Thioctic Acid Derivatives as Building Blocks to Incorporate DNA Oligonucleotides onto Gold Nanoparticles



Abstract
:
1. Introduction

2. Results and Discussion
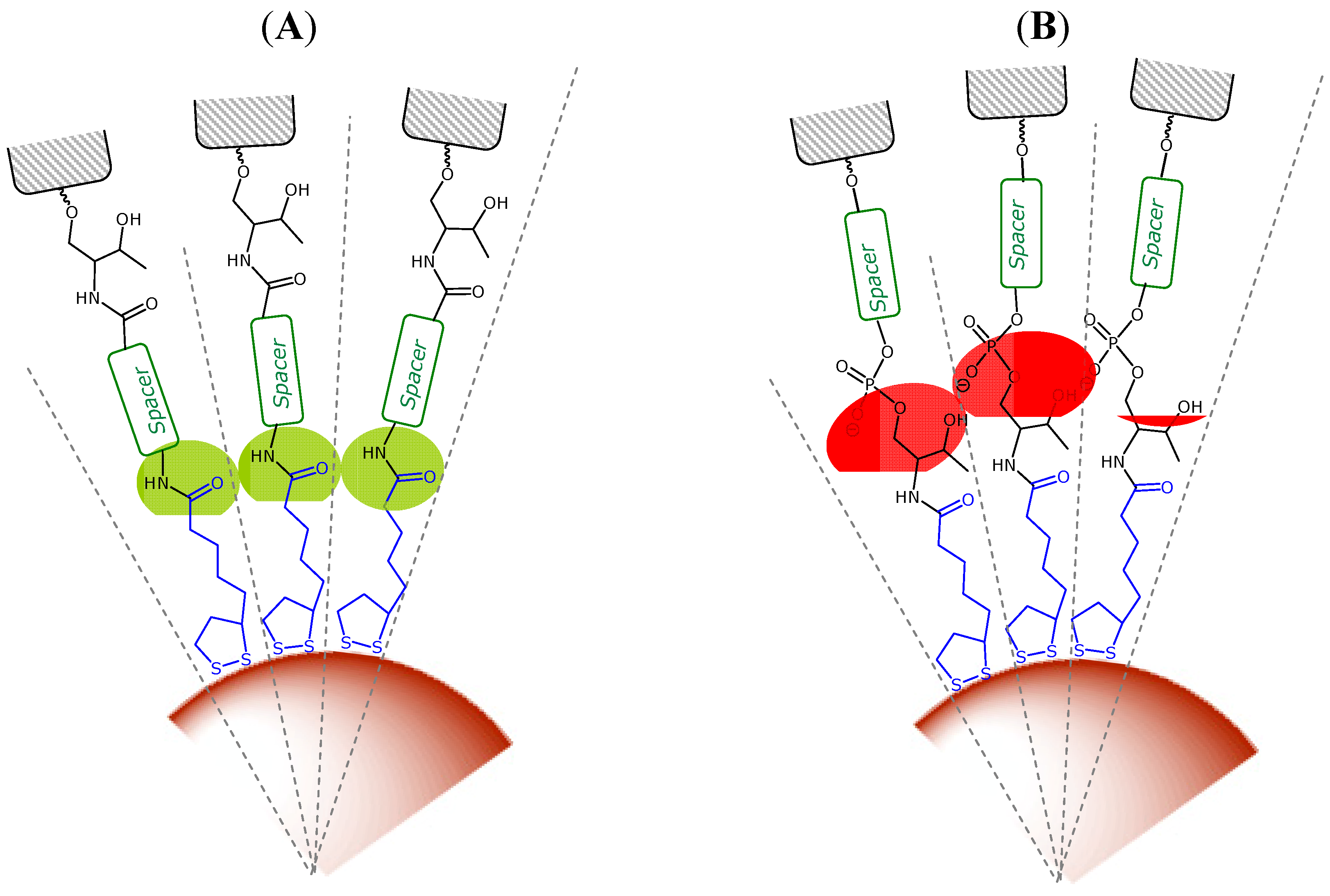
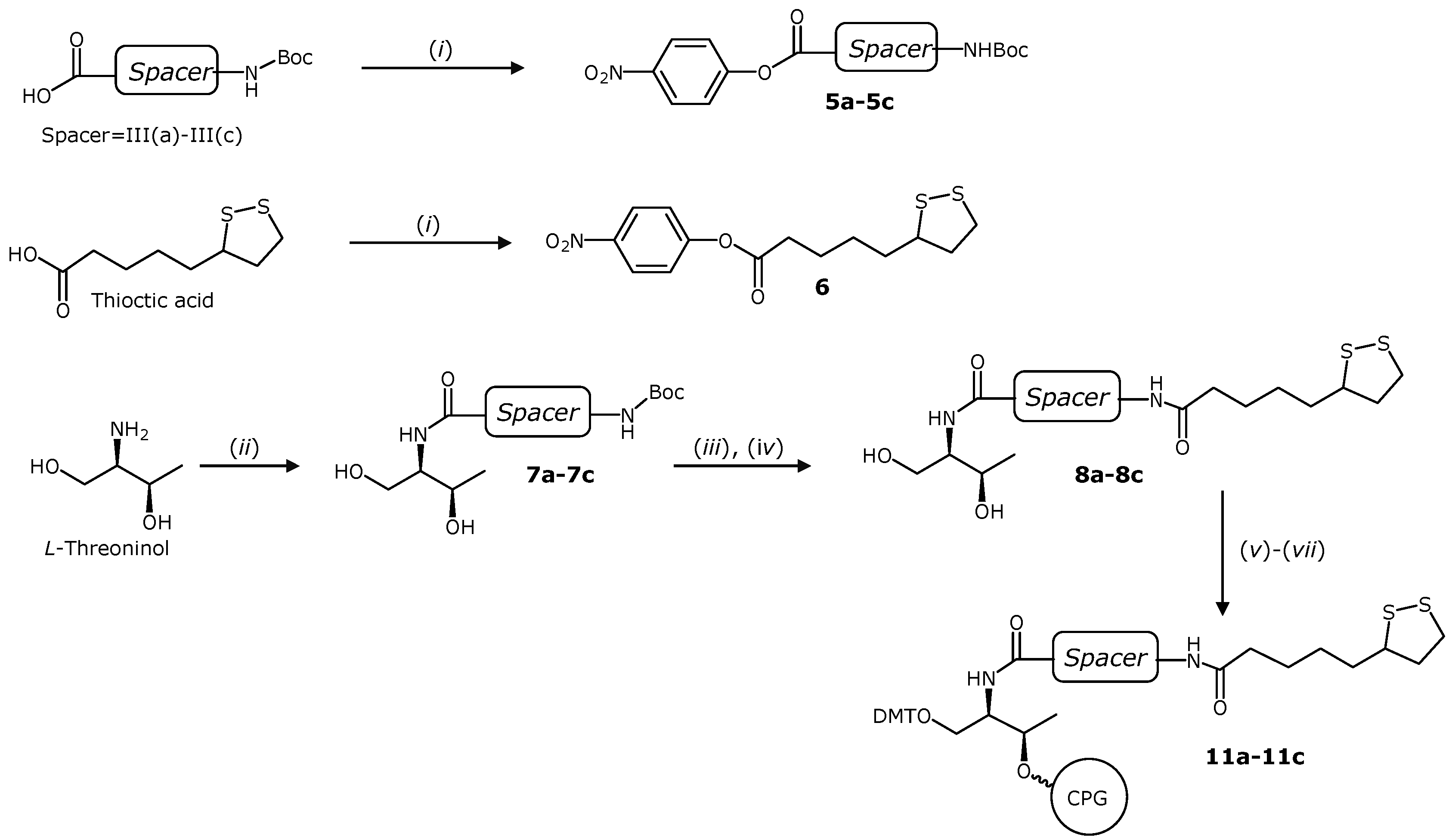
2.1. Synthesis of Solid Supports Functionalized with TA Derivatives


2.2. Synthesis of Novel TA Terminated Oligonucleotides and Alkylthiolated Modified Oligonucleotides

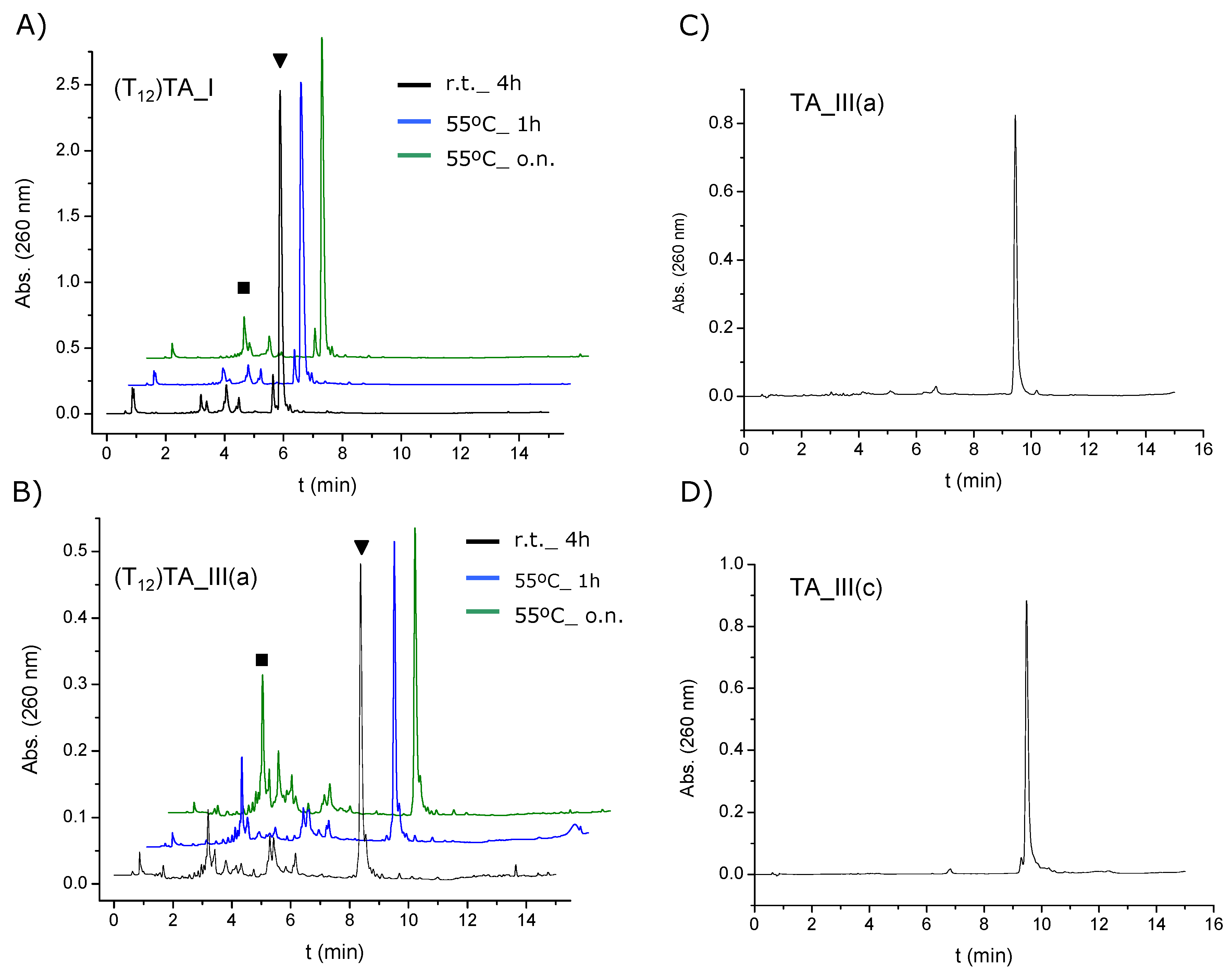
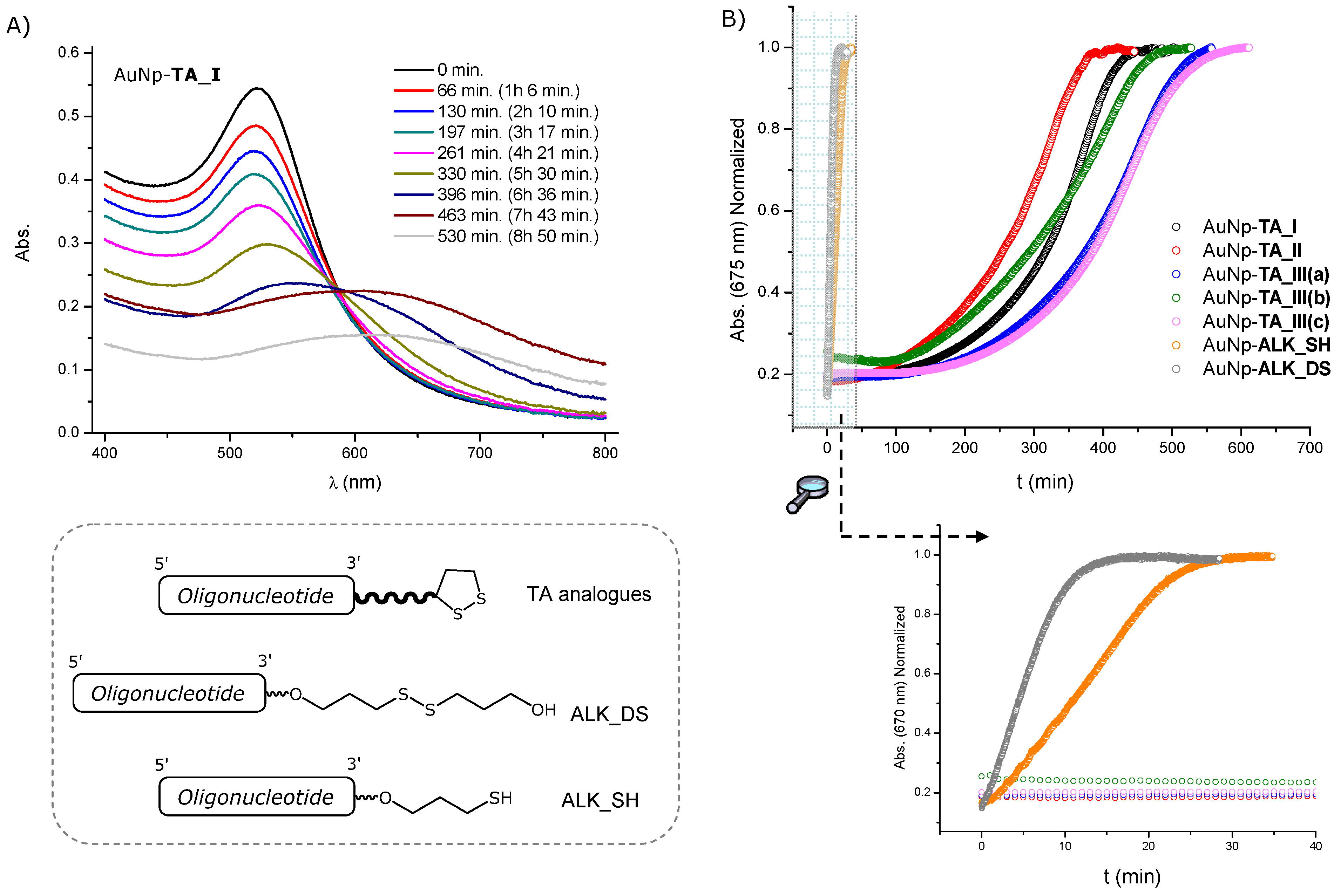
2.3. Surface Coverage Quantification and Stability Tests of Oligonucleotide-Gold Nanoparticle Conjugates

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conjugate | Strands/Particle | Footprint (nm2) | t1/2 (min) |
|---|---|---|---|
| AuNP-TA_I | 94 ± 7 | 3.2 ± 0.2 | 344 ± 12 |
| AuNP-TA_II | 93 ± 13 | 3.2 ± 0.4 | 282 ± 11 |
| AuNP-TA_III(a) | 141 ± 10 | 2.1 ± 0.3 | 444 ± 8 |
| AuNP-TA_III(b) | 103 ± 13 | 2.9 ± 0.4 | 354 ± 10 |
| AuNP-TA_III(c) | 102 ± 12 | 2.8 ± 0.3 | 450 ± 15 |
| AuNP-ALK_DS | 40 ± 2 | 7.4 ± 0.1 | 4 ± 1 |
| AuNP_ALK_SH | 97 ± 7 | 3.0 ± 0.2 | 12 ± 2 |

2.4. Stability of the Thioctic Acid Derivatives to Cathepsin


3. Experimental Section
General Methods and Materials
3.1. 4-(Dithiolan-3-yl)-N-[(1R,2R)-2-hydroxy-1-(hydroxymethyl)propyl]butanamide (1)
3.2. General Procedure for the Synthesis of Amides 7a–7c
3.2.1. tert-Butyl-N-[3-[2-[2-[3-[[4-[[(1R,2R)-2-hydroxy-1-(hydroxymethyl)propyl]amino]-4-oxo-butanoyl]amino]propoxy]ethoxy]ethoxy]propyl] carbamate (7a)
3.2.2. N-[N'-(tert-Butoxycarbonyl)glycylglycylglycyl]-l-threoninol (7b)
3.2.3. tert-Butyl-N-[8-[[(1R,2R)-2-hydroxy-1-(hydroxymethyl)propyl]amino]-8-oxo-octyl]carbamate (7c)
3.3. General Procedure for the Introduction of Lipoic Acid Residue: Synthesis of Compounds 8a–8c
3.3.1. N'-[3-[2-[2-[3-[4-(Dithiolan-3-yl)butanoylamino]propoxy]ethoxy]ethoxy]propyl]-N-[(1R,2R)-2-hydroxy-1-(hydroxymethyl)propyl]butanediamide (8a)
3.3.2. N-[N'-{5-(1,2-Dithiolan-3-yl)pentanoyl}glycylglycylglycyl]-l-threoninol (8b)
3.3.3. 8-[4-(Dithiolan-3-yl)butanoylamino]-N-[(1R,2R)-2-hydroxy-1-(hydroxymethyl)propyl]octan-amide (8c)
3.4. General Procedure for the Preparation of the DMT Protected Compounds 2 and 9a–9c
3.4.1. N-[(1R,2R)-1-[[bis(4-Methoxyphenyl)phenylmethoxy]methyl]-2-hydroxypropyl]-5-(dithiolan-3-yl)pentanamide (2)
3.4.2. N-[(1R,2R)-1-[[bis(4-Methoxyphenyl)phenylmethoxy]methyl]-2-hydroxypropyl]-N'-[3-[2-[2-[3-[5-(dithiolan-3-yl)pentanoylamino]propoxy]ethoxy]ethoxy]propyl]butanediamide (9a)
3.4.3. O1-(4, 4'-Dimetoxytriphenylmethyl)-N-[N'-{5-(1,2-dithiolan-3-yl)pentanoyl}glycylglycylglycyl]-l-threoninol (9b)
3.4.4. N-[(1R,2R)-1-[[bis(4-Methoxyphenyl)phenylmethoxy]methyl]-2-hydroxypropyl]-8-[5-(dithiolan-3-yl)pentanoylamino]octanamide (9c)
3.5. Functionalization of CPG Solid Supports 4 and 11a–11c
3.6. N-[(1R,2R)-1-{[bis(4-Methoxyphenyl)phenylmethoxy]methyl}-2-[2-cyanoethoxydiisopro-pylamino)phosphanyl]oxypropyl]-4-(dithiolan-3-yl)butanamide (12)
3.7. Synthesis of the Model Oligonucleotides (T12)TA_I, and (T12)TA_III(a)–(c)
3.8. Synthesis of 3'-TA-Modified Oligonucleotides and the 5'-Fluorescently Labeled 3'-TA-Modified Oligonucleotide Probes
3.9. Synthesis of a 5'-TA-Modified Oligonucleotide
3.10. Synthesis of 3'-Alkylthiolated-Modified Oligonucleotides and the 5'-Fluorescently Labeled 3'-Alkylthiolated Modified Oligonucleotide Probes
3.11. Functionalization of Gold Nanoparticles
3.12. Stability Tests of Oligonucleotide-Gold Nanoparticle Conjugates
3.13. Oligonucleotide Loading Quantification on Gold Nanoparticles
3.14. Stability of Thioctic Derivatives to Cathepsin
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yeh, Y.-C.; Creran, B.; Rotello, V.M. Gold Nanoparticles: Preparation, properties and applications in bionanotechnology. Nanoscale 2012, 4, 1871–1880. [Google Scholar] [CrossRef]
- Radwan, S.H.; Azzazy, H.M.E. Gold nanoparticles for molecular diagnostics. Expert Rev. Mol. Diagn. 2009, 9, 511–524. [Google Scholar] [CrossRef]
- Saha, K.; Agasti, S.S.; Kim, C.; Li, X.; Rotello, V.M. Gold nanoparticles in chemical and biological sensing. Chem. Rev. 2012, 112, 2739–2779. [Google Scholar] [CrossRef]
- Dykman, L.; Khlebtsov, N. Gold nanoparticles in biomedical applications: Recent advances and perspectives. Chem. Soc. Rev. 2012, 41, 2256–2282. [Google Scholar] [CrossRef]
- Doane, T.L.; Burda, C. The unique role of nanoparticles in nanomedicine: Imaging, drug delivery and therapy. Chem. Soc. Rev. 2012, 41, 2885–2911. [Google Scholar] [CrossRef]
- Giljohann, D.A.; Seferos, D.S.; Daniel, W.L.; Massich, M.D.; Patel, P.C.; Mirkin, C.A. Gold nanoparticles for biology and medicine. Angew. Chem. Int. Ed. 2012, 49, 3280–3294. [Google Scholar]
- Llevot, A.; Astruc, D. Applications of vectorized gold nanoparticles to the diagnosis and therapy of cancer. Chem. Soc. Rev. 2012, 41, 242–257. [Google Scholar] [CrossRef]
- Choi, Y.-E.; Kwak, J.-W.; Park, J.W. Nanotechnology for early cancer detection. Sensors 2010, 10, 428–455. [Google Scholar] [CrossRef]
- Rana, S.; Bajaj, A.; Mout, R.; Rotello, V.M. Monolayer coated gold nanoparticles for delivery applications. Adv. Drug Deliv. Rev. 2012, 64, 200–216. [Google Scholar] [CrossRef]
- Niemeyer, C.M. Nanoparticles, proteins, and nucleic acids: Biotechnology meets materials science. Angew. Chem. Int. Ed. 2001, 40, 4128–4158. [Google Scholar] [CrossRef]
- Lévy, R.; Thanh, N.T.K.; Doty, R.C.; Hussain, I.; Nichols, R.J.; Schiffrin, D.J.; Brust, M.; Fernig, D.G. Rational and combinatorial design of peptide capping ligands for gold nanoparticles. J. Am. Chem. Soc. 2004, 126, 10076–10084. [Google Scholar] [CrossRef]
- Gao, J.; Huang, X.; Liu, H.; Zan, F.; Ren, J. Colloidal stability of gold nanoparticles modified with thiol compounds: Bioconjugation and application in cancer cell imaging. Langmuir 2012, 28, 4464–4471. [Google Scholar]
- Goldstein, D.C.; Thordarson, P.; Peterson, J.R. The bioconjugation of redox proteins to novel electrode materials. Aust. J. Chem. 2009, 62, 1320–1327. [Google Scholar] [CrossRef]
- Wang, H.; Yang, R.; Yang, L.; Tan, W. Nucleic acid conjugated nanomaterials for enhanced molecular recognition. ACS Nano 2009, 3, 2451–2460. [Google Scholar] [CrossRef]
- Cutler, J.I.; Auyeung, E.; Mirkin, C.A. Spherical nucleic acids. J. Am. Chem. Soc. 2012, 134, 1376–1391. [Google Scholar] [CrossRef]
- Hu, R.; Zhang, X.-B.; Kong, R.-M.; Zhao, X.-H.; Jiang, J.; Tan, W. Nucleic acid-functionalized nanomaterials for bioimaging applications. J. Mater. Chem. 2011, 21, 16323–16334. [Google Scholar] [CrossRef]
- Katz, E.; Willner, I. Integrated nanoparticle–biomolecule hybrid systems: Synthesis, properties, and applications. Angew. Chem. Int. Ed. 2004, 43, 6042–6108. [Google Scholar] [CrossRef]
- Li, X.; Guo, J.; Asong, J.; Wolfert, M.A.; Boons, G.-J. Multifunctional surface modification of gold-stabilized nanoparticles by bioorthogonal reactions. J. Am. Chem. Soc. 2011, 133, 11147–11153. [Google Scholar] [CrossRef]
- Stobiecka, M.; Hepel, M. Double-shell gold nanoparticle-based DNA-carriers with poly-L-lysine binding surface. Biomaterials 2011, 32, 3312–3321. [Google Scholar] [CrossRef]
- Cobbe, S.; Connolly, S.; Ryan, D.; Nagle, D.; Eritja, R.; Fitzmaurice, D. DNA-controlled assembly of protein-modified gold nanocrystals. J. Phys. Chem. B 2003, 107, 470–477. [Google Scholar] [CrossRef]
- Mirkin, C.A.; Letsinger, R.L.; Mucic, R.C.; Storhoff, J.J. A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature 1996, 382, 607–609. [Google Scholar] [CrossRef]
- Zhi, L.; Rongchao, J.; Mirkin, C.A.; Letsinger, R.L. Multiplethiol-anchor capped DNA-gold nanoparticle conjugates. Nucleic Acid Res. 2002, 30, 1558–1562. [Google Scholar] [CrossRef]
- Chompoosor, A.; Han, G.; Rotello, V.M. Charge dependence of ligand release and monolayer stability of gold nanoparticles by biogenic thiols. Bioconjug. Chem. 2008, 19, 1342–1345. [Google Scholar] [CrossRef]
- Kang, J.S.; Taton, T.A. Oligothiol graft-copolymer coatings stabilize gold nanoparticles against harsh experimental conditions. Langmuir 2012, 28, 16751–16760. [Google Scholar] [CrossRef]
- Zhang, G.; Yang, Z.; Lu, W.; Zhang, R.; Huang, Q.; Tian, M.; Li, L.; Liang, D.; Li, C. Influence of anchoring ligands and particle size on the colloidal stability and in vivo biodistribution of polyethylene glycol-coated gold nanoparticles in tumor-xenografted mice. Biomaterials 2009, 30, 1928–1936. [Google Scholar] [CrossRef]
- Ghann, W.E.; Aras, O.; Fleiter, T.; Daniel, M.-C. Syntheses and characterization of lisinopril-coated gold nanoparticles as highly stable targeted CT contrast agents in cardiovascular diseases. Langmuir 2012, 28, 10398–10408. [Google Scholar] [CrossRef]
- Oh, E.; Susumu, K.; Blanco-Canosa, J.B.; Medintz, I.L.; Dawson, P.E.; Mattoussi, K. Preparation of stable maleimide-functionalized Au nanoparticles and their use for counting surface ligands. Small 2010, 6, 1273–1278. [Google Scholar] [CrossRef]
- Dougan, J.A.; Reid, A.K.; Graham, D. Thioctic acid modification of oligonucleotides using an H-phosphonate. Tetrahedron Lett. 2010, 51, 5787–5790. [Google Scholar] [CrossRef]
- Dougan, J.A.; Karlsson, C.; Smith, W.E.; Graham, D. Enhanced oligonucleotide-nanoparticle conjugate stability using thioctic acid modified oligonucleotides. Nucleic Acid Res. 2007, 35, 3668–3675. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Walker, M.A. Receptor-mediated and enzyme-dependent targeting of cytotoxic anticancer drugs. Pharm. Ther. 1999, 83, 67–123. [Google Scholar] [CrossRef]
- Trail, P.A.; Willner, D.; Knipe, J.; Henderson, A.J.; Lasch, S.J.; Zoeckler, M.E.; TrailSmith, M.D.; Doyle, T.W.; King, H.D.; Casazza, A.M.; et al. Effect of linker variation on the stability, potency, and efficacy of carcinoma-reactive BR64-doxorubicin immunoconjugates. Cancer Res. 1997, 57, 100–105. [Google Scholar]
- Yan, S.; Sameni, M.; Sloane, B.F. Cathepsin B and human tumor progression. Biol. Chem. 1998, 379, 113–123. [Google Scholar]
- Etrych, T.; Strohalm, J.; Chytil, P.; Cernoch, P.; Starovoytova, L.; Pechar, M.; Ulbrich, K. Biodegradable star HPMA polymer conjugates of doxorubicin for passive tumor targeting. Eur. J. Pharm. Sci. 2011, 42, 527–539. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro. Bioconjug. Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef]
- Harada, M.; Sakakibara, H.; Yano, T.; Suzuki, T.; Okuno, S. Determinants for the drug release from T-0128, camptothecin analogue-carboxymethyl dextran conjugate. J. Control. Release 2000, 69, 399–412. [Google Scholar] [CrossRef]
- Li, M.; Meares, C.F. Synthesis, metal chelate stability studies, and enzyme digestion of a peptide-linked DOTA derivative and its corresponding radiolabeled immunoconjugates. Bioconjug. Chem. 1993, 4, 275–283. [Google Scholar] [CrossRef]
- Hurst, S.J.; Lytton-Jean, A.K.R.; Mirkin, C.A. Maximizing DNA loading on a range of gold nanoparticle sizes. Anal. Chem. 2006, 78, 8313–8318. [Google Scholar] [CrossRef]
- Becker, C.F.W.; Marsac, Y.; Hazarika, P.; Moser, J.; Goody, R.S.; Niemeyer, C.M. Functional immobilization of the small GTPase Rab6A on DNA–gold nanoparticles by using a site-specifically attached poly(ethylene glycol) linker and thiol place- exchange reaction. ChemBioChem 2007, 8, 32–36. [Google Scholar] [CrossRef]
- Singh, N.; Agrawal, A.; Leung, A.K.L.; Sharp, P.A.; Bhatia, S.N. Effect of nanoparticle conjugation on gene silencing by RNA interference. J. Am. Chem. Soc. 2010, 132, 8241–8243. [Google Scholar]
- Shenhar, R.; Rotello, V.M. Nanoparticles: Scaffolds and building blocks. Acc. Chem. Res. 2003, 36, 549–561. [Google Scholar] [CrossRef]
- Liu, Y.; Shipton, M.K.; Ryan, J.; Kaufman, E.D.; Franzen, S.; Feldheim, D.L. Synthesis, stability and cellular internalization of gold nanoparticles containing mixed peptide-poly(ethylene glycol) monolayers. Anal. Chem. 2007, 79, 2221–2229. [Google Scholar] [CrossRef]
- Barrett, L.; Dougan, J.A.; Faulds, K.; Graham, D. Stable dye-labelled oligonucleotide-nanoparticle conjugates for nucleic acid detection. Nanoscale 2011, 3, 3221–3227. [Google Scholar] [CrossRef]
- Kashida, H.; Liang, X.; Asanuma, H. Rational design of functional DNA with a non-ribose acyclic scaffold. Curr. Org. Chem. 2009, 13, 1065–1084. [Google Scholar] [CrossRef]
- Pérez-Rentero, S.; Grijalvo, S.; Ferreira, R.; Eritja, R. Synthesis of oligonucleotides carrying thiol groups using a simple reagent derived from threoninol. Molecules 2012, 17, 10026–10041. [Google Scholar] [CrossRef]
- Pérez-Rentero, S.; Garibotti, A.V.; Eritja, R. Solid-phase synthesis of oligodeoxynucleotides containing N4-[2-(t-butyldisulfanyl)ethyl]-5-methyl-cytosine moieties. Molecules 2010, 15, 5692–5707. [Google Scholar] [CrossRef]
- Taton, T.A. Preparation of gold nanoparticle-DNA conjugates. Curr. Protoc. Nucleic Acid Chem. 2002, 12. [Google Scholar] [CrossRef]
- Demers, L.M.; Mirkin, C.A.; Mucic, R.C.; Reynolds, R.A.; Letsinger, R.L.; Elghanian, R.; Viswanadham, G. A fluorescence-based method for determining the surface coverage and hybridization efficiency of thiol-capped oligonucleotides bound to gold thin films and nanoparticles. Anal. Chem. 2000, 72, 5535–5541. [Google Scholar] [CrossRef]
- Hill, H.D.; Millstone, J.E.; Banholzer, M.J.; Mirkin, C.A. The role radius of curvature plays in thiolated oligonucleotides loading on gold nanoparticles. ACS Nano 2009, 2, 418–424. [Google Scholar]
- Vargas, M.C.; Gianozzi, P.; Selloni, A.; Scoles, G. Coverage-dependent adsorption of CH3S and (CH3S)2 on Au(111): A density functional theory study. J. Phys. Chem. B 2001, 105, 9509–9513. [Google Scholar]
- Ionita, P.; Caragheorgheopol, A.; Gilbert, B.C.; Chechik, V. EPR study of a place-exchange reaction on Au nanoparticles: Two branches of a disulfide molecule do not adsorb adjacent to each other. J. Am. Chem. Soc. 2002, 124, 9048–9049. [Google Scholar] [CrossRef]
- Terrill, R.G.; Postlethwaite, T.A.; Chen, C.-H.; Poon, C.-D.; Terzis, A.; Chen, A.; Hutchison, J.E.; Clark, M.R.; Wignall, G.; Londono, J.D.; et al. Monolayers in three dimensions: NMR, SAXS, thermal, and electron hopping studies of alkanethiol stabilized gold clusters. J. Am. Chem. Soc. 1995, 117, 12537–12548. [Google Scholar] [CrossRef]
- Letsinger, R.L.; Elghanian, R.; Viswanadham, G.; Mirkin, C.A. Use of a steroid cyclic disulphide anchor in constructing gold nanoparticle-oligonucleotide conjugates. Bioconjug. Chem. 2000, 11, 289–291. [Google Scholar] [CrossRef]
- Love, J.C.; Estroff, L.A.; Kriebel, J.K.; Nuzzo, R.G.; Whitesides, G.M. Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem. Rev. 2005, 105, 1103–1169. [Google Scholar] [CrossRef]
- Boal, A.K.; Rotello, V.M. Intra- and intermonolayer hydrogen bonding in amide-functionalized alkanethiol self-assembled monolayers on gold nanoparticles. Langmuir 2000, 16, 9527–9532. [Google Scholar] [CrossRef]
- Kratz, F.; Muller, I.A.; Ryppa, C.; Warnecke, A. Prodrug strategies in anticancer chemotherapy. ChemMedChem 2008, 3, 20–53. [Google Scholar] [CrossRef]
- Gupta, K.C.; Kumar, P.; Bathia, D.; Sharma, A.K. A rapid method for the functionalization of polymer supports for solid phase oligonucleotide synthesis. Nucleos. Nucleot. 1995, 14, 829–832. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 2 and 7c are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Rentero, S.; Grijalvo, S.; Peñuelas, G.; Fàbrega, C.; Eritja, R. Thioctic Acid Derivatives as Building Blocks to Incorporate DNA Oligonucleotides onto Gold Nanoparticles. Molecules 2014, 19, 10495-10523. https://doi.org/10.3390/molecules190710495
Pérez-Rentero S, Grijalvo S, Peñuelas G, Fàbrega C, Eritja R. Thioctic Acid Derivatives as Building Blocks to Incorporate DNA Oligonucleotides onto Gold Nanoparticles. Molecules. 2014; 19(7):10495-10523. https://doi.org/10.3390/molecules190710495
Chicago/Turabian StylePérez-Rentero, Sónia, Santiago Grijalvo, Guillem Peñuelas, Carme Fàbrega, and Ramon Eritja. 2014. "Thioctic Acid Derivatives as Building Blocks to Incorporate DNA Oligonucleotides onto Gold Nanoparticles" Molecules 19, no. 7: 10495-10523. https://doi.org/10.3390/molecules190710495