Microextraction Techniques Used in the Procedures for Determining Organomercury and Organotin Compounds in Environmental Samples

Abstract
:1. Introduction
- control and measuring devices, which ensure the possibility of analysing the prepared samples;
- reference materials which accurately reflect the composition and character of the actual samples tested, which is necessary to ensure a proper system for the quality control and assurance of measurement results;
- analytical methodologies which can be used in testing environmental samples often characterised by a complex matrix composition and low, and sometimes very low, analyte content levels.
- ○
- increasing the level of analyte concentrations in the analysed samples to a higher level than the limit of detection of the analytical technique used;
- ○
- removal of at least a part of interferents which can influence the result of the analysis;
- ○
- replacement or at least simplification of the matrix composition of samples for analysis.
- the use of solvent-free/solvent-loss techniques for preparing samples for analysis;
- the use of microextraction techniques which, due to a reduced scale of the analysis lead to a decrease in the quantity of reagents used, including solvents.
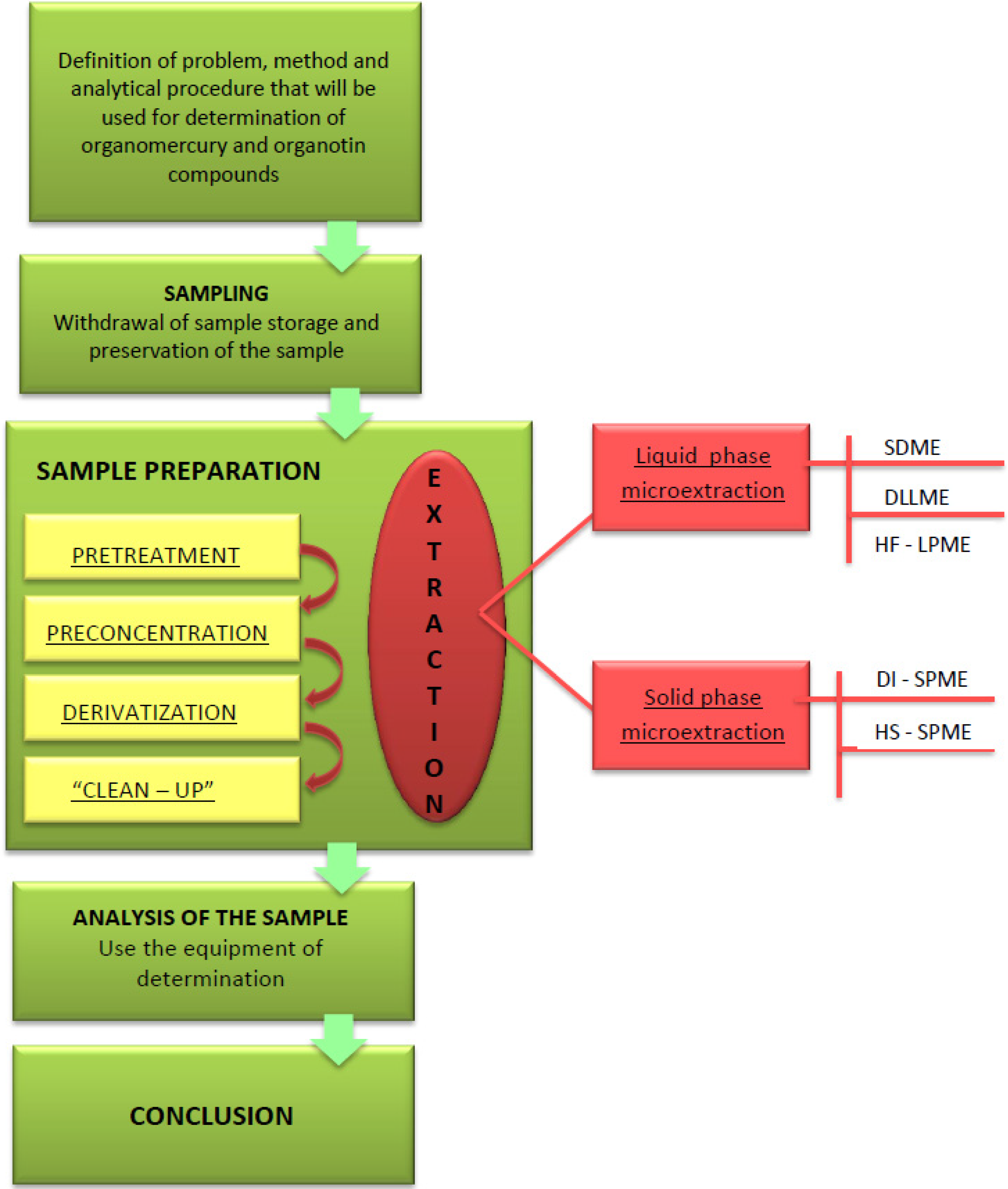
2. Analysis of Organomercury and Organotin Compounds
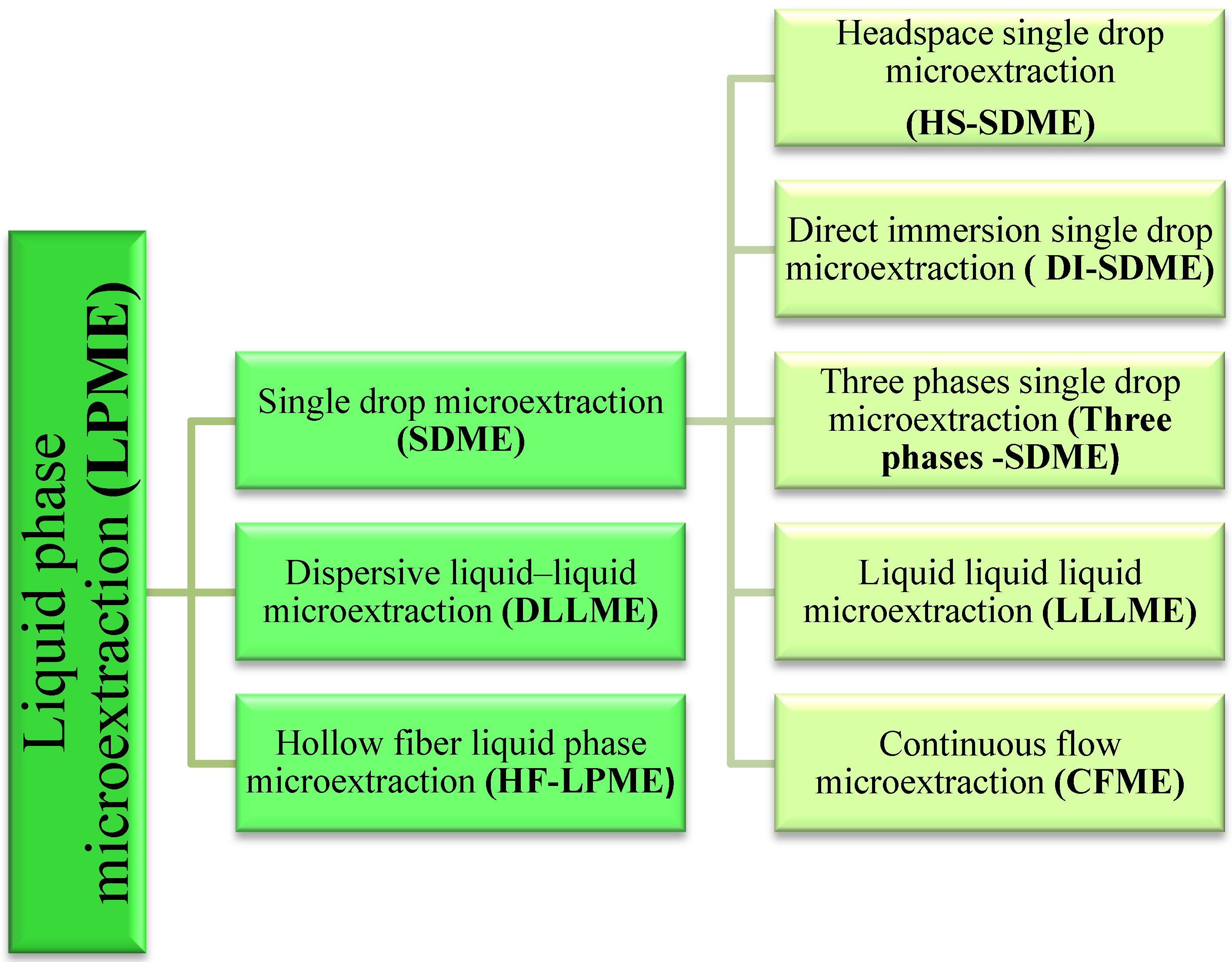
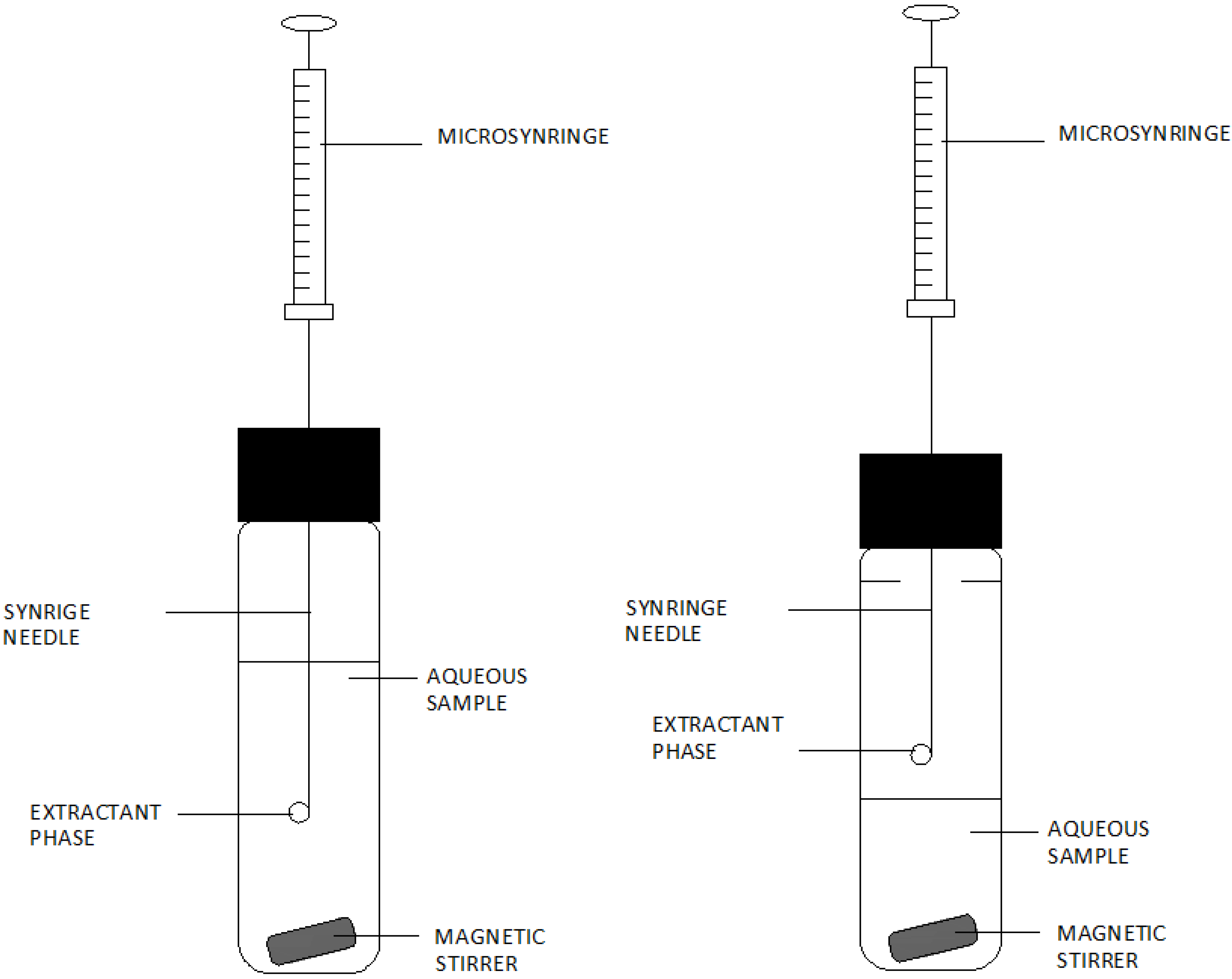
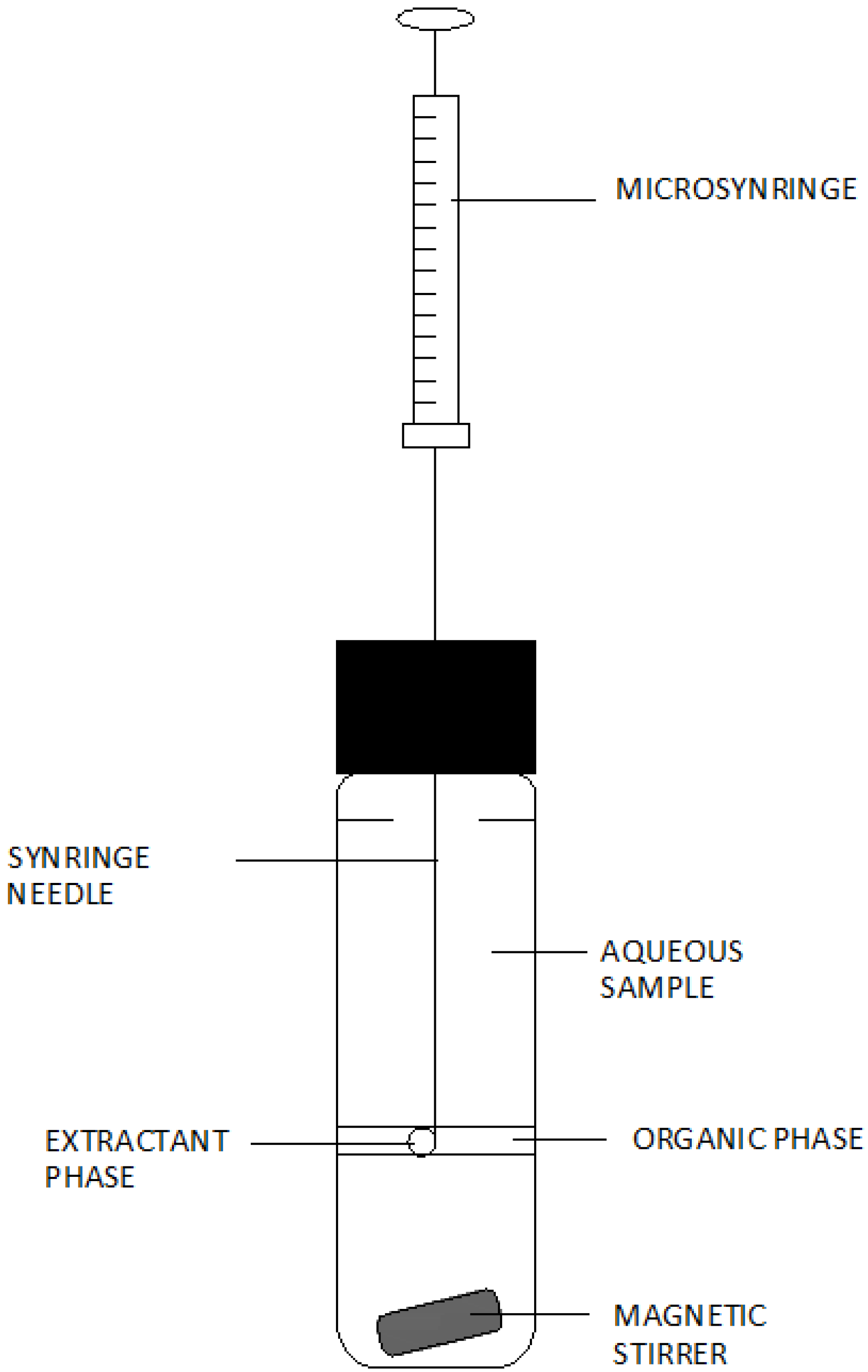
3. Analytical Procedures Using Microextraction Techniques for the Liquid Phase


{kind=link}
{kind=link}
{kind=link}
{kind=link}
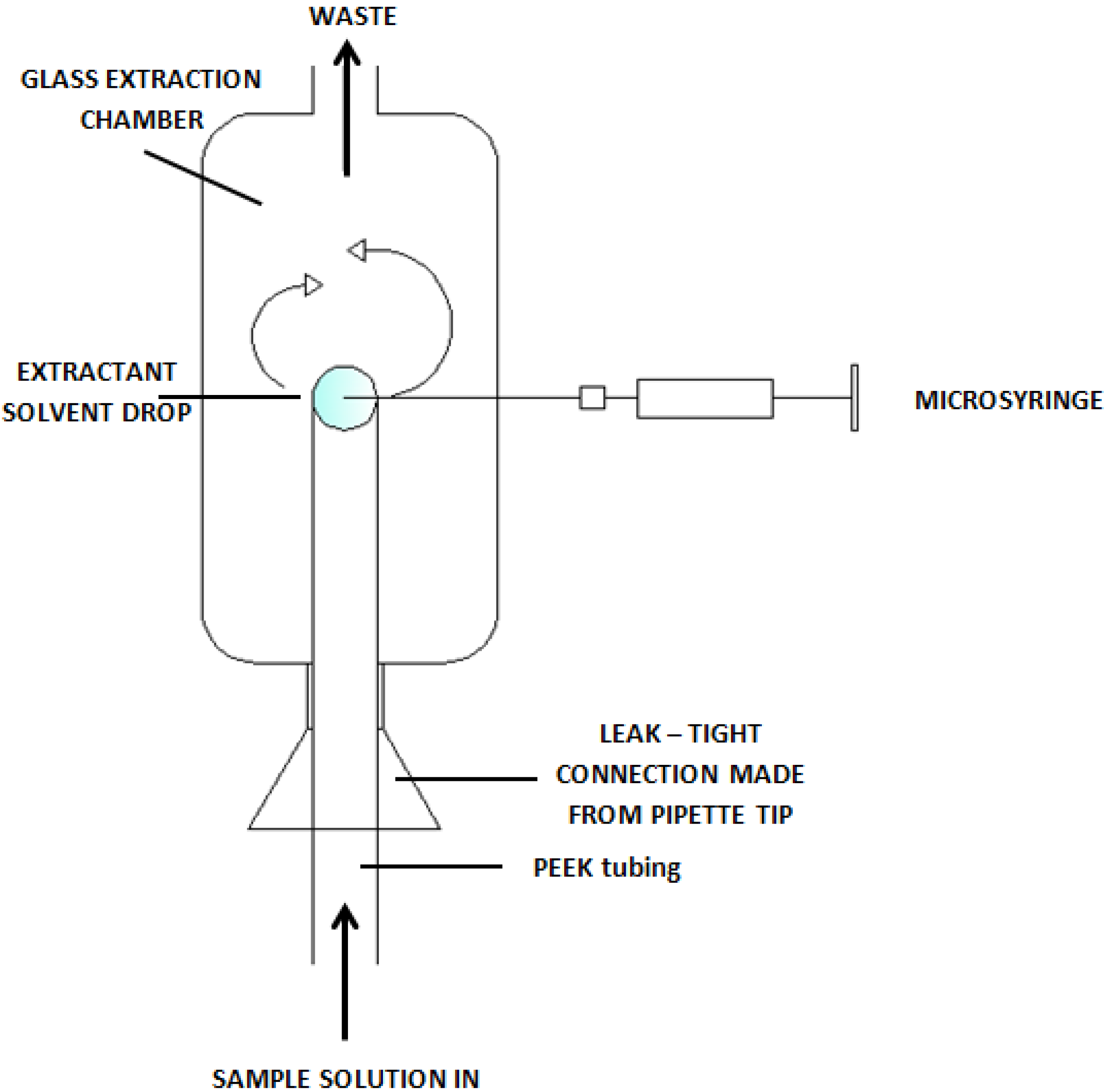{kind=link}
{kind=link}
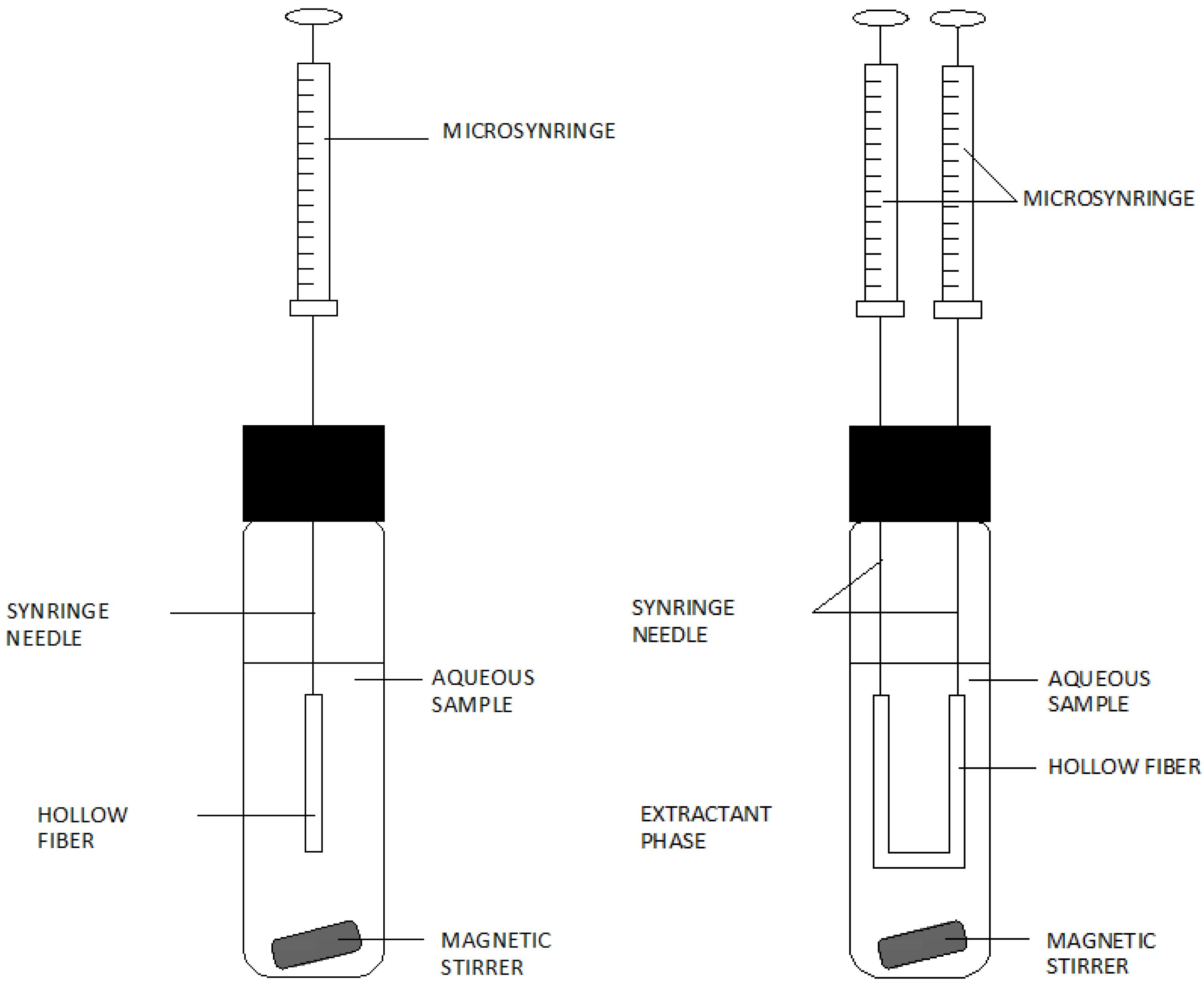{kind=link}
{kind=link}
| Year | Methodological Solution |
|---|---|
| 1995 | First single-drop-based extraction systems |
| 1996 | First drop-in-drop system |
| 1997 | Liquid stage microextraction in a dynamic system The use of microsyringe for supporting the drop |
| 1999 | Liquid-phase microextraction using fibre (LPME) |
| 2001 | Headspace Solid-Phase Microextraction (HS-SDME) |
| 2003 | Using ionic liquids as the extracting agent |
| 2005 | Using water as a solvent in liquid-phase microextraction |
| 2006 | Liquid-phase microextraction using ultrasound as a factor supporting the extraction process |
| 2007 | Liquid-phase microextraction using microwave radiation as a factor supporting the extraction process Automation of the single-drop microextraction process |
| 2008 | Combining microextraction to the liquid phase with flame atomic absorption spectroscopy |
| 2009 | Liquid-phase microextraction using an ionic liquid combined with dispensing a sample to the column using a thermal desorption device |




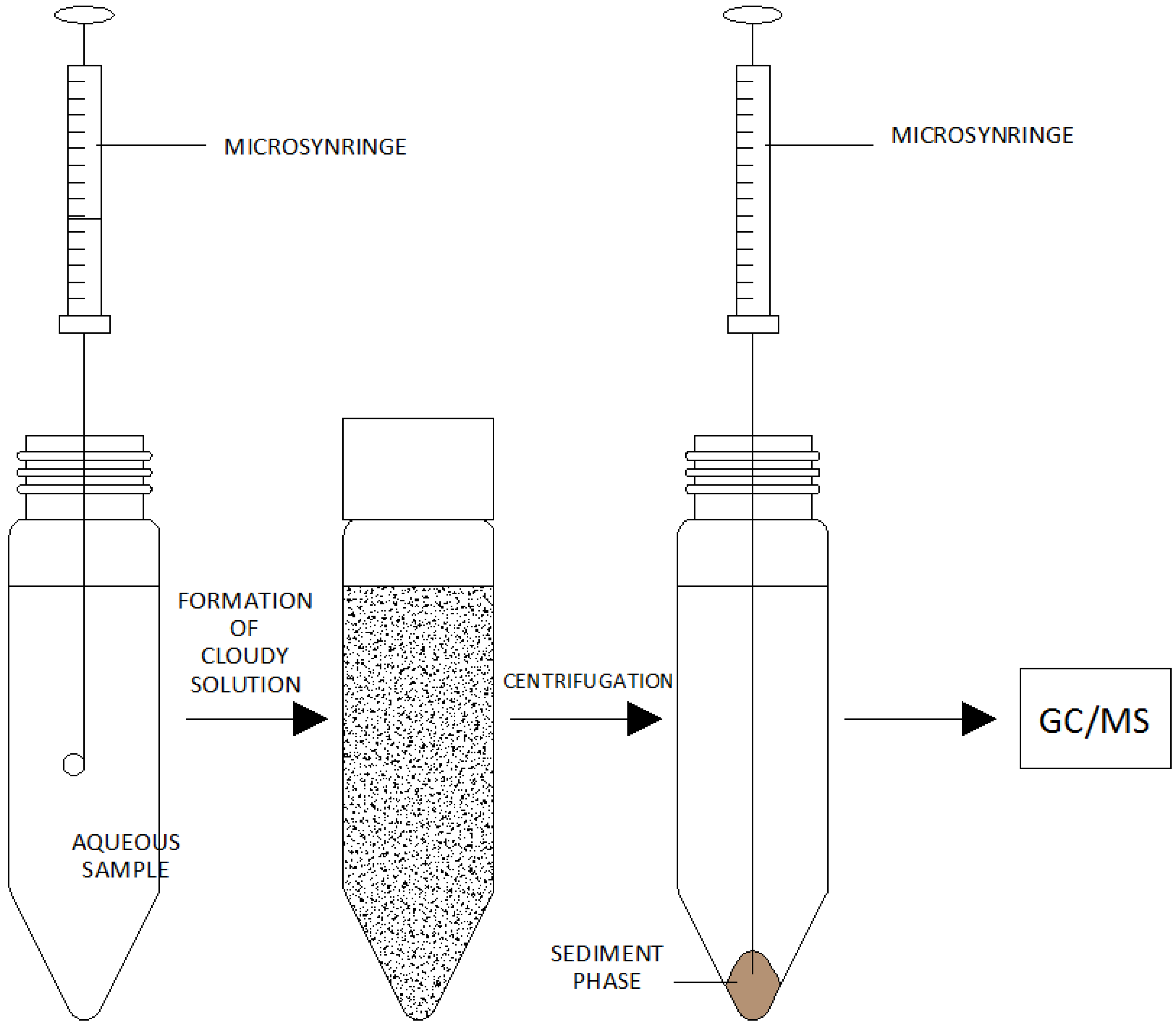
- (a)
- The introduction of an appropriate extraction and dispersing solvent mixture into an aqueous solution of an analyte-containing sample.
- (b)
- Centrifugation of the cloudy solution.


| Sample Type | Species | Method | Derivatization | Fiber/Extraction Time/Extraction Mode Or Extractant Phase/Drop Volume (µL)/Sample Volume (mL)/Extraction Time (min)/Stirring Rate (Flow Rate) | Detection Technique | E.F. | Precision (RSD %) | Detection Limit | Reference | |||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gas condensate | Met2-Hg | SPME | None (direct sampling) | 100 µm PDMS/30 s/HS | MIP-AES | - | - | 20 µg/L | [27] | |||||||||||||||||||||||||||||||||||||||||||
| Water, fish tissue | MetHg | SPME | NaBEt4/acetate buffer pH 4.5 | 100 µm PDMS/5 min/HS | AFS | - | - | 3.0 ng/L | [28] | |||||||||||||||||||||||||||||||||||||||||||
| Water, seawater | TeMT TMT DMT MMT | SPME | NaBEt4/acetic acid buffer pH 4 | 100 µm PDMS/20 min/HS | FPD | - | - | 41 ng/L 15 ng/L 8.4 ng/L 8.6 ng/L | [29] | |||||||||||||||||||||||||||||||||||||||||||
| Surface water, sediment | Alkylmercury Alkyltin | SPME | NaBEt4/acetate buffer pH 5.0 | 100 µm PDMS/10 min/HS | ICP-MS | - | - | 3.7 ng/L 0.38–1.2 ng/L | [30] | |||||||||||||||||||||||||||||||||||||||||||
| Sediment | MBT DBT TBT MetHg | SPME | NaBEt4/acetate buffer pH 5.3 | 100 µm PDMS/10 min/HS | ICP-MS | - | - | 0.34 ng/L 2.1 ng/L 1.1 ng/L 4.3 ng/L | [27,31] | |||||||||||||||||||||||||||||||||||||||||||
| Sediment, sewage sludge | MBT DBT TBT MPhT DPhT TPhT | SPME | NaBEt4/ethanoic acid buffer pH 4.8 | 100 µm PDMS/60 min/LPh | FPD | - | - | 0.031 ng/L 0.007 ng/L 0.006 ng/L 0.114 ng/L 0.167 ng/L 0.583 ng/L | [32] | |||||||||||||||||||||||||||||||||||||||||||
| Slurry of sediment | MBT DBT TBT TeBT | SPME | NaBEt4/acidified with HCl | 100 µm PDMS/45 min/LPh | MIP-AES | - | - | µg/L range | [33] | |||||||||||||||||||||||||||||||||||||||||||
| Soil | MetHg EtHg PhenHg | SPME | Hydride generation (KBH4)/acetate buffer pH 4 | Fused-silica fiber (pretreated with conc. HF acid for 3.5–4 h)/1.5–2 h/HS | AAS (quartz tube) | - | - | Not reported | [27] | |||||||||||||||||||||||||||||||||||||||||||
| Soil | Et2-Hg Met2-Hg | SPME | None (direct sampling) | 100 µm PDMS/20 min/HS | MIP-AES | - | - | 3.5 µg/L | [27] | |||||||||||||||||||||||||||||||||||||||||||
| Environmental, sediment | MBT DBT TBT | SPME | NaBEt4/acetate buffer pH 4 | 100 µm PDMS/60 min/HS | FID | - | - | 10 µg/L 1.2 µg/L 0.9 µg/L | [27] | |||||||||||||||||||||||||||||||||||||||||||
| Body fluids | MBT DBT TBT MetHg Hg2+ | SPME | NaBEt4/acetate buffer pH 5.3 | 100 µm PDMS/10 min/HS | EI-MS-MS | - | - | 9 ng/L 13 ng/L 9 ng/L 22 ng/L 18 ng/L | [34] | |||||||||||||||||||||||||||||||||||||||||||
| Urine | MetHg Hg2+ | SPME | NaBEt4/buffer pH 4 | 100 µm PDMS/15 min/HS | EI-MS | - | - | 303 ng/L 93 ng/L | [35] | |||||||||||||||||||||||||||||||||||||||||||
| Biological samples, sediments | MetHg | SPME | Hydride generation (KBH4)/acetate buffer pH 3 | Fused-silica fiber (pretreated with conc. HF acid for 3.5–4 h)/1.5–2 h/HS | AAS (quartz tube) | - | - | Not reported | [27] | |||||||||||||||||||||||||||||||||||||||||||
| Seawater samples, Sediment sample, Biological samples (fish, crab, prawn) | MeHg | SPME | Na[B(C6H5)4]/acetate buffer pH = 4.5 | 100 µm PDMS/15 min | GC-MS | - | - | 0.02 | [36] | |||||||||||||||||||||||||||||||||||||||||||
| Aqueous samples | Organotin | HS-SPME | NaBEt4 ( in situ)/ammonia/citrate buffer pH 8.5 | 100 mm PDMS | GC-AED | - | - | pg/L ng/L | [37,38] | |||||||||||||||||||||||||||||||||||||||||||
| Organomercury | NaBEt4 ( in situ)/ammonia/citrate buffer pH 5 | CW/PDMS | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Natural water | MBT, TBT, MetHg Hg2+ | HS-SPME | 2% NaBEt4/0.2 M acetic acid and 0.2 M sodium acetate/pH 5.5 | PDMS/30 min | GC-EI-MS | - | - | below ng/L or sub ng/L | [38,39] | |||||||||||||||||||||||||||||||||||||||||||
| Marine sediments | MBT, DBT, TBT | HS-SPME | NaBEt4 ( in situ) | PDMS | GC-MS | - | - | 730–969 pg/g | [37,39,40] | |||||||||||||||||||||||||||||||||||||||||||
| Estuarine superficial sediment | MBT, DBT, TBT | HS-SPME | NaBEt4/1.5 M sodium acetate Buffer/pH 4.3 | 100 mm PDMS/15 min | GC-FID | - | - | - | [37,39,41] | |||||||||||||||||||||||||||||||||||||||||||
| Biological materials and road dust | TMT, DMT, MMT, MBT, DBT, TBT | HS-SPME | - | PDMS/DVB | MC-GC-ICP-TOFMS | - | - | below pg/g | [37,42] | |||||||||||||||||||||||||||||||||||||||||||
| MetHg | CAR/PDMS | 2 pg/g | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Hg2+ | CAR/PDMS | 1.3 pg/g | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Natural water | TMT, DMT, MMT, MetHg Hg2+ | HS-SPME | NaBEt4/buffer pH 5.3 | PDMS µm | GC-MS | - | 5 3 20 14 20 | level ng/L | [39] | |||||||||||||||||||||||||||||||||||||||||||
| DVB/CAR/PDMS 50 µm/30 µm/30 min/5 mL | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Water samples | MeHg DBT TBT | HS-SPME | NaBEt4 | 100 µm PDMS/or 50 µm/30 µm DVB/CAR/PDMS 30 min for MeHg/60 min for DBT and TBT | GC-MS | - | 5 14 20 | 3 ng/L 7 ng/L 16.8 ng/L | [39] | |||||||||||||||||||||||||||||||||||||||||||
| Aqueous samples | MetHg Hg2+ | DI-SPME | - | PDMS | GC-MS | - | - | - | [43] | |||||||||||||||||||||||||||||||||||||||||||
| - | MMT DMT TMT MBT DBT TBT TPT Dioctyltin MetHg EtHg PhenHg Met2-Hg Et2-Hg | SDME | - | [C4MIM][PF6]/[C8MIM][PF6]/5/10/15 (30)/- | ETAAS | 28/18 28/20 90/161 12/14 10/11 15/23 32/24 35/28 5/4 15/13 40/27 15/7 32/14 | - | - | [44] | |||||||||||||||||||||||||||||||||||||||||||
| CV-AFS | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Water | Hg Sn | SDME | NaBH4 in the sample; Pd(II) in the drop | Pd(II)/3/5/3.5/1000 rpm | 72 37 | 8.7 8.2 | 800 90 | [45] | ||||||||||||||||||||||||||||||||||||||||||||
| Water | Hg | SDME | H2Dz in the drop | m-Xylene containing H2Dz/10/ 15/20/300 rpm | ETAAS | 970 | 6.1 | 10 ng/L | [46] | |||||||||||||||||||||||||||||||||||||||||||
| Tuna fish and dogfish muscle | MetHg | SDME | NaBH4 in the sample; Pd(II) in the drop | Pd(II)/3/5/3/300 rpm | ETAAS | 40 | 7 | 5000 | [47] | |||||||||||||||||||||||||||||||||||||||||||
| - | Organotin | HS-SDME | - | Decane/11 min | GC-MS | - | 3.6 | TBT: 3 (Sn) ng/L | [11,14] | |||||||||||||||||||||||||||||||||||||||||||
| Sediment CRM | MBT DBT TBT | HS-SDME | - | Decane/11 min | GC-MS | - | 3.6 | 3 ng/L | [10,11] | |||||||||||||||||||||||||||||||||||||||||||
| Biological, environmental samples | MBT DBT TBT | HS-SDME | - | Decane/5 min | GC-ICP-MS | - | 4.4–10.1 | 0.8–1.8 ng/L | [11] | |||||||||||||||||||||||||||||||||||||||||||
| - | Organomercury | D-SDME | - | [C4MIM][PF6]/15 min | CVAAS | 5–40 | - | - | [14] | |||||||||||||||||||||||||||||||||||||||||||
| - | Organotin | D-SDME | - | [C4MIM][PF6]/15 min | ETAAS | 10–90 | - | - | [14] | |||||||||||||||||||||||||||||||||||||||||||
| - | TBT TPT | D-SDME | - | α,α,α,-Trifluorotoluene/60 min | GC-MS-MS | 140 2.9 | 11 10 | 0.36 ng/l 2.9 ng/L | [14] | |||||||||||||||||||||||||||||||||||||||||||
| River water | Hg | D-SDME | - | Xylene/20 min | ETAAS | 970 | 6.1 | 10 ng/L | [11] | |||||||||||||||||||||||||||||||||||||||||||
| Water samples | MetHg EtHg PhenHg Hg+ | D-SDME | - | [C4MIM][PF6]/20 min | HPLC | 107 31 11 3 | 5.3 3.7 9.4 11.6 | 11.0 ng/L 1.6 ng/L 7.1 ng/L 22.8 ng/L | [11,14] | |||||||||||||||||||||||||||||||||||||||||||
| - | Organotin | DLLME | - | Tetrachloromethane, ethanol/<3 min | GC-FPD | 825–1036 | 2.3–5.9 | 0.2–1 ng/L | [14] | |||||||||||||||||||||||||||||||||||||||||||
| Water samples | MBT DBT TBT | DLLME | butyltin compounds aqueous solution pH = 4.5/NaBEt4 | Tetrachloromethane, methanol/20 min | GC-MS | - | 17 15 9 | 1.7 ng/L 2.5 ng/L 5.9 ng/L | [48] | |||||||||||||||||||||||||||||||||||||||||||
| - | Organomercury | LLLME | - | Toluene, L–cysteine/40 min | CE-UV | 210–324 | 6.1–7.2 | 430–940 ng/L | [14,15] | |||||||||||||||||||||||||||||||||||||||||||
| Water samples | MetHg EtHg PhenHg | HF-LPME | - | Toluene, Na2S2O3/5 min | HPLC-UV | 120 215 350 | 8.9 6.4 6.6 | 3800 ng/L 700 ng/L 300 ng/L | [11] | |||||||||||||||||||||||||||||||||||||||||||
| Human hair, sludge | MetHg | HF-LPME | - | Toluene/0 min | ETAAS | 55 | 11 | 400 ng/L | [11,26] | |||||||||||||||||||||||||||||||||||||||||||
| Human hair, fish sample, dogfish muscle CRM | MetHg EtHg PhenHg | HF-LPME | - | Bromobennzene, L–Cysteine/50 min | LVSS-CE/UV | 3610 3160 4580 | 3.3 3.6 7.5 | 140 ng/L 70 ng/L 30 ng/L | [11] | |||||||||||||||||||||||||||||||||||||||||||
| Fish CRM | MetHg | HF-LPME | - | Toluene/10 min | ETAAS | 55 | 11 | 400 ng/L | [14,26] | |||||||||||||||||||||||||||||||||||||||||||
| Human hair, sludge and dogfish muscle | MetHg | HF-LPME | -/ | Toluene/4/3/10 min/1300 | ETAAS | 55 | 11 | 400 ng/L | [26,49] | |||||||||||||||||||||||||||||||||||||||||||
| thiourea in the lumen of the fibre | Toluene/thiourea 4% (m/v) in 1 mol L−1 HCl | 204 | 13 | 100 ng/L | ||||||||||||||||||||||||||||||||||||||||||||||||
| Fish CRM | MetHg | HF-LLLME | Toluene, thiourea/10 min | ETAAS | 204 | 13 | 100 ng/L | [14,26] | ||||||||||||||||||||||||||||||||||||||||||||
| Fish CRM, water | MetHg EtHg PhenHg | HF-LLLME | - | Polypropylene /toluene (octanol, CCl4)/Na2S2O3/6/3.8/25 min | HPLC-UV | 120–350 | 6.4–8.9 | 3–3.8 ng/mL | [14,16,50] | |||||||||||||||||||||||||||||||||||||||||||
| Dogfish muscle | MeHg EtHg PhHg | HF-LLLME | - | - | On-line FIDS-HPLC | 120 215 350 | 8.9 6.4 6.6 | 10–25 ng/g | [16] | |||||||||||||||||||||||||||||||||||||||||||
| Seawater sample | MeHg EtHg | HF-LLLME | - | - | LC-ICP-MS | 120 215 | 8.9 6.4 | 0.03 ng/mL 0.04 ng/mL | [16] | |||||||||||||||||||||||||||||||||||||||||||
| Fish sample | MeHg | HF-LLLME | - | - | GC-AFS | 120 | 8.9 | 1.2 pg | [16] | |||||||||||||||||||||||||||||||||||||||||||
| Fish tissues | MeHg | HF-LLLME | - | - | GC-ICP-MS | 120 | 8.9 | 2.1 ng/g | [16] | |||||||||||||||||||||||||||||||||||||||||||
4. Analytical Procedures Using Microextraction Techniques for the Stationary Phase
- -
- gas;
- -
- membrane;
- -
- sorbent.
- -
- directly from the tested sample (DI-SPME);
- -
- from the headspace phase (HS-SPME).

- good reproducibility;
- low numerical values of the level of detection;
- reduction of the extraction time;
- reduction of the number of extracted impurities, which, as a result, allows for obtaining chromatograms of considerably better quality.
- extended desorption rate;
- incomplete desorption—memory effect.
| Fibre Cover | Acronym | Thickness of the Film (µm) | Final Determination | Application |
|---|---|---|---|---|
| of Fibre with Non-polar Cover | ||||
| Polydimethylsiloxane | PDMS | 100 30 7 | GC, HPLC | Non-polar organic compounds (Hg0, MetHg, MBT, DBT, TBT, MPhT, DPhT, TPhT), VOCs, PAHs, BTEX |
| Fibre with Polar Cover | ||||
| Polyacrylate | PA | 85 | GC, HPLC | Polar organic compounds, triazine, phosphorganic, pesticides and phenols |
| Fibres with Mixed-Properties Cover | ||||
| Polydimethylsiloxane‒Polydivinylbenzene | PDMS-DVB | 65 60 GC | HPLC | Aromatic hydrocarbons aromatic amines, VOCs, TMT, DMT, MMT, MBT, DBT, TBT |
| Polydimethylsiloxane‒Carboxen | PDMS-CAR | 75 | GC | Gaseous/volatile analytes (Hg0, MeHg), VOCs, hydrocarbons |
| Carbowax‒Polydivinylbenzene | CW-DVB | 65 | GC | Polar organic compounds, alcohols, ketones, nitroaromatic compounds |
| Carbowax-resin with molecular print | CW-TPR | 50 | HPLC | Anion surfactants, aromatic amines |
| Polydimethylsiloxane‒Polydivinylbenzene/Carboxen | PDMS/DVB/CAR | 50/30 | GC | Hg0, MeHg, DBT, TBT |
| Technique | Parameters | References |
|---|---|---|
| LPME |
| [11,12,13,26,69] |
| SPME |
| [52] |
- -
- -
- -
5. Directions for the Development and Possibilities of Using Microextraction Techniques
- -
- modifying already known methodological solutions;
- -
- developing new variants of such techniques, which are characterised by better metrological parameters.
| Advantages | Disadvantages | Technique | References | |
|---|---|---|---|---|
|
| SDME | [11] | |
|
| DI-SDME | [11,12,13,14] | |
|
| HS-SDME | [13] | |
|
| HF-LPME | [11] | |
|
| DLLME | [11,13] | |
| - | LLLME | [15] | |
|
| SPME | [27,29,52,71] | |
| - | CFME | [23] | |
- electromembrane HF(3)ME extraction (EME) of ionized species;
- SME procedures for practical on-line sample analyses;
- solvents less dense than water in DLLME;
- ionic liquids (ILs);
- ultrasound-assisted emulsification for DLLME [19].
6. Conclusions
- -
- high precision and accuracy;
- -
- low numerical value of parameters, such as: the limit of quantification and the value of detection;
- -
- high selectivity [84].
List of abbreviations and acronyms
| Abbreviation Acronym | Explanations |
| LPME | Liquid-Phase Microextraction |
| LLE | Liquid-Liquid Extraction |
| HS-SDME | Headspace Single-Drop Microextraction |
| SDME | Single-Drop microextraction |
| DLLME | Dispersive Liquid-Liquid Microextraction |
| HF-LPME | Hollow-fibre liquid-phase microextraction |
| DI-SDME | Direct Immersion Single-Drop Microextraction |
| LLLME | Liquid-Liquid-Liquid Microextraction |
| CFME | Continuous flow microextraction |
| ETV-ICP-MS | Electrothermal vaporisation Inductively Coupled Plasma Mass Spectrometry |
| CPE | Cloud point extraction |
| GC | Gas Chromatography |
| HPLC | High-performance liquid chromatography |
| CE | Capillary Electrophoresis |
| AS | Atomic Spectroscopy |
| SPME | Solid-Phase Microextraction |
| SPE | Solid-Phase Extraction |
| DI-SPME | Direct Immersion Solid-Phase Microextraction |
| HS-SPME | Headspace Solid-Phase Microextraction |
| MS | Mass Spectrometry |
| ICP | Inductively Coupled Plasma |
| HS-LPME | Headspace Liquid-Phase Microextraction |
| LLLPME | Liquid–Liquid–Liquid-Phase Microextraction |
| EME | Electrokinetic membrane extraction |
Acknowledgments
Conflicts of Interest
References
- Pena-Pereira, F.; Lavilla, I.; Bendicho, C.; Vidal, L.; Canals, A. Speciation of mercury by ionic liquid-based single-drop microextraction combined with high-performance liquid chromatography-photodiode array detection. Talanta 2009, 78, 537–541. [Google Scholar] [CrossRef]
- Pongratz, R.; Heumann, K.G. Production of methylated mercury, lead, and cadmium by marine bacteria as a significant natural source for atmospheric heavy metals in polar regions. Chemosphere 1999, 39, 89–102. [Google Scholar] [CrossRef]
- Rezaee, M.; Assadi, Y.; Milani Hosseini, M.-R.; Aghaee, E.; Ahmadia, F.; Berijani, S. Determination of organic compounds in water using dispersive liquid–liquid microextraction. J. Chromatogr. A 2006, 1116, 1–9. [Google Scholar] [CrossRef]
- Liu, H.; Dasgupta, P.K. Analytical chemistry in a drop. Solvent extraction in a microdrop. Anal. Chem. 1996, 68, 1817–1821. [Google Scholar]
- Oliveira, R.; Santeli, R.E. Occurrence and Chemical Speciation Analysis of Organotin Compounds in the Environment: A Review. Talanta 2010, 82, 9–24. [Google Scholar]
- Pena-Pereira, F.; Lavilla, I.; Bendicho, C. Liquid-phase microextraction techniques within the framework of green chemistry. Trends Anal. Chem. 2010, 29, 617–628. [Google Scholar] [CrossRef]
- Xu, L.; Basheer, C.; Lee, H.K. Developments in single-drop microextraction. J. Chromatogr. A 2007, 1152, 184–192. [Google Scholar] [CrossRef]
- Wardencki, W.; Curyło, J.; Namiesnik, J. Trends in solventless sample preparation techniques for environmental analysis. J. Biochem. Biophys. Methods 2007, 70, 257–288. [Google Scholar]
- Li, Y.; Xionga, Y.; Lianga, Q.; Fanga, C.; Wangb, C. Application of headspace singledrop microextraction coupled with gas chromatography for the determination of short-chain fatty acids in RuO4 oxidation products of asphaltenes. J. Chromatogr. A 2010, 1217, 3561–3566. [Google Scholar]
- Colombini, V.; Bancon-Montigny, C.; Yang, L.; Maxwell, P.; Sturgeon, R.E.; Mester, Z. Headspace single-drop microextration for the detection of organotin compounds. Talanta 2004, 63, 555–560. [Google Scholar] [CrossRef]
- Dadfarnia, S.; Shabani, A.M.H. Recent development in liquid phase microextraction for determination of trace level concentration of metals—A review. Anal. Chim. Acta 2010, 658, 107–119. [Google Scholar] [CrossRef]
- Stepnowski, P.; Synak, E.; Szafranek, B.; Kaczyński, Z. Chapter. 4; Techniki Separacyjne; Uniwersytet Gdański: Gdańsk, Poland, 2010; pp. 69–73. [Google Scholar]
- Sarafraz-Yazdi, A.; Amiri, A. Liquid-phase microextraction. Trends Anal. Chem. 2010, 29, 1–14. [Google Scholar] [CrossRef]
- Pena-Pereira, F.; Lavilla, I.; Bendicho, C. Miniaturized preconcentration methods based on liquid–liquid extraction and their application in inorganic ultratrace analysis and speciation: A review. Spectrochim. Acta B 2009, 64, 1–15. [Google Scholar] [CrossRef]
- Fan, Z.; Liu, X. Determination of methylmercury and phenylmercury in water samples by liquid-liquid-liquid microextraction coupled with capillary electrophoresis. J. Chromatogr. A 2008, 1180, 187–192. [Google Scholar] [CrossRef]
- Xia, L.; Hu, B.; Wu, Y. Hollow fiber-based liquid–liquid–liquid microextraction combined with high-performance liquid chromatography for the speciation of organomercury. J. Chromatogr. A 2007, 1173, 44–51. [Google Scholar] [CrossRef]
- Ma, M.H.; Cantwell, F.F. Solvent microextraction with simultaneous back-extraction for sample cleanup and preconcentration: Quantitative extraction. Anal. Chem. 1998, 70, 3912–3919. [Google Scholar] [CrossRef]
- Ma, M.H.; Cantwell, F.F. Solvent microextraction with simultaneous back-extraction for sample cleanup and preconcentration: Preconcentration into a single Microdrop. Anal. Chem. 1999, 71, 388–393. [Google Scholar] [CrossRef]
- Kokosa, J.M. Advances in solventmicroextraction techniques. Trends Anal. Chem. 2013, 43, 2–13. [Google Scholar] [CrossRef]
- Bjergaard, S.P.; Rasmussen, K.E. liquid-liquid-liquid microextraction for sample preparation of biological fluids prior to capillary electrophoresis. Anal. Chem. 1999, 71, 2650–2656. [Google Scholar] [CrossRef]
- Marlow, M.; Hurtubise, R.J. liquid-liquid-liquid microextraction for the enrichment of polycyclic aromatic hydrocarbon metabolites investigated with fluorescence spectroscopy and capillary electrophoresis. Anal. Chim. Acta 2004, 526, 41–49. [Google Scholar] [CrossRef]
- Zhao, L.M.; Lee, H.K. Determination of phenols in water using liquid phase microextraction with back extraction combined with high-performance liquid chromatography. J. Chromatogr. A 2001, 931, 95–105. [Google Scholar] [CrossRef]
- Liu, W.; Lee, H.K. Continuous-flow microextraction exceeding 1000-fold concentration of dilute analytes. Anal. Chem. 2000, 72, 4462–4467. [Google Scholar] [CrossRef]
- Kozani, R.R.; Assad, Y.; Shemirani, F.; Milani Hosseini, M.R.; Jamali, M.R. Part-per-trillion determination of chlorobenzenes in water using dispersive liquid-liquid microextraction combined gas chromatography-electron capture detection. Talanta 2007, 72, 387–393. [Google Scholar] [CrossRef]
- Rasmussen, K.E.; Pedersen-Bjergaard, S. Developments in hollow fibrebased, liquid-phase microextraction. Trends Anal. Chem. 2004, 23, 1–10. [Google Scholar] [CrossRef]
- Jiang, H.; Hu, B.; Chen, B.; Zu, W. Hollow fiber liquid phase microextraction combined with graphite furnace atomic absorption spectrometry for the determination of methylmercury in human hair and sludge samples. Spectrochim. Acta B 2008, 63, 770–776. [Google Scholar] [CrossRef]
- Mestera, Z.; Sturgeon, R.; Pawliszyn, J. Solid phase microextraction as a tool for trace element speciation. Spectrochim. Acta B 2001, 56, 233–260. [Google Scholar] [CrossRef]
- Cai, Y.; Monsalud, S.; Furton, K.G.; Jaffé, R.; Jones, R.D. Determination of methylmercury in fish and aqueous samples using solid-phase microextraction followed by gas chromatography-atomic fluorescence spectrometry. Appl. Organomet. Chem. 1998, 12, 565–569. [Google Scholar] [CrossRef]
- Nerín, C.; Salafranca, J.; Aznar, M.; Batlle, R. Critical review on recent developments in solventless techniques for extraction of analytes. Anal. Bioanal. Chem. 2009, 393, 809–833. [Google Scholar] [CrossRef]
- De Smaele, T.; Moens, L.; Sandra, P.; Dams, R. Determination of organometallic compounds in surface water and sediment samples with SPME–CGC–ICPMS. Mikrochim. Acta 1999, 130, 241–251. [Google Scholar] [CrossRef]
- Bin, H.; Guibin, J. Analysis of organomercuric species in soils from orchards and wheat fields by capillary gas chromatography on-line coupled with atomic absorption spectrometry after in situ hydride generation and headspace solid phase microextraction. Fresenius J. Anal. Chem. 1999, 365, 615–618. [Google Scholar] [CrossRef]
- Aguerre, S.; Bancon Montigny, C.; Lespes, G.; Potin Gautier, M. Solid phase microextraction (SPME): A new procedure for the control of butyl- and phenyl-tin pollution in the environment by GC-FPD. Analyst 2000, 125, 263–268. [Google Scholar] [CrossRef]
- Tutschku, S.; Mothes, S.; Wennrich, R. Preconcentration and determination of Sn- and Pb-organic species in environmental samples by SPME and GC-AED. Fresenius J. Anal. Chem. 1996, 354, 587–591. [Google Scholar]
- Dunemann, L.; Hajimiragha, H.; Begerow, J. Simultaneous determination of Hg(II) and alkylated Hg, Pb, and Sn species in human body fluids using SPME–GC/MS-MS. Fresenius J. Anal. Chem. 1999, 363, 466–468. [Google Scholar] [CrossRef]
- Guidotti, M. Determination of urinary mercury and methylmercury by solid phase microextraction and GC/MS. J. High Resolut. Chromatogr. 1998, 21, 665–666. [Google Scholar] [CrossRef]
- Mishra, S.; Tripathi, R.M.; Bhalke, S.; Shukla, V.K.; Puranik, V.D. Determination of methylmercury and mercury(II) in a marine ecosystem using solid-phase microextraction gas chromatography-mass spectrometry. Anal. Chim. Acta 2005, 551, 192–198. [Google Scholar] [CrossRef]
- Kaur, V.; Malik, A.K.; Verma, N. Applications of solid phase microextraction for the determination of metallic and organometallic species. J. Sep. Sci. 2006, 29, 333–345. [Google Scholar]
- Mothes, S.; Wennrich, R. Coupling of SPME and GC-AED for the determination of organometallic compounds. Mikrochim. Acta 2000, 135, 91–95. [Google Scholar] [CrossRef]
- Centineo, G.; Blanco González, E.; Sanz-Medel, A. Multielemental speciation analysis of organometallic compounds of mercury, lead and tin in natural water samples by headspace-solid phase microextraction followed by gas chromatography–mass spectrometry. J. Chromatogr. A 2004, 1034, 191–197. [Google Scholar]
- Cardellicchio, N.; Giandomenico, S.; Decataldo, A.; di Leo, A. Speciation of butyltin compounds in marine sediments with headspace solid phase microextraction and gas chromatography-mass spectrometry. Fresenius J. Anal. Chem. 2001, 369, 510–515. [Google Scholar] [CrossRef]
- Arambarri, I.; Gracia, R.; Millan, E. Assessment of tin and butyltin species in estuarine superficial sediments from Gipuzkoa, Spain. Chemosphere 2003, 51, 643–649. [Google Scholar] [CrossRef]
- Jitaru, P.; Goenaga, I.; Heidi, A.; Freddy, C. Simultaneous multi-elemental speciation analysis of organometallic compounds by solid-phase microextraction and multicapillary gas chromatography hyphenated to inductively coupled plasma-time-of-flight-mass spectrometry. J. Anal. At. Spectrom. 2004, 19, 867–875. [Google Scholar] [CrossRef]
- Ouyang, G.; Pawliszyn, J. SPME in environmental analysis. Anal. Bioanal. Chem. 2006, 386, 1059–1073. [Google Scholar] [CrossRef]
- Liu, J.; Chi, Y.; Jiang, G. Screening the extractability of some typical environmental pollutants by ionic liquids in liquid-phase microextraction. J. Sep. Sci. 2005, 28, 87–91. [Google Scholar] [CrossRef]
- Gil, S.; de Loos-Vollebregt, M.T.C.; Bendicho, C. Optimization of a single-drop microextraction method for multielemental determination by electrothermal vaporization inductively coupled plasma mass spectrometry following in situ vapor generation. Spectrochim. Acta B 2009, 64, 208–214. [Google Scholar] [CrossRef]
- Bagheri, H.; Naderi, M. Immersed single-drop microextraction-electrothermal vaporization atomic absorption spectroscopy for the trace determination of mercury in water samples. J. Hazard. Mater. 2009, 165, 353–358. [Google Scholar] [CrossRef]
- Gil, S.; Fragueiro, S.; Lavilla, I.; Bendicho, C. Determination of methylmercury by electrothermal atomic absorption spectrometry using headspace single-drop microextraction with in situ hydride generation. Spectrochim. Acta B 2005, 60, 145–150. [Google Scholar] [CrossRef]
- Šmitienė, V.; Baškirova, I.; Vičkačkaitė, V. Speciation of butyltins by dispersive liquid-liquid microextraction and gas chromatography-mass Spectrometry. Chemija 2013, 24, 210–216. [Google Scholar]
- Pena-Pereira, F.; Lavilla, I.; Bendicho, C. Liquid-phase microextraction approaches combined with atomic detection: A critical review. Anal. Chem. Acta 2010, 669, 1–16. [Google Scholar] [CrossRef]
- Xiao, Q.; Hu, B.; He, M. Speciation of butyltin compounds in environmental and biological samples using headspace single drop microextraction coupled with gas chromatography-induc tively coupled plasma mass spectrometry. J. Chromatogr. A 2008, 1211, 135–141. [Google Scholar] [CrossRef]
- Pawliszyn, J. New directions in sample preparation for analysis of organic compounds. Trends Anal. Chem. 1995, 14, 113–122. [Google Scholar]
- Banel, A.; Zygmunt, B. Application of combination of solid phase microextraction and gas chromatography for determination of volatile fatty acids in environmental and related samples. Ecol. Chem. Eng. 2008, 15, 7–28. [Google Scholar]
- Psillakis, E.; Kalogerakis, N. Hollow-fibre liquid-phase microextraction of phthalate esters from water. J. Chromatogr. A 2003, 999, 145–153. [Google Scholar] [CrossRef]
- Psillakis, E.; Kalogerakis, N. Application of solvent microextraction to the analysis of nitroaromatic explosives in water samples. J. Chromatogr. A 2001, 907, 211–219. [Google Scholar] [CrossRef]
- Psillakis, E.; Kalogerakis, N. Solid-phase microextraction versus single-drop microextraction for the analysis of nitroaromatic explosives in water samples. J. Chromatogr. A 2001, 938, 113–120. [Google Scholar] [CrossRef]
- Mester, Z.; Sturgeon, R. Trace element speciation using solid phase microextraction. Spectrochim. Acta B 2005, 60, 1243–1269. [Google Scholar] [CrossRef]
- Alpendurada, M.F. Solid-phase microextraction: A promising technique for sample preparation in environmental analysis. J. Chromatogr. A 2000, 889, 3–14. [Google Scholar] [CrossRef]
- Sporkert, F.; Pragst, F. Use of headspace solid-phase microextraction (HS-SPME) in hair analysis for organic compounds. Forensic Sci. Int. 2000, 107, 129–148. [Google Scholar] [CrossRef]
- Eisert, R.; Pawliszyn, J. Automated in-tube solid-phase microextraction coupled to high-performance liquid chromatography. Anal. Chem. 1997, 60, 3140–3147. [Google Scholar] [CrossRef]
- Eisert, R.; Levsen, K. Development of a prototype system for quasi-continuous analysis of organic contaminants in surface or sewage water based on in-line coupling of solid-phase microextraction to gas chromatography. J. Chromatogr. A 1996, 737, 59–65. [Google Scholar] [CrossRef]
- Peñalver, A.; Pocurull, E.; Borrull, F.; Marceè, R.M. Trends in solid-phase microextraction for determining organic pollutants in environmental samples. Trends Anal. Chem. 1999, 18, 557–568. [Google Scholar]
- Jinno, K.; Muramatsu, T.; Saito, Y.; Kiso, Y.; Magdic, S.; Pawliszyn, J. Analysis of pesticides in environmental water samples by solid-phase micro-extraction–High-performance liquid chromatography. J. Chromatogr. A 1996, 754, 137–144. [Google Scholar]
- Boyd-Boland, A.A.; Pawliszyn, J.B. Solid-phase microextraction coupled with high-performance liquid chromatography for the determination of alkylphenol ethoxylate surfactants in water. Anal. Chem. 1996, 68, 1521–1529. [Google Scholar] [CrossRef]
- Li, S.; Weber, S.G. Determination of barbiturates by solid-phase microextraction and capillary electrophoresis. Anal. Chem. 1997, 69, 1217–1222. [Google Scholar] [CrossRef]
- Whang, C.W.; Pawliszyn, J. Solid phase microextraction coupled to capillary electrophoresis. Anal. Commun. 1998, 35, 353–356. [Google Scholar] [CrossRef]
- Darrach, M.R.; Chutjian, A.; Plett, G.A. Trace explosives signatures from World War II unexploded undersea ordnance. Environ. Sci. Technol. 1998, 32, 1354–1358. [Google Scholar] [CrossRef]
- Pan, L.; Pawliszyn, J. Derivatization/solid-phase microextraction: New approach to polar analytes. Anal. Chem. 1997, 69, 196–205. [Google Scholar] [CrossRef]
- Field, J.A. Coupling chemical derivatization reactions with supercritical fluid extraction. J. Chromatogr. A 1997, 785, 239–249. [Google Scholar] [CrossRef]
- Theis, A.L.; Waldack, A.J.; Hansen, S.M.; Jeannot, M.A. Headspace solvent microextraction. Anal. Chem. 2001, 73, 5651–5654. [Google Scholar] [CrossRef]
- Stashenko, E.E.; Martỉnez, J.R. Derivatization and solid-phasemicroextraction. Trends Anal. Chem. 2004, 23, 553–561. [Google Scholar] [CrossRef]
- Grinberg, P.; Campos, R.C.; Mester, Z.; Sturgeon, R.E. Solid phase microextraction capillary gas chromatography combined with furnace atomization plasma emission spectrometry for speciation of mercury in fish tissues. Spectrochim. Acta B 2003, 58, 427–441. [Google Scholar] [CrossRef]
- Tutschku, S.; Schantz, M.M.; Horvat, M.; Logar, M.; Akagi, H.; Emons, H.; Levenson, M.; Wise, S.A. Certification of the methylmercury content in SRM 2977 mussel tissue (organic contaminants and trace elements) and SRM 1566b oyster tissue. Fresenius J. Anal. Chem. 2001, 369, 364–369. [Google Scholar] [CrossRef]
- Gorecki, T.; Pawliszyn, J. Determination of tetraethyllead and inorganic lead in water by solid phase microextraction gas chromatography. Anal. Chem. 1996, 68, 3008–3014. [Google Scholar]
- Moens, L.; de Smaele, T.; Dams, R.; VandenBroeck, P.; Sandra, P. Sensitive, simultaneous determination of organomercury, -lead, and -tin compounds with headspace solid phase microextraction capillary gas chromatography combined with inductively coupled plasma mass spectrometry. Anal. Chem. 1997, 69, 1604–1611. [Google Scholar] [CrossRef]
- Yu, X.M.; Pawliszyn, J. Speciation of alkyllead and inorganic lead by derivatization with deuterium-labeled sodium tetraethylborate and SPME–GC/MS. Anal. Chem. 2000, 72, 1788–1792. [Google Scholar] [CrossRef]
- Yu, X.M.; Yuan, H.D.; Gorecki, T.; Pawliszyn, J. Determination of lead in blood and urine by SPME/GC. Anal. Chem. 1999, 71, 2998–3002. [Google Scholar] [CrossRef]
- Mester, Z.; Lam, J.; Sturgeon, R.; Pawliszyn, J. Determination of methylmercury by solid-phase microextraction inductively coupled plasma mass spectrometry: A new sample introduction method for volatile metal species. J. Anal. At. Spectrom. 2000, 15, 837–842. [Google Scholar] [CrossRef]
- Vercauteren, J.; de Meester, A.; de Smaele, T.; Vanhaecke, F.; Moens, L.; Dams, R.; Sandra, P. Headspace solidphase microextraction—capillary gas chromatography—ICP mass spectrometry for the determination of the organotin pesticide fentin in environmental samples. J. Anal. At. Spectrom. 2000, 15, 651–656. [Google Scholar] [CrossRef]
- Stashenko, E.E.; Martỉnez, J.R. Sampling flower scent for chromatographic analysis. J. Sep. Sci. 2008, 31, 2022–2031. [Google Scholar] [CrossRef]
- He, Y.; Lee, H.K. Continuous flow microextraction combined with high-performance liquid chromatography for the analysis of pesticides in natural waters. J. Chromatogr. A 2006, 1122, 7–12. [Google Scholar] [CrossRef]
- Xia, L.; Li, X.; Wu, Y.; Hu, B.; Chen, R. Ionic liquids based single drop microextraction combined with electrothermal vaporization inductively coupled plasma mass spectrometry for determination of Co, Hg and Pb in biological and environmental samples. Spectrochim. Acta B 2008, 63, 1290–1296. [Google Scholar] [CrossRef]
- Li, S.; Cai, S.; Hu, W.; Chen, H.; Liu, H. Ionic liquid-based ultrasound-assisted dispersive liquid-liquid microextraction combined with electrothermal atomic absorption spectrometry for a sensitive determination of cadmium in water samples. Spectrochim. Acta B 2009, 64, 666–671. [Google Scholar] [CrossRef]
- Dong, S.; Hu, Q.; Yang, Z.; Liu, R.; Huang, G.; Huang, T. An ionic liquid-based ultrasound assisted dispersive liquid–liquid microextraction procedure followed by HPLC for the determination of low concentration of phytocides in soil. Microchem. J. 2013, 110, 221–226. [Google Scholar] [CrossRef]
- Stanisz, E.; Werner, J.; Matusiewicz, H. Mercury species determination by task specific ionic liquid-based ultrasoundassisted dispersive liquid-liquid microextraction combined with cold vapour generation atomic absorption spectrometry. Microchem. J. 2013, 110, 28–35. [Google Scholar] [CrossRef]
© 2014 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rutkowska, M.; Dubalska, K.; Konieczka, P.; Namieśnik, J. Microextraction Techniques Used in the Procedures for Determining Organomercury and Organotin Compounds in Environmental Samples. Molecules 2014, 19, 7581-7609. https://doi.org/10.3390/molecules19067581
Rutkowska M, Dubalska K, Konieczka P, Namieśnik J. Microextraction Techniques Used in the Procedures for Determining Organomercury and Organotin Compounds in Environmental Samples. Molecules. 2014; 19(6):7581-7609. https://doi.org/10.3390/molecules19067581
Chicago/Turabian StyleRutkowska, Małgorzata, Kinga Dubalska, Piotr Konieczka, and Jacek Namieśnik. 2014. "Microextraction Techniques Used in the Procedures for Determining Organomercury and Organotin Compounds in Environmental Samples" Molecules 19, no. 6: 7581-7609. https://doi.org/10.3390/molecules19067581