 ng/g | [78] |
| DLLME | GC-FID | sediment | PAHs | SFE: 1.2 g s.s. + 50 µL MeOH (PM); SFE at T 313 K, p 253.2 bar, static textr 10 min, dynamic textr 30 min, CO2 F = 0.5 mL/min; collected in 1 mL ACN in ice bath | pressure, temperature, static & dynamic extraction time | 1.0 mL extract (DS) + 16 µL ClBz (ES) inj. into 5 mL w.; centrif.; direct injection | η 67.8%–98.9%; RSD < 10.3%; LOD 200 ng/g | [79] |
| DLLME | GC-FID | soil sediment | 7 organo-phosphor. pesticides | SFE: 1.2 g s.s. . + 50 µL MeOH (PM); SFE at T 60 °C, p 150 bar, static textr 10 min, dynamic textr 30 min, CO2 F = 0.5 mL/min; collected in 1 mL ACN in ice bath | pressure, temperature, static & dynamic extraction time | 1.0 mL extract (DS) + 17 µL CCl4 (ES) inj. into 5 mL w.; centrif.; direct injection | η 80%–100%; RSD < 75%; LOD 1–9 ng/g | [80] |
| DLLME | GC-FID | soil | 2 nitrotoluenes | SFE: 2 g s.s. + 150 µL MeOH (PM); SFE at T 35 °C, p 350 atm, static textr 10 min, dynamic textr 30 min, CO2 F = 0.4 mL/min; collected in 1 mL MeOH in ice bath | central composite design to optimize SFE parameters: T, pressure, VPM, dynamic textr | 1.0 mL extract (DS) + 20 µL CCl4 (ES) inj. into 5.0 mL w. (3% NaCl); centrif.; direct injection | η 80%–84%; RSD < 6.5%; LOD 0.12 µg/g | [81] |
| DLLME | GC-MS | sediment | hydroxylated PAHs | SWE: 10 g s.s. + 2 g diatomaceous earth; PLE with w. pH 3.0 + 20% ACN (OM) 10 min at 150 °C & 1500 psi; purged with N2, collected 11 mL extract | type and V of organic modifier for SWE, pH, T, pressure, extr. time | 100 µL ClBz (ES) inj. into 11 mL extract (20% ACN as DS); VA 30 s; centrif.; sedim. phase evaporated, added 50 µL MTBSTFA to derivatize, evaporated, rediss. in 100 µL AC | η 57.63%–91.07%; RSD < 11.07%; LOD 0.0139–0.2334 ng/g; comparison with SWE-SPE - all parameters better for SWE-DLLME | [82] |
| DLLME | GC-MS | pyrolysis solid residue | 15 aromatic volatiles | extracted s.s. (extr. with CH2Cl2) and raw s.s. leached with 0.001 M CaCl2 sol. (leach test ISO/TS 21268-2) | not given | 0.5 mL AC (DS) + 50 µL CCl4 (ES) inj. into 5.0 mL leachate; centrif.; direct injection | LOD 1.02–24.6 ng/L a; compared with static HS and HS-SPME (both lower LODs) | [83] |
| DLLME | GC-MS | pyrolysis solid residue | 11 alkylphenols | extracted s.s. (extr. with CH2Cl2) and raw s.s. leached with 0.001 M CaCl2 sol. (leach test ISO/TS 21268-2) | not given | 1.0 mL AC (DS) + 15 µL TtCEt (ES) inj. into 4.0 mL leachate + NaCl (15%); centrif.; direct injection | η 61.9%–101.4%; RSD < 8.0%; LOD 0.07–0.17 µg/L b | [84] |
| DLLME | GC-MS | particul. matter in seawater | 8 UV filters | unfiltered seawater, US 15 min; pH adj. to 2.5 with acetic a.; filtered | US time | 250 µL AC (DS) + 50 µL CHCl3 (ES) inj. into 5.0 mL sample; centrif.; direct injection | η 88%–117% (2 levels); RSD < 14%; LOD 10–30 ng/L a | [85] |
| DLLME & in-syringe back-extract. | HPLC-UV | soil sediment | 5 chlorophenols | MWE: 1.2 g s.s. + 2 mL w. (pH 10.0); MWE 90 s, cooling, diluted to 5 mL with w., pH adj. to 6.0; centrif., filtered | Vsolv, pHsolv, MWE time | 1.0 mL AC (DS) + 37 µL ClBz (ES) inj. into 5.0 mL extract; centrif.; 20 µL sedim. phase in syringe, then 20 µL w. (pH 12.0), plunger moving 5 min; w. phase injected | η 66.1%–82.0%; RSD < 7.6%; LOD 0.5–2.0 ng/g; chromatograms free of interferences | [86] |
| USA-EME | HPLC-DAD | soil | triazine herbicides | 10 g s.s. + 10 mL w.; mech. shaking 40 min; filtered, diluted to 10.0 mL with w. | not given | 5.0 mL extract + 100 µL ClBz,; US 3 min at 25 °C; centrif.; sedim. phase evaporated, rediss. in 20 µL MeOH | η 82.6%–92% (2 levels); RSD < 4.3%; LOD 0.1–0.5 ng/g | [87] |
| ATPS | HPLC-UV | soil | 2 phytohormones | 10 g s.s. + 30 mL MeOH/w. (80:20); US 20 min; centrif.; repeat; filtered, evaporated, rediss. in 10 mL MeOH/w. (80:20, pH 3) | not given | 1.0 mL solution + 0.6 g [BMIM]Br + 0.75 g K2HPO4; stirred 10 min at 30 °C; centrif.; upper phase collected, direct injection | η 86%–102%; RSD < 5.3%; LOD 2–10 ng/g; compared to direct HF-LPME | [88] |
| CAE-ME | HPLC-DAD | sediment | PAHs, alkyl-phenols, paraben | MWE: 0.1 g s.s. + 3 mL 40 mM CTAB solution; MWE for 6 min at 90 °C and 140 W, cooled; centrif., filtered | T, MW power, CTAB solution V and concentration | 2 mL solution + 200 µL ACN + 46 µL Li-NTf2 0.5 g/mL; VA 3 min; heated 2 min at 65 °C; centrif.; sedimented droplet dil. to 100 µL with ACN, VA | η 92.8%–95.7% (2 levels); RSD < 19.3%; LOQ 0.02–0.36 µg/g; several interferences from the sample co-extracted | [89] |
| in-situ LPME with IL-based surfactant | HPLC-DAD | sediment | PAHs, alkyl-phenols, paraben | MWE: 0.1 g s.s. + 3–5 mL 40 mM C16MIm-Br sol.; MWE for 6 min at 90 °C and 140 W, cooled; centrif., filtered | T, type of ILS, ILS solution V and concentration | 4 mL solution + 800 µL ACN + 92 µL Li-NTf2 0.5 g/mL; heated 5 min at 65 °C; VA 3 min; centrif.; sedimented droplet (≈90 µL) dil. to 200 µL with ACN, VA | η (2 levels) 91.1%–127%; RSD < 19%; LOQ 0.04–1.0 µg/g | [90] |
| HF-LPME; DLLME | GC-FPD | soil | 6 organosulfur pesticides | 5 g s.s. + 10 mL w.; US 40 min; centrif.; used for HF-LPME or filtered (2×), diluted 25× with w. for DLLME | not given | HF-LPME: 5.0 mL extract, 5 µL o-xylene (ES) in 1-cm HF; stirring 35 min at 1200 rpm; direct injectionDLLME: 0.8 mL MeOH (DS) + 10 µL CCl4 (ES) inj. into 5.0 mL solution; centrif.; direct injection | HF-LPME: η 81.7%–109.2%; RSD < 9.6%; DLLME: η 87.8%–100.6%; RSD < 9.0%; LOD not given for soil samples Comparison: DLLME faster & higher capacity, HF-LPME more robust & simple for complex samples | [91] |
| HF-LPME | GC-MS | sediment | 12 OCPs 8 PCBs | MWE: 1 g s.s. + 10 mL w.; MWE at 600 W for 20 min at 80 °C; supernatant diluted to 10 mL | T, extraction time | 10 mL extract, 5 µL toluene (ES) in 1.3-cm HF; stirring 20 min at 700 rpm; direct injection | η 73%–111% (OCP) 86–110 % (PCB); RSD < 20%; LOD 0.07–0.70 ng/g | [92] |
| HF-LLLME | LC-ESI-MS | dried sewage sludge | NSAIDs | PHWE: 0.5 g s.s. + 20 g sea sand, PLE with 0.01 M NaOH 5 min (5 cyc.) at 120 °C & 100 bar, flush V 60 %; purged with N2, collected 90 mL extract adj. pH to 1.5 and diluted to 100 mL | pH of solvent, T, number of cycles, flush volume | 100 mL extract, DHE in 10-cm HF wall (ES) & 25 µL 0.1 M (NH4)2CO3 (AS) in HF lumen; stirring 120 min at 600 rpm; direct injection | η (PHWE) 101%–109% (spike), 38.9%–90.3%(native); η (HF-LPME) 23.6%–30.3%; RSD < 20%; LOD 0.4–3.7 ng/g; only small matrix effect in ESI | [93] |
| HF-LLLME | LC-ESI-MS | dried sewage sludge | SSRIs | PHWE: 0.5 g s.s. + 20 g sea sand; PLE with 0.05 M H3PO4 pH 2 for 5 min (5 cyc.) at 120 °C & 100 bar, flush V 90%; purged with N2, collected 90 mL extract adj. pH to 12.4 and diluted to 100 mL | pH of solvent, T, number of cycles, flush volume | 100 mL extract, DHE in 10-cm HF wall (ES) & 0.1 M (NH4)H2PO4 pH 2.1 (AS) in HF lumen; stirring 8 h; direct injection | η (PHWE) 67%–83%(spike) 72.2%–85.8%(native); η (HF-LPME) 29%–47%; RSD < 20.8 %; LOD 6 ng/g; comparison to direct HF-LLLME method (without PHWE) | [94] |
| HF-LLLME | LC-MS/MS | sewage sludge | SSRIs and metabolites | 1 g s.s. + 1.1 L w. + 20 µL HCOOH; stirred 16 h at 900 rpm; filtered, diluted 1:100 or 1:20 | not given | solution + IS + 10 mL 5 M NaOH, DHE in 28-cm HF wall (ES) & 20 µL w.+HCOOH pH 2 (AS) in HF lumen; stirring 2 h at 800 rpm; direct injection | η 26.2%–71.4%; RSD < 24.6%(SSRI), < 51% (metab.); LOD not given | [95] |
| DI-SDME | AP-MALDI-MS | soil | antibiotic monensin | 5 g s.s. + 15 mL w. (10% NaCl); shaking 5 min, US 5 min; centrif., repeat; supernatants collected | not given | 20.0 mL solution + 10% NaCl, 1.5 µL CHCl3/toluene (1:1) drop immersed for 10 min at 240 rpm; direct injection | η 74.5%–82.8% (3 levels); RSD < 6.5%; LOD 12.4 ng/mL b | [96] |
| HS-SDME | GC-FID | fire debris | fire accelerants | 20x20 cm piece of textile soaked with accelerant, ignited; debris + 100 mL w., mixed 3 min; centrif., filtered | sample volume | 10 mL filtrate stirred at 1500 rpm, 2.5 µL benzyl alcohol drop exposed to HS for 20 min; direct injection | LOD 0.15 mg/L a | [97] |
| ESy | GC-ECD GC-MS | soil | OCPs | 1 g s.s. + 10 mL w./ACN (8:2); US 15 min; centrif.; supernatant + 70 µL conc. H3PO4 + 100 mg Cu granules; US 15 min; filtered | ACN addition to extr. solvent | 3 mL filtrate flushed through donor side ESy at 100 µL/min; acceptor phase: n-undecane; direct injection | compared with SE and PLE: comparable results, less solvent (~4 mL vs. 420 mL-SE or 18 mL-PLE) and time (1.5 h vs. 4 h-SE or 0.85 h-PLE), less s.s. | [98] |
3.2. LPME Combined with Environmentally-Friendly Extraction
| LPME | Analytical techn. | Sample | Analytes | Preparation of solid sample | LPME procedure | Optimization of extraction conditions | Method performance | Ref. |
|---|---|---|---|---|---|---|---|---|
| DLLME | GC-FPD | soil | chlorpyrifos | "soil solution" a | 1.5 mL MeOH (DS) + 40 µL 1-dodecanol (ES) inj. into 25 mL solution at 40 °C; kept still 5 min, added 0.5 g NaCl, shaken; centrif.; ice bath to solidify drop, rinsed with ice w., diss. in 60 µL EtAc | extraction solvent type & V, disperser solvent type & V, mass of NaCl, extr. time | η 84%–103% (2 levels); RSD < 6.4%; LOD 0.084–0.52 ng/mL b | [99] |
| US-DLLME | GC-MS | house dust | TBBPA | Kimwipe sprayed with MeOH/AC (1:3), 1 min wiping of 100 cm2 area | 1 cm2 Kimwipe + 800 µL w. + 100 µL MeOH/AC (1:3, DS) + 30 µL ClBz (ES) + 1 drop HClconc + 50 µL Ac anhydride; US 5 min; centrif.; sedim. phase added Ac anhydr., IS & CH2Cl2 to 56 µL; US 5 min; heated 60 °C for 5 min, injected | disperser solv. type & V, extraction solvent type & V, swabbing material | η 104%–106%; RSD < 18%; LOD 2.5 ng/mL; compared with SPE | [100] |
| HF-LPME | GC-FID | soil | 6 PAHs | 1 g s.s. + 7 mL AC + 15 mL w.; shaking 30 s | 22 mL suspension, 8 µL octane (ES) + IS in 6.5-cm HF; stirring 8 min at 1350 rpm; direct injection | extraction time; HF: stirring rate, VAC, extr. time, VES | η 2.9%–6.2% (PF 80.1–170.7); RSD < 23.3%; LOD 130–220 ng/g | [101] |
| HF-LPME | HPLC-UV | soil | 2 phytohormones | none | 5 g s.s. + 14 mL NaCl sol. (340 g/L), 10 µL [BMIM]PF6 (ES) in 2.5-cm HF as solvent bar; stirring 50 min at 900 rpm and 25 °C; direct injection | Vsolv, extr. time, T, NaCl concentration, stirring rate | η 40%–60%; RSD < 7.9%; LOD 5–30 ng/g; compared to ATPS | [88] |
| HF-LPME | GC-MS | soil | 8 triazines | prepared slurry: 20 g s.s./mL w., added NaCl to 10% | 3 µL toluene (ES) in 1.3-cm HF in soil slurry; extr. for 20 min at 1000 rpm | extraction solvent, extraction time, stirring rate, addition of NaCl, pH, humic acids | η not given; RSD < 5%; LOD not given; compared with SDME (drop unstable) & SPME (poorer precision) | [102] |
| HF-LPME | GC-MS | soil | 4 chlorophenols | 30 mg s.s. + 15 mL w. + IS | 15 mL suspension + 15 mL pH 1 buffer sol., 15 µL 1-octanol (ES) in 5.0-cm HF, stirring 80 min at 1100 rpm; direct injection | pH & ionic strength of donor sol., stirring rate, extraction time | η 90.52%–106.47%; RSD < 5.13%; LOD not given for soil; compared with SPME - with HF-LPME less interference | [103] |
| HF-LPME, dynamic | GC-MS | soil | methylphenols chloro-benzenes, chlorinated pesticides | 1 g s.s.+ 4 mL AC/w.(40:60).; US (5 min); stirring at 1000 rpm (40 min) | 4 mL suspension, 3 µL toluene (ES) in 1.3-cm HF, stirring 4 min at 200 rpm, dynamic extr. (syringe plunger); direct injection | type & ratio org. solv.: w. in suspension; HF: extr. solvent, extr. time, plunger speed, ionic strength, HA conc. | η 92%–100%; RSD < 13.0%; LOD 50–100 ng/g; comparison with SPME | [104] |
| HF-LLLME | LC-ESI-MS | sewage sludge | SSRIs | 0.25–1 g s.s. + 50 mL w., pH adj. to 12.4 | 50 mL suspension, DHE in 20-cm HF wall (ES) & 10 µL 0.1 M (NH4)H2PO4 pH 2.1 (AS) in HF lumen; stirring 6 h; direct injection | pH of sample suspension, acceptor solvent & pH, extraction time | η 5%–19% (PF 221-995); RSD < 18.4%; LOD 1–12 ng/g; comparison to HF-LLLME after PHWE | [94] |
| HF-LLLME | LC-ESI-MS | sewage sludge | 4 NSAIDs | 0.5–1.5 g s.s. + 50 mL w., stirred 17 h at 660 rpm, pH adj. to 1.5 | 50 mL slurry, DHE in 18-cm HF wall (ES) & 10 µL 0.1 M (NH4)2CO3 pH 9 (AS) in HF lumen; stirring 4 h at 660 rpm; direct injection | extraction time | η not given; RSD < 17.7%; LOD not given | [105] |
| DHF-HS-LPME | GC-MS | soil | 6 PAHs | 1 g s.s. + 1 mL w.; heated at 90 °C for 10 min | 3 µL 1-octanol (ES) in 1.5-cm HF in headspace over soil slurry heated at 40 °C for 10 min at 400 rpm, dynamic extr. (5 s dwell time); direct injection | extraction solvent, dwell time, number of cycles, extraction time, T, addition of w. & NaCl to s.s. | η not given; RSD < 14.6%; LOD 5.9–76 ng/g | [106] |
| HS-SDME | GC-FID | drilling mud | C6-C12 hydrocarbons | drilling mud with water left to separate | 5 mL supernatant in vial, a drop (1.5 µL) of n-hexadecane (ES) + IS suspended from needle tip in HS; extr. for 30 min at 1000 rpm; direct injection | extraction solvent type & V, ionic strength of sample, stirring rate, extraction time | clean chromatograms with no intereferences; other data not given for drilling mud | [107] |
| HS-LPME | GC-ECD | soil | 5 chlorobenzenes | 1 g s.s. + 1.5 mL w.; heated for 30 min at 40 °C before extr. | 2 µL toluene (ES) into 10-µL microsyringe, 5 µL HS at 40 °C withdrawn at 1µL/s, expelled, 5 s waiting, repeat 25 times; direct injection | extraction solvent type & V, HS sampling volume, withdrawal rate, number of cycles | η not given; RSD < 17.7%; LOD 6–14 ng/g; compared with HS-SPME | [44] |
| CF-SDME (GF-HS-LPME c) | GC-MS | sediment | PAHs | not given | continuous gas flow SDME in a home-designed apparatus: sample heated at 80 °C, extr. for 20 min into 2 µL dodecane (ES) in the gas channel, gas flow rate 2.7 mL/min | gas flow rate, position of solvent drop, i.d. gas outlet channel, extraction time, sample T, extr. solvent T | η not given; RSD < 19.7%; LOD 0.020–8.0 ng d | [108] |
4. LPME without Previous Extraction of Solid or Semisolid Samples
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ramos, L. Critical overview of selected contemporary sample preparation techniques. J. Chromatogr. A 2012, 1221, 84–98. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, Zh.; Wang, X.; Qiu, Ch. Sample preparation. J. Chromatogr. A 2008, 1184, 191–219. [Google Scholar] [CrossRef]
- Raynie, D.E. Modern extraction techniques. Anal. Chem. 2010, 82, 4911–4916. [Google Scholar] [CrossRef]
- Yazdi, A.S. Surfactant-based extraction methods. Trends Anal. Chem. 2011, 30, 918–929. [Google Scholar] [CrossRef]
- Rocha, D.L.; Batista, A.D.; Rocha, F.R.P.; Donati, G.L.; Nóbrega, J.A. Greening sample preparation in inorganic analysis. Trends Anal. Chem. 2013, 45, 79–92. [Google Scholar] [CrossRef]
- Miró, M.; Hansen, E.H. On-line sample processing involving microextraction techniques as a front-end to atomic spectrometric detection for trace metal assays: A review. Anal. Chim. Acta 2013, 782, 1–11. [Google Scholar]
- Andruch, V.; Balogh, I.S.; Kocúrová, L.; Šandrejová, J. The present state of coupling of dispersive liquid-liquid microextraction with atomic absorption spectrometry. J. Anal. At. Spectrom. 2013, 28, 19–32. [Google Scholar] [CrossRef]
- Sarafraz-Yazdi, A.; Amiri, A. Liquid phase microextraction. Trends Anal. Chem. 2010, 29, 1–14. [Google Scholar] [CrossRef]
- Andruch, V.; Burdel, M.; Kocúrová, L.; Šandrejová, J.; Balogh, I.S. Application of ultrasonic irradiation and vortex agitation in solvent microextraction. Trends Anal. Chem. 2013, 49, 1–19. [Google Scholar]
- Ghambarian, M.; Yamini, Y.; Esrafili, A. Developments in hollow fiber based liquid-phase microextraction: Principles and applications. Microchim. Acta 2012, 177, 271–294. [Google Scholar] [CrossRef]
- Mahugo-Santana, C.; Sosa-Ferrera, Z.; Torres-Padrón, M.E.; Santana-Rodríguez, J.J. Application of new approaches to liquid-phase microextraction for the determination of emerging pollutants. Trends Anal. Chem. 2011, 30, 731–748. [Google Scholar] [CrossRef]
- Han, D.; Row, K.H. Trends in liquid-phase microextraction, and its application to environmental and biological samples. Microchim. Acta 2012, 176, 1–22. [Google Scholar] [CrossRef]
- Trujillo-Rodríguez, M.J.; Rocío-Bautista, P.; Pino, V.; Afonso, A.M. Ionic liquids in dispersive liquid-liquid microextraction. Trends Anal. Chem. 2013, 51, 87–106. [Google Scholar] [CrossRef]
- Herrera-Herrera, A.V.; Asensio-Ramos, M.; Hernández-Borges, J.; Rodríguez-Delgado, M.A. Dispersive liquid-liquid microextraction for determination of organic analytes. Trends Anal. Chem. 2010, 29, 728–751. [Google Scholar] [CrossRef]
- Chimuka, L.; Michel, M.; Cukrowska, E.; Buszewski, B. Advances in sample preparation using membrane-based liquid-phase microextraction techniques. Trends Anal. Chem. 2011, 30, 1781–1792. [Google Scholar] [CrossRef]
- Zgoła-Grześkowiak, A.; Grześkowiak, T. Dispersive liquid-liquid microextraction. Trends Anal. Chem. 2011, 30, 1382–1399. [Google Scholar] [CrossRef]
- Yan, H.; Wang, H. Recent development and applications of dispersive liquid-liquid microextraction. J. Chromatogr. A 2013, 1295, 1–15. [Google Scholar] [CrossRef]
- Rezaee, M.; Yamini, Y.; Faraji, M. Evolution of dispersive liquid-liquid microextraction method. J. Chromatogr. A 2010, 1217, 2342–2357. [Google Scholar] [CrossRef]
- Bosch Ojeda, C.; Sánchez Rojas, F. Separation and preconcentration by dispersive liquid-liquid microextraction procedure: Recent applications. Chromatographia 2011, 74, 651–679. [Google Scholar] [CrossRef]
- Abadi, M.D.M.; Ashraf, N.; Chamsaz, M.; Shemirani, F. An overview of liquid phase microextraction approaches combined with UV–Vis spectrophotometry. Talanta 2012, 99, 1–12. [Google Scholar] [CrossRef]
- Pinto, M.I.; Sontag, G.; Bernardino, R.J.; Noronha, J.P. Pesticides in water and the performance of the liquid-phase microextraction based techniques. A review. Microchem. J. 2010, 96, 225–237. [Google Scholar] [CrossRef]
- Jeannot, M.A.; Przyjazny, A.; Kokosa, J.M. Single drop microextraction — Development, applications and future trends. J. Chromatogr. A 2010, 1217, 2326–2336. [Google Scholar] [CrossRef]
- Lambropoulou, D.A.; Albanis, T.A. Liquid-phase micro-extraction techniques in pesticide residue analysis. J. Biochem. Biophys. Meth. 2007, 70, 195–228. [Google Scholar] [CrossRef]
- Jeannot, M.A.; Cantwell, F.F. Solvent microextraction into a single drop. Anal. Chem. 1996, 68, 2236–2240. [Google Scholar] [CrossRef]
- Kokosa, J.M. Advances in solvent microextraction techniques. Trends Anal. Chem. 2013, 43, 2–13. [Google Scholar] [CrossRef]
- Zanjani, M.R.K.; Yamini, Y.; Shariati, S.; Jönsson, J.Å. A new liquid-phase microextraction method based on solidification of floating organic drop. Anal. Chim. Acta 2007, 585, 286–293. [Google Scholar] [CrossRef]
- Pedersen-Bjergaard, S.; Rasmussen, K.E. Liquid-liquid-liquid microextraction for sample preparation of biological fluids prior to capillary electrophoresis. Anal. Chem. 1999, 71, 2650–2656. [Google Scholar] [CrossRef]
- Rasmussen, K.E.; Pedersen-Bjergaard, S.; Krogh, M.; Ugland, H.G.; Grønhaug, T. Development of a simple in-vial liquid-phase microextraction device for drug analysis compatible with capillary gas chromatography, capillary electrophoresis and high-performance liquid chromatography. J. Chromatogr. A 2000, 873, 3–11. [Google Scholar] [CrossRef]
- Rezaee, M.; Assadi, Y.; Hosseini, M.R.M.; Aghaee, E.; Ahmadi, F.; Berijani, S. Determination of organic compounds in water using dispersive liquid–liquid microextraction. J. Chromatogr. A 2006, 1116, 1–9. [Google Scholar] [CrossRef]
- Fernandes, V.C.; Subramanian, V.; Mateus, N.; Domingues, V.F.; Delerue-Matos, C. The development and optimization of a modified single-drop microextraction method for organochlorine pesticides determination by gas chromatography-tandem mass spectrometry. Microchim. Acta 2012, 178, 195–202. [Google Scholar] [CrossRef]
- Ma, M.; Cantwell, F.F. Solvent microextraction with simultaneous back-extraction for sample cleanup and preconcentration: Preconcentration into a single microdrop. Anal. Chem. 1999, 71, 388–393. [Google Scholar] [CrossRef]
- Kocúrová, L.; Balogh, I.S.; Šandrejová, J.; Andruch, V. Recent advances in dispersive liquid-liquid microextraction using organic solvents lighter than water. A review. Microchem. J. 2012, 102, 11–17. [Google Scholar] [CrossRef]
- Guo, L.; Lee, H.K. Low-density solvent-based solvent demulsification dispersive liquid–liquid microextraction for the fast determination of trace levels of sixteen priority polycyclic aromatic hydrocarbons in environmental water samples. J. Chromatogr. A 2011, 1218, 5040–5046. [Google Scholar] [CrossRef]
- Liu, W.; Lee, H.K. Continuous-flow microextraction exceeding 1000-fold concentration of dilute analytes. Anal. Chem. 2000, 72, 4462–4467. [Google Scholar] [CrossRef]
- Hansson, E.; Hakkarainen, M. Multiple headspace single-drop microextraction-a new technique for quantitative determination of styrene in polystyrene. J. Chromatogr. A 2006, 1102, 91–95. [Google Scholar] [CrossRef]
- Esrafili, A.; Yamini, Y.; Ghambarian, M.; Moradi, M. Dynamic three-phase hollow fiber microextraction based on two immiscible organic solvents with automated movement of the acceptor phase. J. Sep. Sci. 2011, 34, 98–106. [Google Scholar] [CrossRef]
- Jiang, X.; Lee, H.K. Solvent bar microextraction. Anal. Chem. 2004, 76, 5591–5596. [Google Scholar] [CrossRef]
- Zhang, J.; Su, T.; Lee, H.K. Development and application of microporous hollow fiber protected liquid-phase microextraction via gaseous diffusion to the determination of phenols in water. J. Chromatogr. A 2006, 1121, 10–15. [Google Scholar] [CrossRef]
- Anthemidis, A.N.; Mitani, C.; Balkatzopoulou, P.; Tzanavaras, P.D. On-line micro-volume introduction system developed for lower density than water extraction solvent and dispersive liquid–liquid microextraction coupled with flame atomic absorption spectrometry. Anal. Chim. Acta 2012, 733, 34–37. [Google Scholar] [CrossRef]
- Zhou, Q.; Bai, H.; Xie, G.; Xiao, J. Temperature-controlled ionic liquid dispersive liquid phase micro-extraction. J. Chromatogr. A 2008, 1177, 43–49. [Google Scholar] [CrossRef]
- Yiantzi, E.; Psillakis, E.; Tyrovola, K.; Kalogerakis, N. Vortex-assisted liquid–liquid microextraction of octylphenol, nonylphenol and bisphenol-A. Talanta 2010, 80, 2057–2062. [Google Scholar] [CrossRef]
- Papadopoulou, A.; Román, I.P.; Canals, A.; Tyrovola, K.; Psillakis, E. Fast screening of perfluorooctane sulfonate in water using vortex-assisted liquid–liquid microextraction coupled to liquid chromatography–mass spectrometry. Anal. Chim. Acta 2011, 691, 56–61. [Google Scholar] [CrossRef]
- Farajzadeh, M.A.; Afshar Mogaddam, M.R. Air-assisted liquid–liquid microextraction method as a novel microextraction technique; Application in extraction and preconcentration of phthalate esters in aqueous sample followed by gas chromatography–flame ionization detection. Anal. Chim. Acta 2012, 728, 31–38. [Google Scholar] [CrossRef]
- Shen, G.; Lee, H.K. Headspace liquid-phase microextraction of chlorobenzenes in soil with gas chromatography-electron capture detection. Anal. Chem. 2003, 75, 98–103. [Google Scholar]
- Ouyang, G.; Zhao, W.; Pawliszyn, J. Automation and optimization of liquid-phase microextraction by gas chromatography. J. Chromatogr. A 2007, 1138, 47–54. [Google Scholar] [CrossRef]
- Esrafili, A.; Yamini, Y.; Ghambarian, M.; Moradi, M.; Seidi, S. A novel approach to automation of dynamic hollow fiber liquid-phase microextraction. J. Sep. Sci. 2011, 34, 957–964. [Google Scholar] [CrossRef]
- Pedersen-Bjergaard, S.; Rasmussen, K.E. Electrokinetic migration across artificial liquid membranes. New concept for rapid sample preparation of biological fluids. J. Chromatogr. A 2006, 1109, 183–190. [Google Scholar] [CrossRef]
- Yamini, Y.; Seidi, S.; Rezazadeh, M. Electrical field-induced extraction and separation techniques: Promising trends in analytical chemistry – A review. Anal. Chim. Acta 2014, 814, 1–22. [Google Scholar] [CrossRef]
- Bello-López, M.Á.; Ramos-Payán, M.; Ocaña-González, J.A.; Fernández-Torres, R.; Callejón-Mochón, M. Analytical applications of hollow fiber liquid phase microextraction (HF-LPME): A review. Anal. Lett. 2012, 45, 804–830. [Google Scholar] [CrossRef]
- Nitiyanontakit, S.; Varanusupakul, P.; Miró, M. Hybrid flow analyzer for automatic hollow-fiber-assisted ionic liquid-based liquid-phase microextraction with in-line membrane regeneration. Anal. Bioanal. Chem. 2013, 405, 3279–3288. [Google Scholar] [CrossRef]
- Yan, X.; Yang, C.; Ren, C.; Li, D. Importance of extracting solvent vapor pressure in headspace liquid-phase microextraction. J. Chromatogr. A 2008, 1205, 182–185. [Google Scholar] [CrossRef]
- Zhang, J.; Lee, H.K. Headspace ionic liquid-based microdrop liquid-phase microextraction followed by microdrop thermal desorption-gas chromatographic analysis. Talanta 2010, 81, 537–542. [Google Scholar] [CrossRef]
- Chisvert, A.; Román, I.P.; Vidal, L.; Canals, A. Simple and commercial readily-available approach for the direct use of ionic liquid-based single-drop microextraction prior to gas chromatography. Determination of chlorobenzenes in real water samples as model analytical application. J. Chromatogr. A 2009, 1216, 1290–1295. [Google Scholar] [CrossRef]
- Holopainen, S.; Nousiainen, M.; Anttalainen, O.; Sillanpää, M.E.T. Sample-extraction methods for ion-mobility spectrometry in water analysis. Trends Anal. Chem. 2012, 37, 124–134. [Google Scholar] [CrossRef]
- Sitko, R.; Kocot, K.; Zawisza, B.; Feist, B.; Pytlakowska, K. Liquid-phase microextraction as an attractive tool for multielement trace analysis in combination with X-ray fluorescence spectrometry: An example of simultaneous determination of Fe, Co, Zn, Ga, Se and Pb in water samples. J. Anal. At. Spectrom. 2011, 26, 1979–1985. [Google Scholar] [CrossRef]
- Gonzálvez, A.; Garrigues, S.; Armenta, S.; de la Guardia, M. Headspace-liquid phase microextraction for attenuated total reflection infrared determination of volatile organic compounds at trace levels. Anal. Chem. 2010, 82, 3045–3051. [Google Scholar] [CrossRef]
- Psillakis, E.; Kalogerakis, N. Developments in single-drop microextraction. Trends Anal. Chem. 2002, 21, 53–63. [Google Scholar]
- Ghambarian, M.; Yamini, Y.; Esrafili, A.; Yazdanfar, N.; Moradi, M. A new concept of hollow fiber liquid–liquid–liquid microextraction compatible with gas chromatography based on two immiscible organic solvents. J. Chromatogr. A 2010, 1217, 5652–5658. [Google Scholar] [CrossRef]
- Melwanki, M.B.; Fuh, M.R. Dispersive liquid–liquid microextraction combined with semi-automated in-syringe back extraction as a new approach for the sample preparation of ionizable organic compounds prior to liquid chromatography. J. Chromatogr. A 2008, 1198–1199, 1–6. [Google Scholar] [CrossRef]
- Hu, J.; Fu, L.; Zhao, X.; Liu, X.; Wang, H.; Wang, X.; Dai, L. Dispersive liquid–liquid microextraction combined with gas chromatography–electron capture detection for the determination of polychlorinated biphenyls in soils. Anal. Chim. Acta 2009, 640, 100–105. [Google Scholar] [CrossRef]
- Mudiam, M.K.R.; Ch, R.; Chauhan, A.; Manickam, N.; Jain, R.; Murthy, R.C. Optimization of UA-DLLME by experimental design methodologies for the simultaneous determination of endosulfan and its metabolites in soil and urine samples by GC–MS. Anal. Methods 2012, 4, 3855–3863. [Google Scholar] [CrossRef]
- Leng, G.; Lui, G.; Chen, Y.; Yin, H.; Dan, D. Vortex-assisted extraction combined with dispersive liquid–liquid microextraction for the determination of polycyclic aromatic hydrocarbons in sediment by high performance liquid chromatography. J. Sep. Sci. 2012, 35, 2796–2804. [Google Scholar] [CrossRef]
- Fu, L.; Liu, X.; Hu, J.; Zhao, X.; Wang, H.; Huang, C.; Wang, X. Determination of two pesticides in soils by dispersive liquid–liquid microextraction. Chromatographia 2009, 70, 1697–1701. [Google Scholar] [CrossRef]
- Dong, S.; Hu, Q.; Yang, Z.; Liu, R.; Huang, G.; Huang, T. An ionic liquid-based ultrasound assisted dispersive liquid–liquid microextraction procedure followed by HPLC for the determination of low concentration of phytocides in soil. Microchem. J. 2013, 110, 221–226. [Google Scholar] [CrossRef]
- Asensio-Ramos, M.; Hernández-Borges, J.; Borges-Miquel, T.M.; Rodríguez-Delgado, M.Á. Ionic liquid-dispersive liquid-liquid microextraction for the simultaneous determination of pesticides and metabolites in soils using high-performance liquid chromatography and fluorescence detection. J. Chromatogr. A 2011, 1218, 4808–4816. [Google Scholar] [CrossRef]
- Asensio-Ramos, M.; Hernández-Borges, J.; Ravelo-Pérez, L.M.; Afonso, M.M.; Palenzuela, J.A.; Rodríguez-Delgado, M.Á. Dispersive liquid–liquid microextraction of pesticides and metabolites from soils using 1,3-dipentylimidazolium hexafluorophosphate ionic liquid as an alternative extraction solvent. Electrophoresis 2012, 33, 1449–1457. [Google Scholar] [CrossRef]
- Zhang, S.; Yin, X.; Yang, Q.; Wang, C.; Wang, Z. Determination of some sulfonylurea herbicides in soil by a novel liquid-phase microextraction combined with sweeping micellar electrokinetic chromatography. Anal. Bioanal. Chem. 2011, 401, 1071–1081. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, C.; Liu, Z.; Wu, C.; Zeng, X.; Wen, J.; Wang, Z. Dispersive solid-phase extraction followed by dispersive liquid–liquid microextraction for the determination of some sulfonylurea herbicides in soil by high-performance liquid chromatography. J. Chromatogr. A 2009, 1216, 5504–5510. [Google Scholar] [CrossRef]
- Fontana, A.R.; Lana, N.B.; Martinez, L.D.; Altamirano, J.C. Ultrasound-assisted leaching-dispersive solid-phase extraction followed by liquid–liquid microextraction for the determination of polybrominated diphenyl ethers in sediment samples by gas chromatography–tandem mass spectrometry. Talanta 2010, 82, 359–366. [Google Scholar] [CrossRef]
- Lana, N.B.; Berton, P.; Covaci, A.; Atencio, A.G.; Ciocco, N.F.; Altamirano, J.C. Ultrasound leaching–dispersive liquid–liquid microextraction based on solidification of floating organic droplet for determination of polybrominated diphenyl ethers in sediment samples by gas chromatography–tandem mass spectrometry. J. Chromatogr. A 2013, 1285, 15–21. [Google Scholar] [CrossRef]
- Wang, X.D.; Zhao, X.N.; Liu, X.J.; Li, Y.Y.; Fu, L.Y.; Hu, J.; Huang, C.J. Homogeneous liquid–liquid extraction combined with gas chromatography–electron capture detector for the determination of three pesticide residues in soils. Anal. Chim. Acta 2008, 620, 162–169. [Google Scholar] [CrossRef]
- Abdollahzadeh, Y.; Yamini, Y.; Jabbari, A.; Esrafili, A.; Rezaee, M. Application of ultrasound-assisted emulsification microextraction followed by gas chromatography for determination of organophosphorus pesticides in water and soil samples. Anal. Methods 2012, 4, 830–837. [Google Scholar] [CrossRef]
- Xiao, Q.; Hu, B.; Duan, J.; He, M.; Zu, W. Analysis of PBDEs in soil, dust, spiked lake water, and human serum samples by hollow fiber-liquid phasemicroextraction combined with GC-ICP-MS. J. Am. Soc. Mass Spectrom. 2007, 18, 1740–1748. [Google Scholar] [CrossRef]
- Lambropoulou, D.A.; Albanis, T.A. Sensitive trace enrichment of environmental antiandrogen vinclozolin from natural waters and sediment samples using hollow fiber liquid-phase microextraction. J. Chromatogr. A 2004, 1061, 11–18. [Google Scholar] [CrossRef]
- Asensio-Ramos, M.; Hernández-Borges, J.; González-Hernández, G.; Rodríguez-Delgado, M.Á. Hollow-fiber liquid-phase microextraction for the determination of pesticides and metabolites in soils and water samples using HPLC and fluorescence detection. Electrophoresis 2012, 33, 2184–2191. [Google Scholar] [CrossRef]
- Lambropoulou, D.A.; Psillakis, E.; Albanis, T.A.; Kalogerakis, N. Single-drop microextraction for the analysis of organophosphorous insecticides in water. Anal. Chim. Acta 2004, 516, 205–211. [Google Scholar] [CrossRef]
- Wang, H.; Yan, H.; Qiao, J. Miniaturized matrix solid-phase dispersion combined with ultrasound-assisted dispersive liquid–liquid microextraction for the determination of three pyrethroids in soil. J. Sep. Sci. 2012, 35, 292–298. [Google Scholar] [CrossRef]
- Wu, Q.; Li, Y.; Wang, C.; Liu, Z.; Zang, X.; Zhou, X.; Wang, Z. Dispersive liquid–liquid microextraction combined with high performance liquid chromatography–fluorescence detection for the determination of carbendazim and thiabendazole in environmental samples. Anal. Chim. Acta 2009, 638, 139–145. [Google Scholar] [CrossRef]
- Rezaee, M.; Yamini, Y.; Moradi, M.; Saleh, A.; Faraji, M.; Naeeni, M.H. Supercritical fluid extraction combined with dispersive liquid–liquid microextraction as a sensitive and efficient sample preparation method for determination of organic compounds in solid samples. J. Supercrit. Fluid 2010, 55, 161–168. [Google Scholar] [CrossRef]
- Naeeni, M.H.; Yamini, Y.; Rezaee, M. Combination of supercritical fluid extraction with dispersive liquid–liquid microextraction for extraction of organophosphorus pesticides from soil and marine sediment samples. J. Supercrit. Fluid 2011, 57, 219–226. [Google Scholar] [CrossRef]
- Jowkarderis, M.; Raofie, F. Optimization of supercritical fluid extraction combined with dispersive liquid–liquid microextraction as an efficient sample preparation method for determination of 4-nitrotoluene and 3-nitrotoluene in a complex matrix. Talanta 2012, 88, 50–53. [Google Scholar] [CrossRef]
- Wang, X.; Lin, L.; Luan, T.; Yang, L.; Tam, N.F.Y. Determination of hydroxylated metabolites of polycyclic aromatic hydrocarbons in sediment samples by combining subcritical water extraction and dispersive liquid–liquid microextraction with derivatization. Anal. Chim. Acta 2012, 753, 57–63. [Google Scholar] [CrossRef]
- Bernardo, M.S.; Gonçalves, M.; Lapa, N.; Barbosa, R.; Mendes, B.; Pinto, F.; Gulyurtlu, I. Determination of aromatic compounds in eluates of pyrolysis solid residues using HS-GC–MS and DLLME–GC–MS. Talanta 2009, 80, 104–108. [Google Scholar] [CrossRef]
- Bernardo, M.; Gonçalves, M.; Lapa, N.; Mendes, B. Determination of alkylphenols in eluates from pyrolysis solid residues using dispersive liquid–liquid microextraction. Chemosphere 2010, 79, 1026–1032. [Google Scholar] [CrossRef]
- Benedé, J.L.; Chisvert, A.; Salvador, A.; Sánchez-Quiles, D.; Tovar-Sánchez, A. Determination of UV filters in both soluble and particulate fractions of seawaters by dispersive liquid–liquid microextraction followed by gas chromatography–mass spectrometry. Anal. Chim. Acta 2014, 812, 50–58. [Google Scholar]
- Naeeni, M.H.; Yamini, Y.; Rezaee, M.; Seidi, S. Microwave-assisted extraction combined with dispersive liquid–liquid microextraction as a new approach to determination of chlorophenols in soil and sediments. J. Sep. Sci. 2012, 35, 2469–2475. [Google Scholar] [CrossRef]
- Wu, Q.H.; Li, Z.; Wu, C.X.; Wang, C.; Wang, Z. Application of ultrasound-assisted emulsification microextraction for the determination of triazine herbicides in soil samples by high performance liquid chromatography combined with LC-fluorescence detection. Microchim. Acta 2010, 170, 59–65. [Google Scholar] [CrossRef]
- Dong, S.Y.; Yang, Z.; Zhang, P.H.; Hu, Q.; Huang, T.L. Comparative study of hollow-fiber liquid-phase micro-extraction and an aqueous two-phase system for determination of phytohormones in soil. Anal. Bioanal. Chem. 2012, 403, 1743–1749. [Google Scholar] [CrossRef]
- Delgado, B.; Pino, V.; Ayala, J.H.; Afonso, A.M.; González, V. A novel preconcentration strategy for extraction methods based on common cationic surfactants: An alternative to classical coacervative extraction. J. Chromatogr. A 2012, 1257, 9–18. [Google Scholar] [CrossRef]
- Delgado, B.; Pino, V.; Anderson, J.L.; Ayala, J.H.; Afonso, A.M.; González, V. An in-situ extraction–preconcentration method using ionic liquid-based surfactants for the determination of organic contaminants contained in marine sediments. Talanta 2012, 99, 972–983. [Google Scholar] [CrossRef]
- Xiong, J.; Hu, B. Comparison of hollow fiber liquid phase microextraction and dispersive liquid–liquid microextraction for the determination of organosulfur pesticides in environmental and beverage samples by gas chromatography with flame photometric detection. J. Chromatogr. A 2008, 1193, 7–18. [Google Scholar] [CrossRef]
- Basheer, C.; Obbard, J.P.; Lee, H.K. Analysis of persistent organic pollutants in marine sediments using a novel microwave assisted solvent extraction and liquid-phase microextraction technique. J. Chromatogr. A 2005, 1068, 221–228. [Google Scholar] [CrossRef]
- Saleh, A.; Larsson, E.; Yamini, Y.; Jönsson, J.Å. Hollow fiber liquid phase microextraction as a preconcentration and clean-up step after pressurized hot water extraction for the determination of non-steroidal anti-inflammatory drugs in sewage sludge. J. Chromatogr. A 2011, 1218, 1331–1339. [Google Scholar] [CrossRef]
- Sagristà, E.; Cortés, J.M.; Larsson, E.; Salvadó, V.; Hidalgo, M.; Jönsson, J.Å. Comparison of two extraction methods for the determination of selective serotonin reuptake inhibitors in sewage sludge by hollow fiber liquid-phase microextraction. J. Sep. Sci. 2012, 35, 2460–2468. [Google Scholar] [CrossRef]
- Vasskog, T.; Bergersen, O.; Anderssen, T.; Jensen, E.; Eggen, T. Depletion of selective serotonin reuptake inhibitors during sewage sludge composting. Waste Managem. 2009, 29, 2808–2815. [Google Scholar] [CrossRef]
- Sekar, R.; Wu, H.F. Quantitative method for analysis of monensin in soil, water, and urine by direct combination of single-drop microextraction with atmospheric pressure matrix-assisted laser desorption/ionization mass spectrometry. Anal. Chem. 2006, 78, 6306–6313. [Google Scholar] [CrossRef]
- Sanagi, M.M.; Basri, R.S.; Miskam, M.; Ibrahim, W.A.W.; Ahmad, U.K.; Aboul-Enein, H.Y. Headspace single drop microextraction for the analysis of fire accelerants in fire debris samples. Anal. Lett. 2010, 43, 2257–2266. [Google Scholar] [CrossRef]
- Barri, T.; Bergström, S.; Hussen, A.; Norberg, J.; Jönsson, J.Å. Extracting syringe for determination of organochlorine pesticides in leachate water and soil-water slurry: A novel technology for environmental analysis. J. Chromatogr. A 2006, 1111, 11–20. [Google Scholar] [CrossRef]
- Xiong, J.; Guan, Z.; Zhou, G.; Tang, X.; Lv, Y.; Wang, H. Determination of chlorpyrifos in environmental water samples by dispersive liquid–liquid microextraction with solidification of a floating organic drop followed by gas chromatography with flame photometry detection. Anal. Methods 2012, 4, 3246–3250. [Google Scholar] [CrossRef]
- Di Napoli-Davis, G.; Owens, J.E. Quantitation of tetrabromobisphenol-A from dust sampled on consumer electronics by dispersed liquid-liquid microextraction. Environ. Pollut. 2013, 180, 274–280. [Google Scholar] [CrossRef]
- King, S.; Meyer, J.S.; Andrews, A.R.J. Screening method for polycyclic aromatic hydrocarbons in soil using hollow fiber membrane solvent microextraction. J. Chromatogr. A 2002, 982, 201–208. [Google Scholar] [CrossRef]
- Shen, G.; Lee, H.K. Hollow fiber-protected liquid-phase microextraction of triazine herbicides. Anal. Chem. 2002, 74, 648–654. [Google Scholar] [CrossRef]
- Chung, L.W.; Lee, M.R. Evaluation of liquid-phase microextraction conditions for determination of chlorophenols in environmental samples using gas chromatography–mass spectrometry without derivatization. Talanta 2008, 76, 154–160. [Google Scholar] [CrossRef]
- Hou, L.; Lee, H.K. Determination of pesticides in soil by liquid phase microextraction and gas chromatography–mass spectrometry. J. Chromatogr. A 2004, 1038, 37–42. [Google Scholar] [CrossRef]
- Sagristà, E.; Larsson, E.; Ezoddin, M.; Hidalgo, M.; Salvadó, V.; Jönsson, J.Å. Determination of non-steroidal anti-inflammatory drugs in sewage sludge by direct hollow fiber supported liquid membrane extraction and liquid chromatography-mass spectrometry. J. Chromatogr. A 2010, 1217, 6153–6158. [Google Scholar] [CrossRef]
- Jiang, X.; Basheer, C.; Zhang, J.; Lee, H.K. Dynamic hollow fiber-supported headspace liquid-phase microextraction. J. Chromatogr. A 2005, 1087, 289–294. [Google Scholar] [CrossRef]
- Fang, C.; Xiong, Y.; Liang, Q.; Li, Y.; Peng, P. Optimization of headspace single-drop microextraction technique for extraction of light hydrocarbons (C6–C12) and its potential applications. Org. Geochem. 2011, 42, 316–322. [Google Scholar] [CrossRef]
- Yang, C.; Qiu, J.; Ren, C.; Piao, X.; Li, X.; Wu, X.; Li, D. Gas flow headspace liquid phase microextraction. J. Chromatogr. A 2009, 1216, 7694–7599. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Prosen, H. Applications of Liquid-Phase Microextraction in the Sample Preparation of Environmental Solid Samples. Molecules 2014, 19, 6776-6808. https://doi.org/10.3390/molecules19056776
Prosen H. Applications of Liquid-Phase Microextraction in the Sample Preparation of Environmental Solid Samples. Molecules. 2014; 19(5):6776-6808. https://doi.org/10.3390/molecules19056776
Chicago/Turabian StyleProsen, Helena. 2014. "Applications of Liquid-Phase Microextraction in the Sample Preparation of Environmental Solid Samples" Molecules 19, no. 5: 6776-6808. https://doi.org/10.3390/molecules19056776