3-Aminothiophene-2-Acylhydrazones: Non-Toxic, Analgesic and Anti-Inflammatory Lead-Candidates

 , ,
, ,
Abstract
:
1. Introduction


2. Results and Discussion
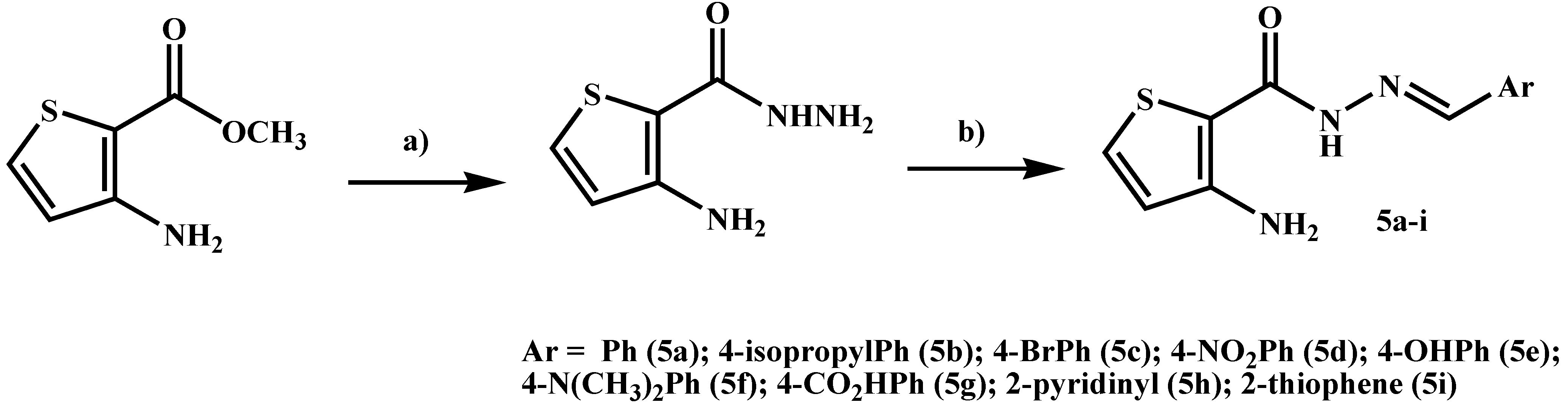
2.1. Chemistry

2.2. In Silico Drug-Like Profile and Toxicity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Theoretical Toxicity Risks * | |||
|---|---|---|---|---|
| Mutagenic | Tumorigenic | Irritant | Reproductive Effects | |
| Dipyrone | 3 | 3 | 1 | 3 |
| Dexamethasone | 1 | 1 | 1 | 1 |
| Indomethacin | 1 | 1 | 1 | 1 |
| 5a | 1 | 1 | 1 | 1 |
| 5b | 1 | 1 | 3 | 1 |
| 5c | 1 | 1 | 1 | 1 |
| 5d | 1 | 1 | 1 | 1 |
| 5e | 1 | 1 | 2 | 2 |
| 5f | 1 | 3 | 1 | 1 |
| 5g | 1 | 1 | 1 | 1 |
| 5h | 3 | 1 | 1 | 1 |
| 5i | 1 | 1 | 1 | 1 |
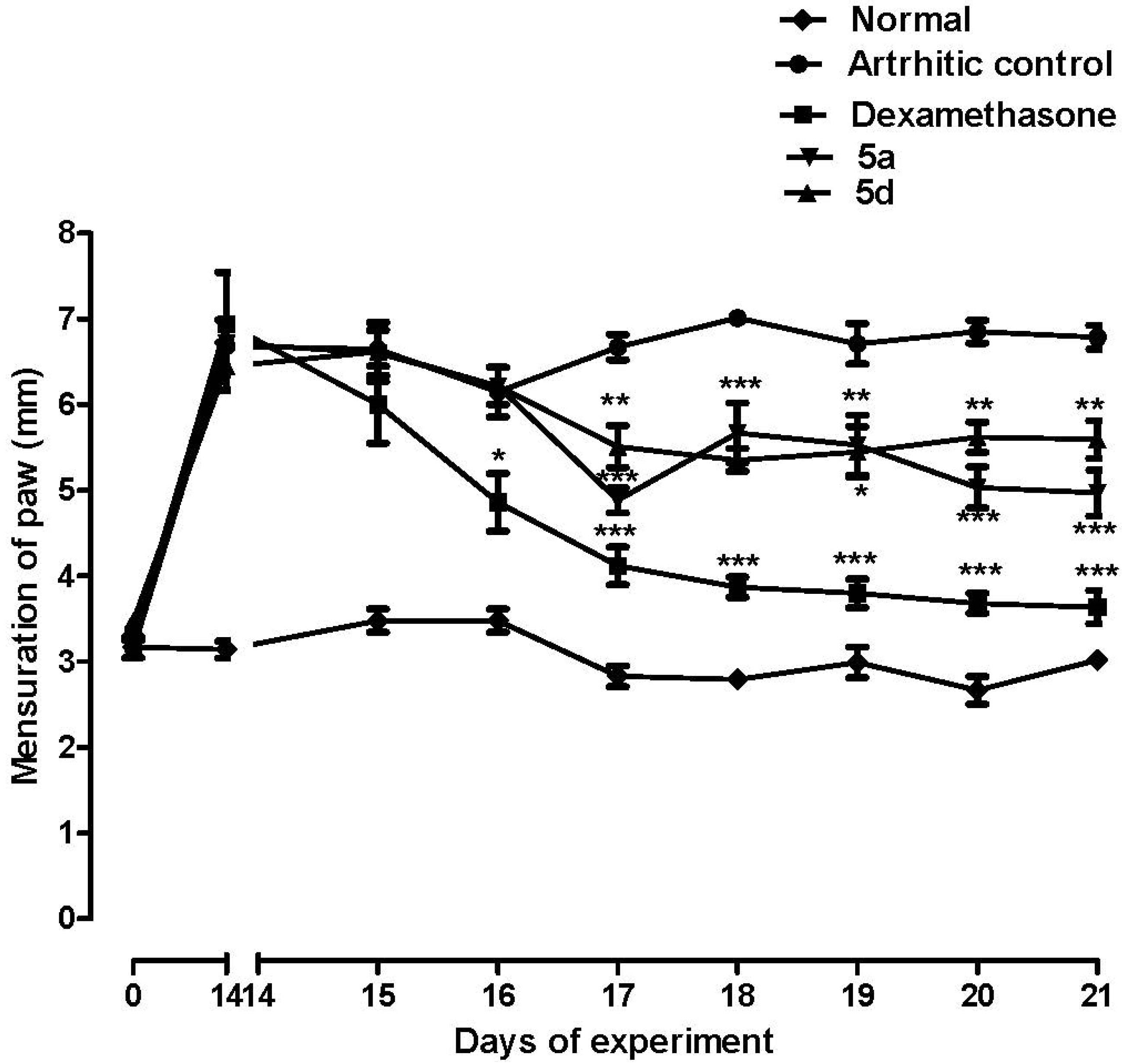
2.3. Biological Assays
| Compound | ID50 (µmol/Kg ± S.E.M.) | Emax (% ± S.E.M.) |
|---|---|---|
| Dipyrone | 11.4 ± 4.9 | 75.2 ± 5.8 |
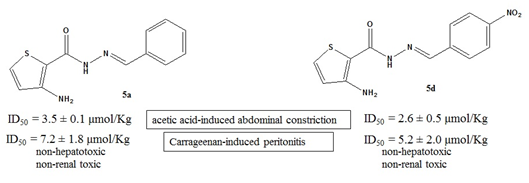
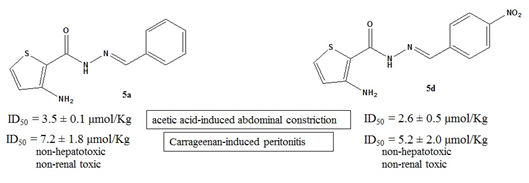
| 5a | 3.5 ± 0.1 | 72.6 ± 4.1 |
| 5b | 6.1 ± 2.2 | 64.3 ± 8.5 |
| 5c | 2.3 ± 0.4 | 76.7 ± 5.2 |
| 5d | 2.6 ± 0.5 | 81.0 ± 5.7 |
| 5e | >100 | - |
| 5f | 32.8 ± 17.3 | 74.5 ± 9.6 |
| 5g | >100 | - |
| 5h | 2.5 ± 0.4 | 84.1 ± 5.6 |
| 5i | 7.8 ± 3.2 | 86.8 ± 5.2 |

| Compound | ID50 (µmol/Kg ± S.E.M.) | Emax (% ± S.E.M.) |
|---|---|---|
| Indomethacin | 3.3 ± 1.0 | 73.8 ± 5.1 |
| 5a | 7.2 ± 1.8 | 69.7 ± 12.1 |
| 5b | 15.9 ± 6.3 | 73.9 ± 11.7 |
| 5c | 8.5 ± 4.2 | 72.7 ± 5.9 |
| 5d | 5.2 ± 2.0 | 70.3 ± 5.2 |
| 5e | >100 | - |
| 5f | 8.8 ± 6.1 | 62.4 ± 5.8 |
| 5g | 15.3 ± 12.5 | 75.1 ± 1.6 |
| 5h | 20.2 ± 17.2 | 76.4 ± 10.1 |
| 5i | >100 | - |




3. Experimental
3.1. General Information
3.2. General Procedure for the Preparation of 3-Aminothiophene-2-Carbohydrazide [23]
3.3. General Procedure for the Preparation of 3-Aminothiophene-2-Carbohydrazide Derivatives 5a–i
3.4. In Silico Toxicological Evaluation and Drug-Like Profile
3.5. Analgesic and Anti-Inflammatory Murine Models
3.5.1. Animals
3.5.2. Acetic Acid-Induced Abdominal Constriction Test
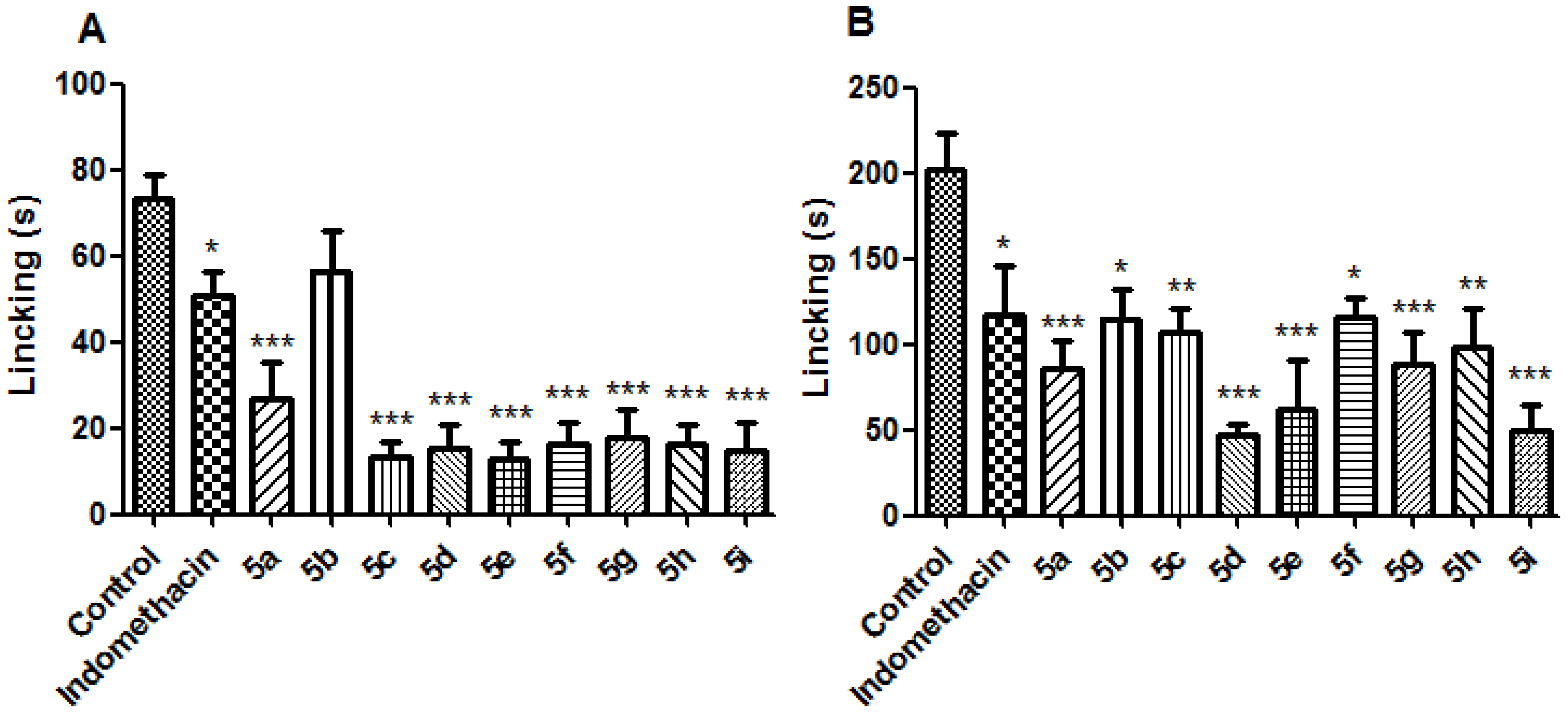
3.5.3. Formalin Test
3.5.4. Carrageenan-Induced Peritonitis
3.5.5. Arthritis Model Induced by Complete Freund’s Adjuvant (CFA) in Rats
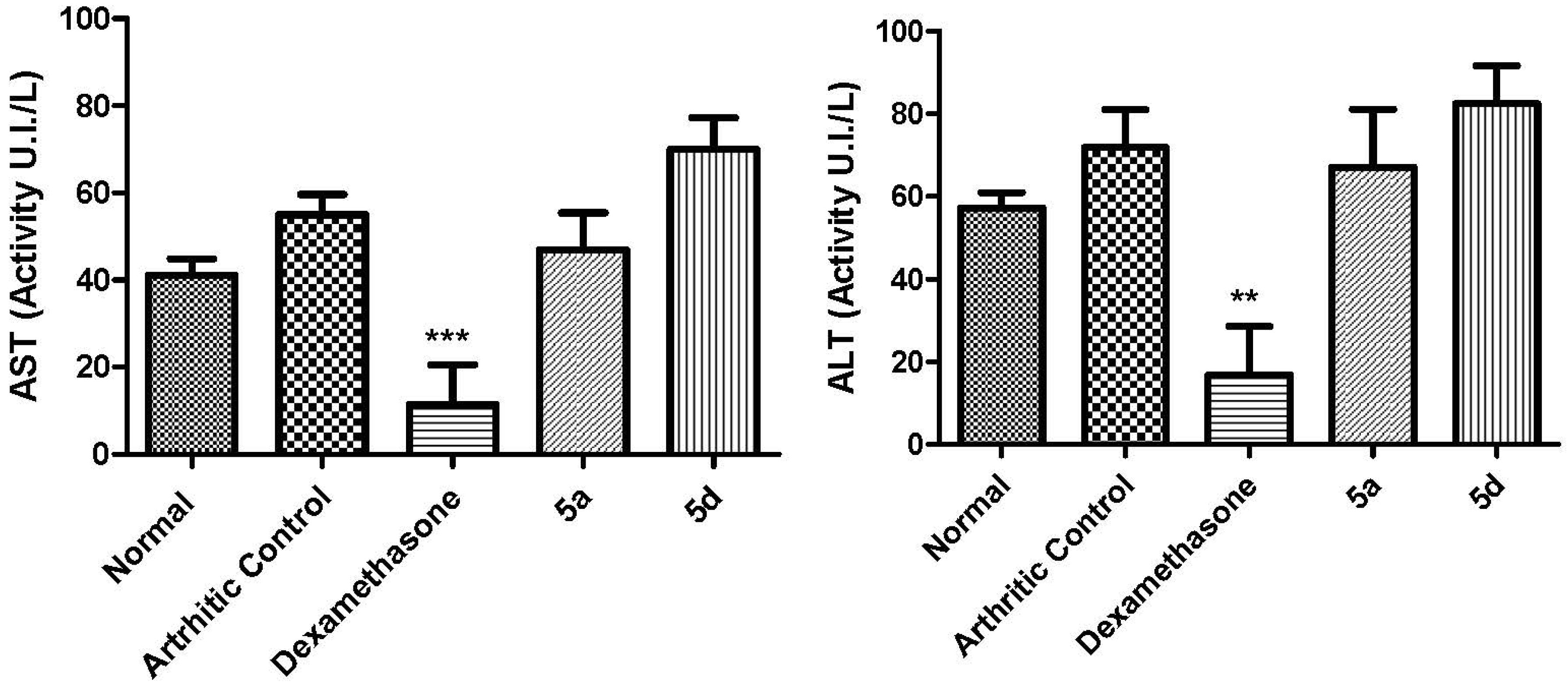
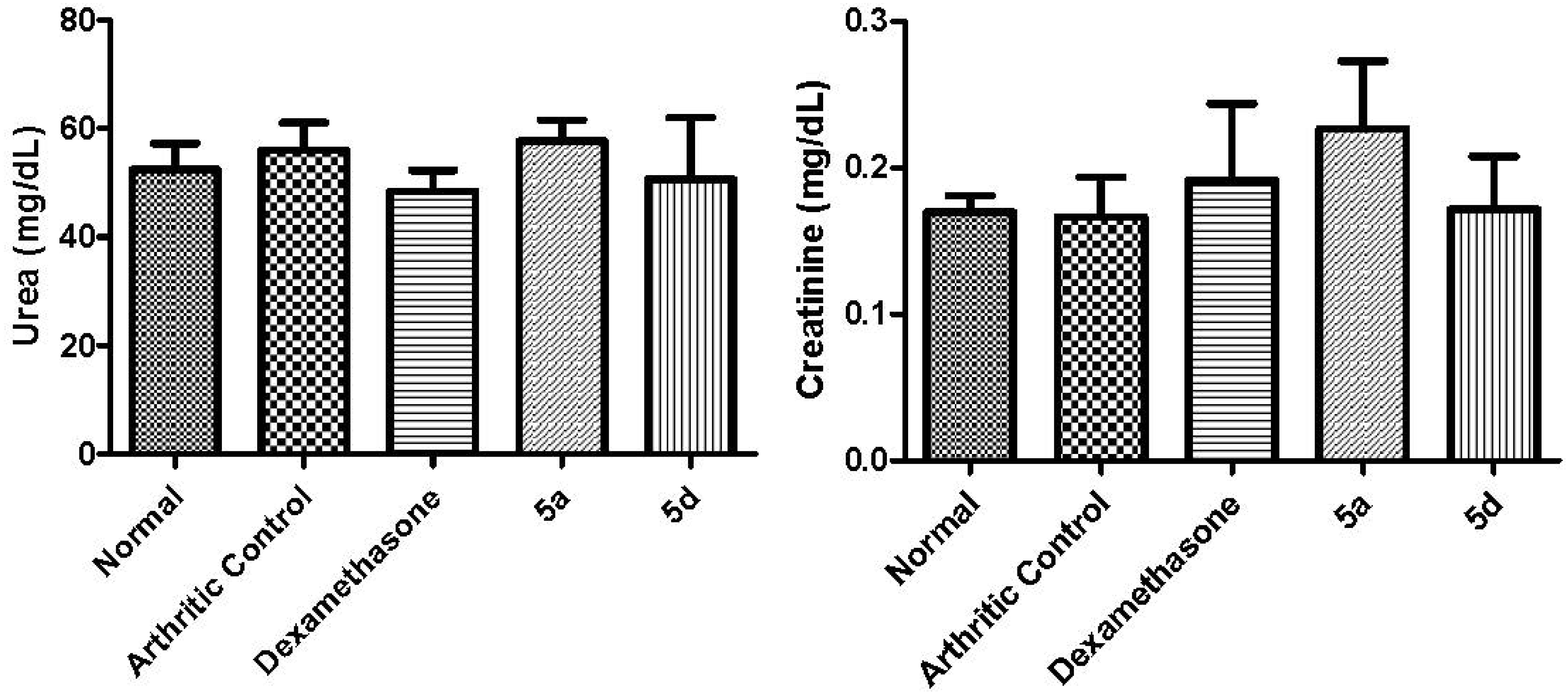
3.5.6. Biochemical Measurements


3.5.7. Macroscopic Analyses of the Stomach

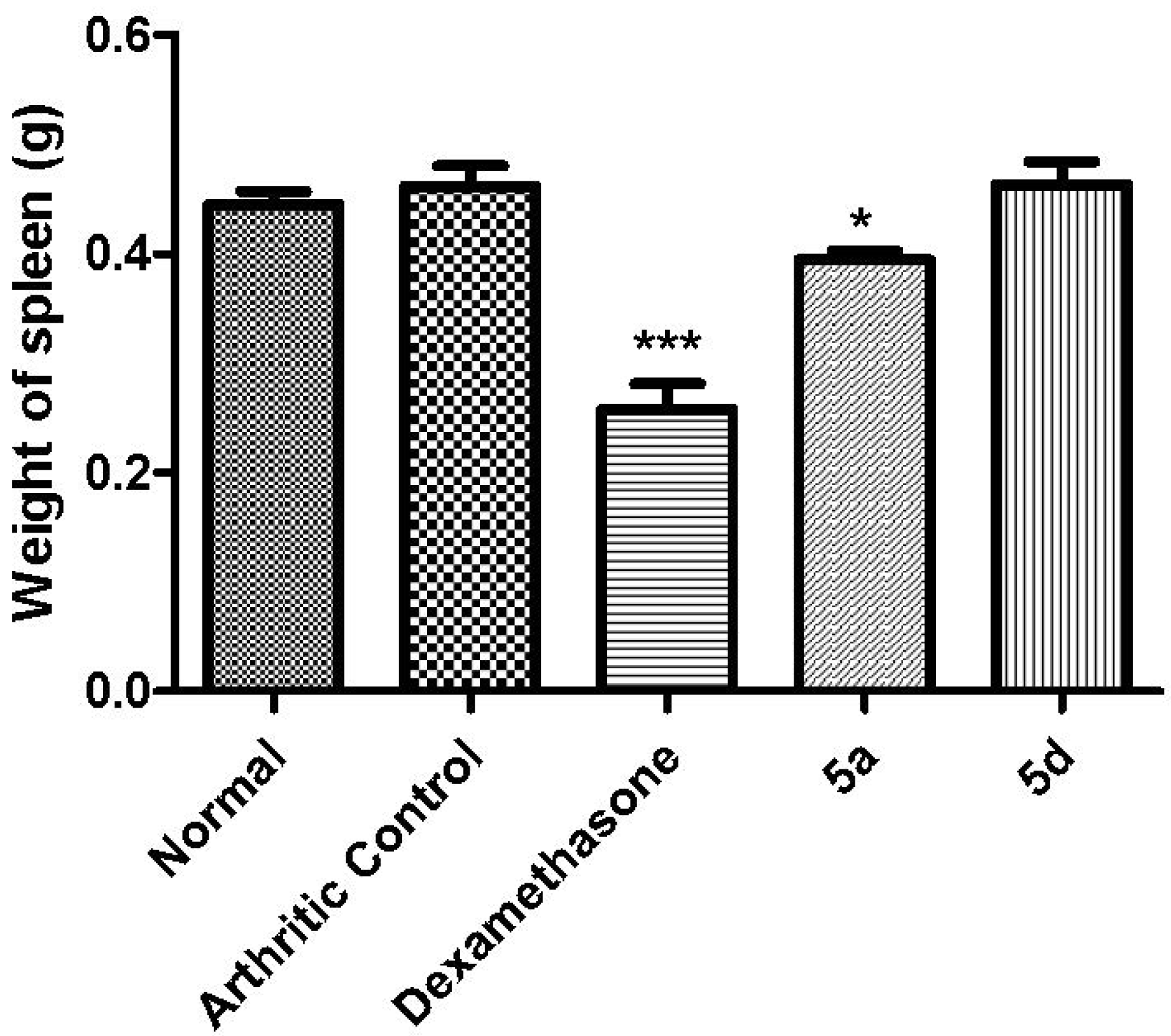
3.5.8. Spleen Weight
3.5.9. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Majno, G.; Joris, I. Cells, Tissues and Disease, 2nd ed.; Oxford University Press: Oxford, UK, 2004. [Google Scholar]
- Kumar, V.; Cotran, R.S.; Robbins, S.L. Robbins Basic Pathology, 9th ed.; Elsevier: Philadelphia, PA, USA, 2013. [Google Scholar]
- Nathan, C. Points of control in inflammation. Nature 2002, 420, 846–885. [Google Scholar] [CrossRef]
- Gilroy, D.W.; Lawrence, T.; Perretti, M.; Rossi, A.G. Inflammatory resolution: New opportunities for drug discovery. Nat. Rev. Drug Discov. 2004, 3, 401–416. [Google Scholar] [CrossRef]
- Turini, M.E.; DuBois, R.N. Cyclooxygenase-2: A therapeutic target. Annu. Rev. Med. 2002, 53, 35–57. [Google Scholar] [CrossRef]
- Marnett, L.J.; Kalgutkar, A.S. Cyclooxygenase 2 inhibitors: Discovery, selectivity and the future. In Trends Pharmacol. Sci.; 1999; Volume 20, pp. 465–469. [Google Scholar]
- Hale, K.K.; Trollinger, D.; Rihanek, M.; Manthey, C.L. Differential expression and activation of p38 mitogen-activated protein kinase α, β, γ and δ in inflammatory cell lineages. J. Immunol. 1999, 162, 4246–4252. [Google Scholar]
- Philip, C. Protein kinases—The major drug targets of the twenty-first century. Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar]
- Chandrasekharan, N.V.; Dai, H.; Roos, K.L.; Evanson, N.K.; Tomsik, J.; Elton, T.S.; Simmons, D.L. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: Cloning, structure, and expression. Proc. Natl. Acad. Sci. USA 2002, 21, 13926–13931. [Google Scholar]
- Kajal, A.; Bala, S.; Sharma, N.; Kamboj, S.; Saini, V. Therapeutic potential of hydrazones as anti-inflammatory agents. Int. J. Med. Chem. 2014, 2014, 1–11. [Google Scholar]
- Duarte, C.M.; Barreiro, E.J.; Fraga, C.A.M. Privileged structures: A useful concept for the rational design of new lead drugs candidates. Mini Rev. Med. Chem. 2007, 7, 1108–1119. [Google Scholar] [CrossRef]
- Tributino, J.L.M.; Duarte, C.D.; Correa, R.S.; Doriguetto, A.C.; Ellena, J.; Romeiro, N.C.; Castro, N.G.; Miranda, A.L.P.; Barreiro, E.J.; Fraga, C.A.M. Novel 6-methanesulfonamide-3,4-methylenedioxyphenyl-N-acylhydrazones: Orally effective anti-inflammatory drug candidates. Bioorg. Med. Chem. 2009, 17, 1125–1131. [Google Scholar] [CrossRef]
- Da Silva, Y.K.; Augusto, C.V.; de Castro Barbosa, M.L.; de Albuquerque Melo, G.M.; de Queiroz, A.C.; de Lima Matos Freire Dias, T.; Júnior, W.B.; Barreiro, E.J.; Lima, L.M.; Alexandre-Moreira, M.S. Synthesis and pharmacological evaluation of pyrazine N-acylhydrazone derivatives designed as novel analgesic and anti-inflammatory drug candidates. Bioorg. Med. Chem. 2010, 18, 5007–5015. [Google Scholar] [CrossRef]
- Kheradmand, A.; Navidpour, L.; Shafaroodi, H.; Saeedi-Motahar, G.; Shafiee, A. Design and synthesis of niflumic acid-based N-acylhydrazone derivatives as novel anti-inflammatory and analgesic agents. Med. Chem. Res. 2013, 22, 2411–2420. [Google Scholar] [CrossRef]
- Kaplancikli, Z.A; Altintop, M.D.; Özdemir, A.; Turan-Zitouni, Khan G.S.I.; Tabanca, N. Synthesis and biological evaluation of some hydrazone derivatives as anti-inflammatory agents. Lett. Drug Des. Discov. 2012, 9, 310–315. [Google Scholar] [CrossRef]
- Asif, M.; Husain, A. Analgesic, Anti-inflammatory, and antiplatelet profile of hydrazones containing synthetic molecules. J. Appl. Chem. 2013, 2013, 1–7. [Google Scholar] [CrossRef]
- Rollas, S.; Küçükgüzel, Ş.G. Biological activities of hydrazone derivatives. Molecules 2007, 12, 1910–1939. [Google Scholar] [CrossRef]
- Zhai, X.; Huang, Q.; Jiang, N.; Wu, D.; Zhou, H.; Gong, P. Discovery of hybrid dual N-acylhydrazone and diaryl urea derivatives as potent antitumor agents: Design, synthesis and cytotoxicity evaluation. Molecules 2013, 18, 2904–2923. [Google Scholar] [CrossRef]
- Lima, M.L.; Barreiro, E.J. Bioisosterism: A useful strategy for molecular modification and drug design. Curr. Med. Chem. 2005, 12, 23–49. [Google Scholar] [CrossRef]
- Lima, P.C.; Lima, L.M.; da Silva, K.C.; Léda, P.H.; de Miranda, A.L.; Fraga, C.A.; Barreiro, E.J. Synthesis and analgesic activity of novel N-acylhydrazones and isosters, derived from natural safrole. Eur. J. Med. Chem. 2000, 35, 187–203. [Google Scholar] [CrossRef]
- Kummerle, A.E.; Raimundo, J.M.; Leal, C.M.; da Silva, G.S.; Balliano, T.L.; Pereira, M.A.; de Simone, C.A.; Sudo, R.T.; Zapata-sudo, G.; Fraga, C.A.M.; et al. Studies towards the identification of putative bioactive conformation of potent vasodilatador arylidene N-acylhydrazone derivatives. Eur. J. Med. Chem. 2009, 44, 4004–4009. [Google Scholar] [CrossRef]
- Huddleston, P.R.; Barker, J.M.; Adamczewska, Y.Z.; Wood, M.L.; Holmes, D. Synthesis and chemistry of some Thieno[3,2-d]-l,2,3-triazin-4(3H)-ones. J. Chem. Res. 1993, 72–73. [Google Scholar]
- Reinecke, M.G.; Woodrow, T.A.; Brown, E.S. Pyrazolo[3,4-c]pyridazines from hydrazine and aminothiophenecarboxylates. J. Org. Chem. 1992, 57, 1018–1021. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; Von-Korff, M.; Reich, J.R.; Rufener, C. OSIRIS, an entirely in-house developed drug discovery informatics system. J. Chem. Inf. Model. 2009, 49, 232–246. [Google Scholar] [CrossRef]
- OSIRIS Property Explorer. Available online: http://www.organic-chemistry.org/prog/peo/ (accessed on 9 June 2014).
- Coolier, H.O.J.; Dinnen, L.C.; Schneider, C. The abdominal constriction response and its supression by analgesic drugs in mice. Br. J. Pharmacol. 1968, 32, 285–310. [Google Scholar]
- Hunskaar, S.; Hole, K. The formalin test in mice: Dissociation between inflammatory pain. Pain 1987, 30, 103–114. [Google Scholar] [CrossRef]
- Ferrándiz, M.L.; Alcaraz, M.J. Antiinflammatory activity and inhibition of arachidonic acidmetabolism by flavonoids. Inflamm. Res. 1991, 32, 283–288. [Google Scholar]
- Newbould, B.B. Chemotherapy of arthritis induced in rats by mycobacterium adjuvant. Br. J. Pharmacol. 1963, 45, 375–333. [Google Scholar]
- Cioli, V.; Putzolu, S.; Rossi, V.; Barcellona, P.S.; Corradino, C. The Role of direct contact in the production of gastrointestinal ulcers by anti-inflammatory drugs in rats. Toxicol. Appl. Pharmacol. 1979, 50, 283–289. [Google Scholar] [CrossRef]
- Descotes, J.; Choquet-Kastylevsky, G.; van Ganse, E.; Vial, T. Responses of the immune system to injury. Toxicol. Pathol. 2000, 28, 479–481. [Google Scholar] [CrossRef]
- Kishimoto, C.; Thorp, K.A.; Abelmann, W.H. Immunosuppression with high doses of cyclophosphamide reduces the severity of myocarditis but increases the mortality in murine Coxsackievirus B3 myocarditis. Circulation 1990, 82, 982–989. [Google Scholar] [CrossRef]
- US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). Guidance for Industry: Immunotoxicology Evaluation of Investigational New Drugs. Available online: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079239.pdf (accessed on 9 June 2014).
- Sample Availability: Samples of the compounds 5a–i are available from the authors.
© 2014 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Silva, Y.K.C.d.; Reyes, C.T.M.; Rivera, G.; Alves, M.A.; Barreiro, E.J.; Moreira, M.S.A.; Lima, L.M. 3-Aminothiophene-2-Acylhydrazones: Non-Toxic, Analgesic and Anti-Inflammatory Lead-Candidates. Molecules 2014, 19, 8456-8471. https://doi.org/10.3390/molecules19068456
Silva YKCd, Reyes CTM, Rivera G, Alves MA, Barreiro EJ, Moreira MSA, Lima LM. 3-Aminothiophene-2-Acylhydrazones: Non-Toxic, Analgesic and Anti-Inflammatory Lead-Candidates. Molecules. 2014; 19(6):8456-8471. https://doi.org/10.3390/molecules19068456
Chicago/Turabian StyleSilva, Yolanda Karla Cupertino da, Christian Tadeo Moreno Reyes, Gildardo Rivera, Marina Amaral Alves, Eliezer J. Barreiro, Magna Suzana Alexandre Moreira, and Lídia Moreira Lima. 2014. "3-Aminothiophene-2-Acylhydrazones: Non-Toxic, Analgesic and Anti-Inflammatory Lead-Candidates" Molecules 19, no. 6: 8456-8471. https://doi.org/10.3390/molecules19068456