Synthesis and Cytotoxicity Evaluation of Naphthalimide Derived N-Mustards


Abstract
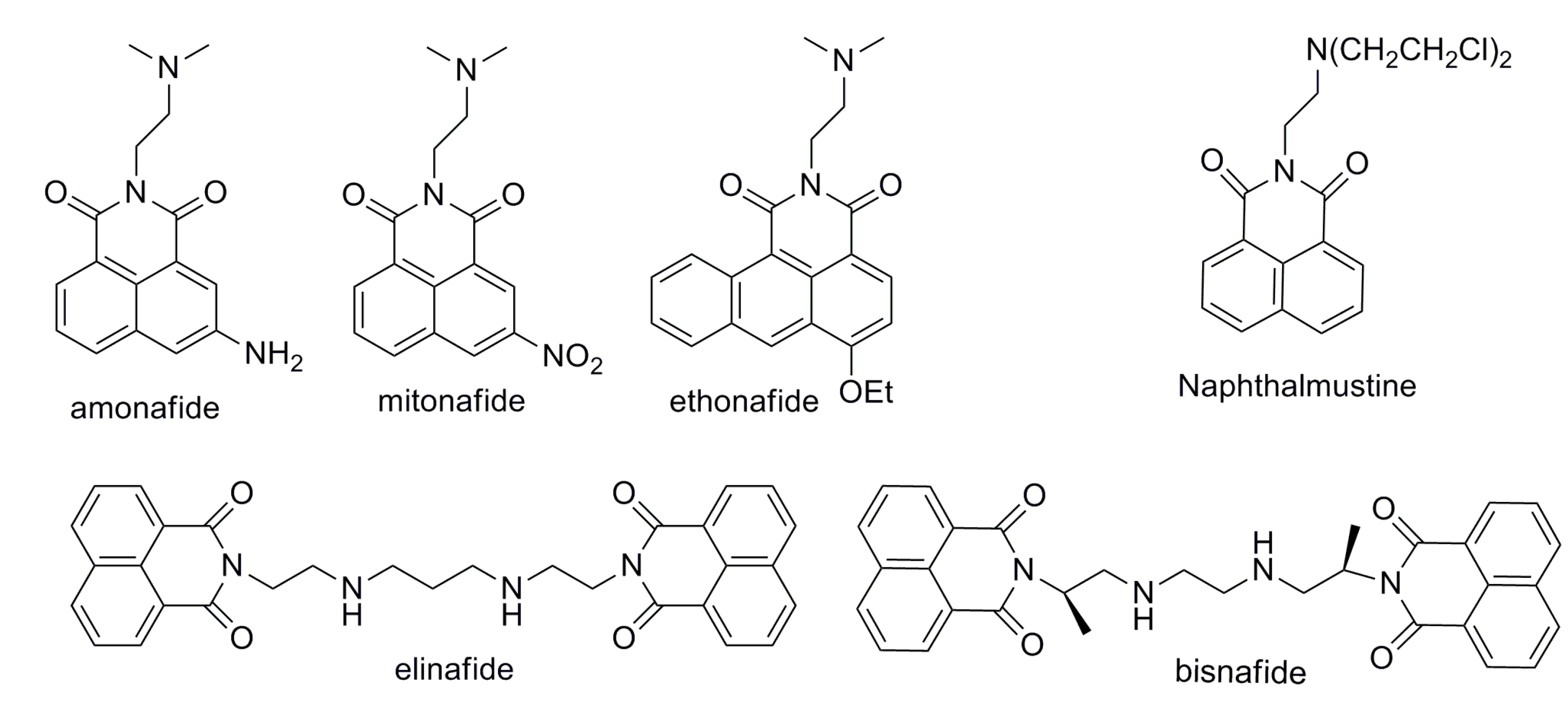
:1. Introduction


2. Results and Discussion
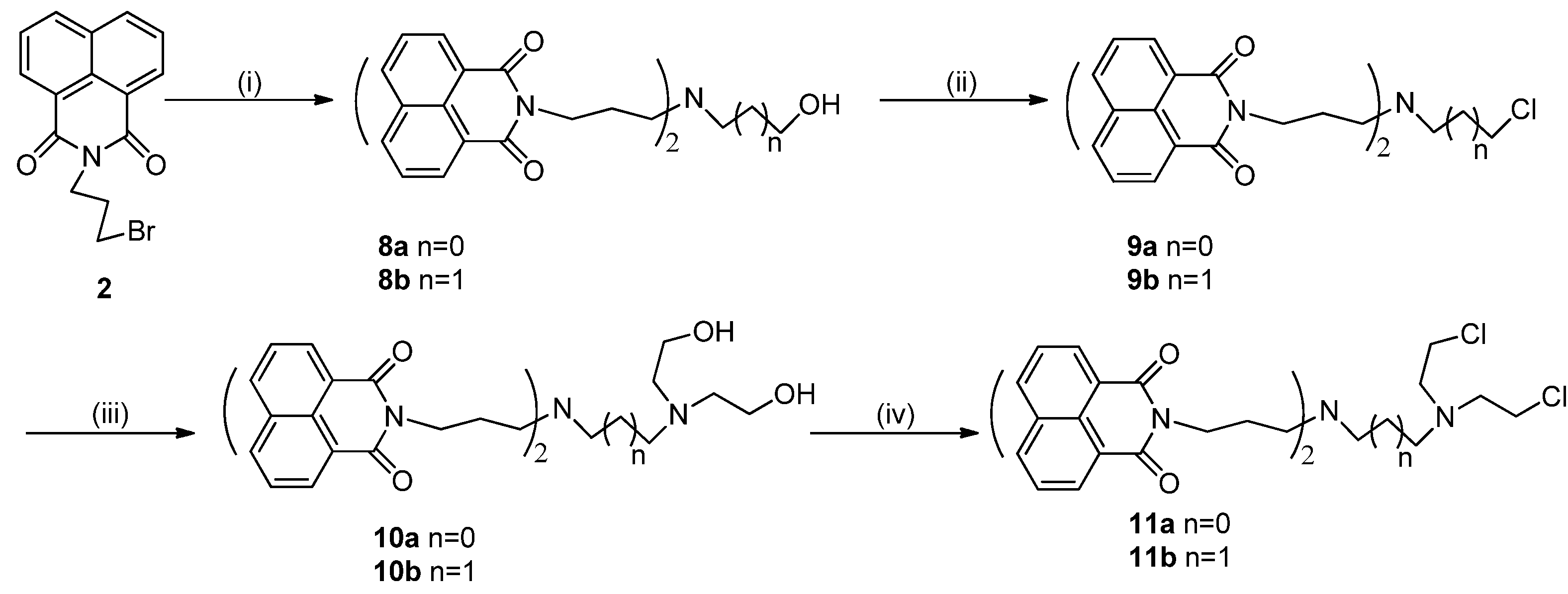
2.1. Chemistry


2.2. Biological Assay
2.2.1. Cytotoxic Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound b | IC5 (μM) | ||||
|---|---|---|---|---|---|
| HCT-116 | PC-3 | U87MG | Hep G2 | SK-OV-3 | |
| 6a | 73.77 ± 2.54 | 225.80 ± 6.41 | 40.80 ± 3.14 | 42.81 ± 26.39 | 63.35 ± 5.42 |
| 6b | 6.96 ± 1.64 | 69.97 ± 10.14 | 10.24 ± 0.19 | 9.39 ± 4.37 | 12.76 ± 1.40 |
| 7a | 33.29 ± 3.13 | 73.10 ± 14.61 | 10.77 ± 0.07 | 15.38 ± 2.88 | 35.31 ± 5.69 |
| 7b | 2.42 ± 0.12 | 3.02 ± 0.34 | 1.95 ± 0.07 | 1.40 ± 0.07 | 3.59 ± 0.70 |
| 10a | 1.30 ± 0.09 | 2.98 ± 0.08 | 1.77 ± 0.14 | 1.25 ± 0.16 | 2.33 ± 0.28 |
| 10b | 2.80 ± 0.36 | 3.33 ± 0.17 | 2.04 ± 0.13 | 0.82 ± 0.06 | 2.43 ± 0.13 |
| 11a | 31.54 ± 18.66 | 96.35 ± 64.69 | 24.88 ± 3.17 | 31.76 ± 18.21 | 15.40 ± 6.22 |
| 11b | 1.01 ± 0.06 | 0.35 ± 0.03 | 0.39 ± 0.03 | 0.89 ± 0.15 | 0.61 ± 0.04 |
| Amonafide | 4.81 ± 0.54 | 3.89 ± 0.28 | 2.60 ± 0.32 | 1.23 ± 0.07 | 5.10 ± 0.76 |
| Compound b | IC5 (μM) | ||||
|---|---|---|---|---|---|
| HCT-116 | PC-3 | U87MG | Hep G2 | SK-OV-3 | |
| 3a | 13.45 ± 0.88 | 45.82 ± 3.13 | 15.39 ± 1.39 | 22.60 ± 8.29 | 24.18 ± 4.64 |
| 3b | 12.52 ± 1.47 | 49.17 ± 6.69 | 14.25 ± 1.19 | 12.17 ± 0.65 | 17.48 ± 1.68 |
| 4a | 12.00 ± 0.46 | 25.54 ± 2.24 | 14.08 ± 0.86 | 9.43 ± 2.39 | 15.07 ± 2.17 |
| 4b | 23.72 ± 2.53 | 67.16 ± 8.79 | 27.50 ± 5.02 | 18.80 ± 1.91 | 21.05 ± 3.09 |
| 5a | 8.44 ± 0.68 | 7.40 ± 0.64 | 3.33 ± 0.37 | 4.20 ± 0.20 | 16.71 ± 1.47 |
| 5b | 81.69 ± 3.79 | 64.16 ± 14.12 | 36.89 ± 13.15 | 31.19 ± 1.09 | 102.58 ± 6.99 |
| 8a | 0.78 ± 0.06 | 1.83 ± 0.07 | 0.77 ± 0.02 | 0.71 ± 0.10 | 0.89 ± 0.03 |
| 8b | 0.77 ± 0.05 | 1.38 ± 0.18 | 0.65 ± 0.02 | 0.56 ± 0.02 | 0.87 ± 0.03 |
| 9b | 2.07 ± 0.24 | 14.60 ± 0.53 | 2.26 ± 0.37 | 1.04 ± 0.07 | 5.29 ± 0.31 |
| amonafide | 4.81 ± 0.54 | 3.89 ± 0.28 | 2.60 ± 0.32 | 1.23 ± 0.07 | 5.10 ± 0.76 |

2.2.2. Fluorescence Studies

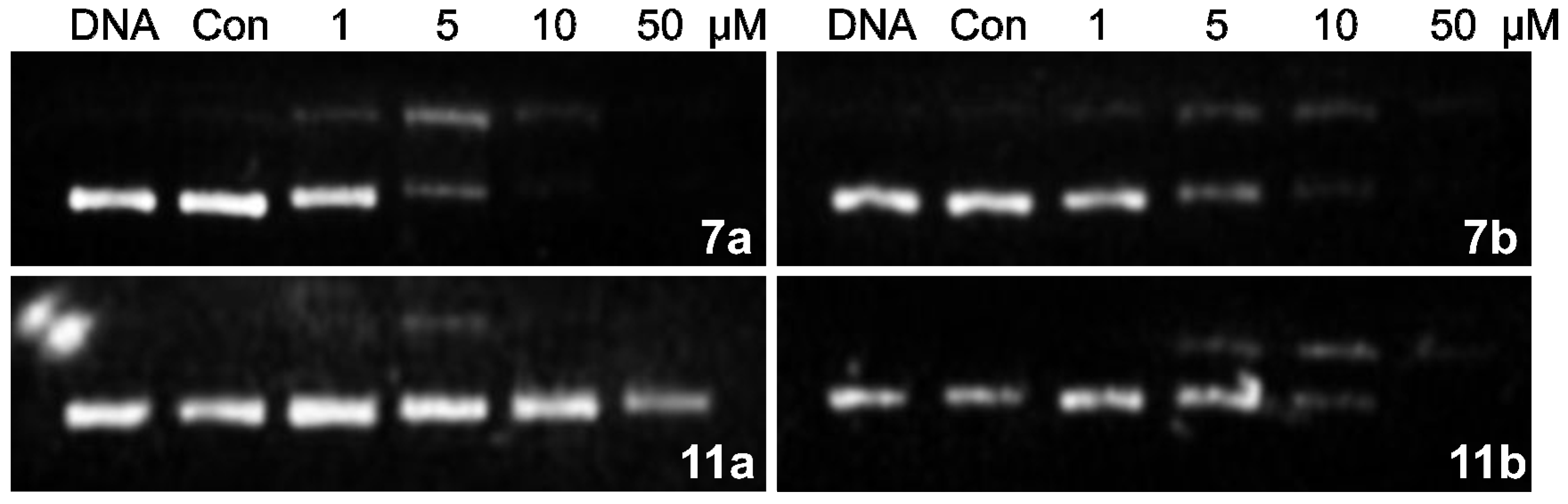
2.2.3. DNA Interstrand Cross-Linking Assay
2.2.4. Flow Cytometry Assay

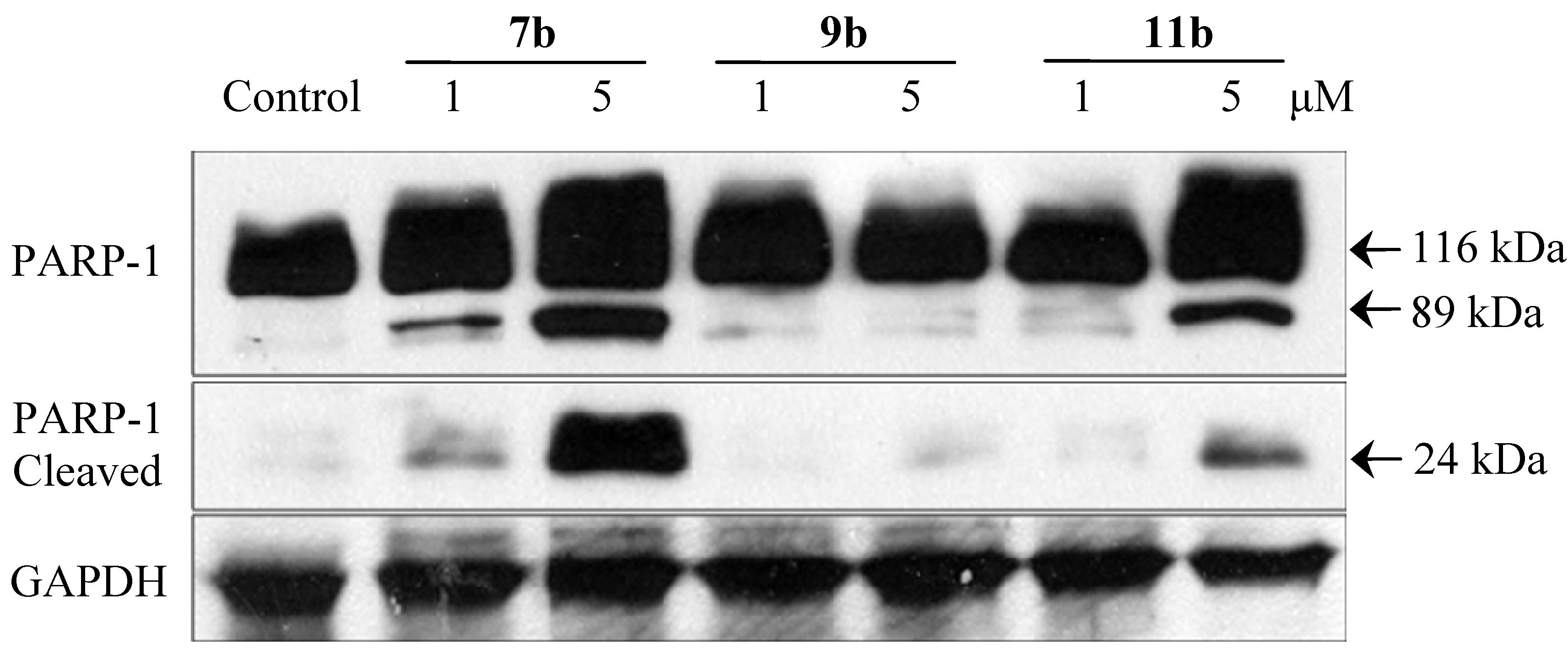
2.2.5. Immunoblotting Assay

3. Experimental Section
3.1. General Information
3.2. Synthesis
3.3. Fluorescence Study Experiment
3.4. Agarosegel Cross-Linking Assay
3.5. In Vitro Cytotoxicity Evaluation
3.6. Flow Cytometry Assay
3.7. Immunoblotting Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Neidle, S.; Thurston, D.E. Chemical approaches to the discovery and development of cancer therapies. Nat. Rev. Cancer 2005, 5, 285–296. [Google Scholar] [CrossRef]
- Chu, P.M.; Chen, L.H.; Chen, M.T.; Ma, H.I.; Su, T.L.; Hsieh, P.C.; Chien, C.S.; Jiang, B.H.; Chen, Y.C.; Lin, Y.H.; et al. Targeting autophagy enhances BO-1051-induced apoptosis in human malignant glioma cells. Cancer Chemother. Pharmacol. 2012, 69, 621–633. [Google Scholar] [CrossRef]
- Kakadiya, R.; Dong, H.; Kumar, A.; Narsinh, D.; Zhang, X.; Chou, T.C.; Lee, T.C.; Shah, A.; Su, T.L. Potent DNA-directed alkylating agents: Synthesis and biological activity of phenyl N-mustard-quinoline conjugates having a urea or hydrazinecarboxamide linker. Bioorg. Med. Chem. 2010, 18, 2285–2299. [Google Scholar] [CrossRef]
- Jiang, Y.; Han, J.; Yu, C.; Vass, S.O.; Searle, P.F.; Browne, P.; Knox, R.J.; Hu, L. Design, synthesis, and biological evaluation of cyclic and acyclic nitrobenzylphosphoramide mustards for E. coli nitroreductase activation. J. Med. Chem. 2006, 49, 4333–4343. [Google Scholar] [CrossRef]
- Duan, J.X.; Jiao, H.L.; Kaizerman, J.; Stanton, T.; Evans, J.W.; Lan, L.; Lorente, G.; Banica, M.; Jung, D.; Wang, J.W.; et al. Potent and highly selective hypoxia-activated achiral phosphoramidate mustards as anticancer drugs. J. Med. Chem. 2008, 51, 2412–2420. [Google Scholar] [CrossRef]
- Kapuriya, N.; Kapuriya, K.; Dong, H.; Zhang, X.; Chou, T.C.; Chen, Y.T.; Lee, T.C.; Lee, W.C.; Tsai, T.H.; Naliapara, Y.; et al. Novel DNA-directed alkylating agents: Design, synthesis and potent antitumor effect of phenyl N-mustard-9-anilinoacridine conjugates via a carbamate or carbonate linker. Bioorg. Med. Chem. 2009, 17, 1264–1275. [Google Scholar] [CrossRef]
- Maze, R.; Carney, J.P.; Kelley, M.R.; Glassner, B.J.; Williams, D.A.; Samson, L. Increasing DNA repair methyltransferase levels via bone marrow stem cell transduction rescues mice from the toxic effects of 1,3-bis(2-chloroethyl)-1-nitrosourea, a chemotherapeutic alkylating agent. Proc. Natl. Acad. Sci. USA 1996, 93, 206–210. [Google Scholar]
- Suzukake, K.; Vistica, B.P.; Vistica, D.T. Dechlorination of l-phenylalanine mustard by sensitive and resistant tumor cells and its relationship to intracellular glutathione content. Biochem. Pharmacol. 1983, 32, 165–167. [Google Scholar] [CrossRef]
- Su, T.L.; Lin, Y.W.; Chou, T.C.; Zhang, X.; Bacherikov, V.A.; Chen, C.H.; Liu, L.F.; Tsai, T.J. Potent antitumor 9-anilinoacridines and acridines bearing an alkylating N-mustard residue on the acridine chromophore: Synthesis and biological activity. J. Med. Chem. 2006, 49, 3710–3718. [Google Scholar]
- Kapuriya, N.; Kapuriya, K.; Zhang, X.; Chou, T.C.; Kakadiya, R.; Wu, Y.T.; Tsai, T.H.; Chen, Y.T.; Lee, T.C.; Shah, A.; et al. Synthesis and biological activity of stable and potent antitumor agents, aniline nitrogen mustards linked to 9-anilinoacridines via a urea linkage. Bioorg. Med. Chem. 2008, 16, 5413–5423. [Google Scholar] [CrossRef]
- Kohler, B.; Su, T.L.; Chou, T.C.; Jiang, X.J.; Watanabe, K.A. Synthesis of cyclopentanthraquinones: Analogs of mitomycin C. J. Org. Chem. 1993, 58, 1680–1686. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Beria, I.; Cozzi, P.; Geroni, C.; Espinosa, A.; Gallo, M.A.; Entrena, A.; Bingham, J.P.; Hartley, J.A.; Romagnoli, R. Cinnamoyl nitrogen mustard derivatives of pyrazole analogues of tallimustine modified at the amidino moiety: Design, synthesis, molecular modeling and antitumor activity studies. Bioorg. Med. Chem. 2004, 12, 3911–3921. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Romagnoli, R.; Giovanna Pavani, M.; del Carmen Nunez, M.; Bingham, J.P.; Hartley, J.A. Benzoyl and cinnamoyl nitrogen mustard derivatives of benzoheterocyclic analogues of the tallimustine: Synthesis and antitumour activity. Bioorg. Med. Chem. 2002, 10, 1611–1618. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Romagnoli, R.; Guadix, A.E.; Pineda de las Infantas, M.J.; Gallo, M.A.; Espinosa, A.; Martinez, A.; Bingham, J.P.; Hartley, J.A. Design, synthesis, and biological activity of hybrid compounds between uramustine and DNA minor groove binder distamycin A. J. Med. Chem. 2002, 45, 3630–3638. [Google Scholar] [CrossRef]
- Pors, K.; Paniwnyk, Z.; Ruparelia, K.C.; Teesdale-Spittle, P.H.; Hartley, J.A.; Kelland, L.R.; Patterson, L.H. Synthesis and biological evaluation of novel chloroethylaminoanthraquinones with potent cytotoxic activity against cisplatin-resistant tumor cells. J. Med. Chem. 2004, 47, 1856–1859. [Google Scholar] [CrossRef]
- Denny, W.A. DNA minor groove alkylating agents. Curr. Med. Chem. 2001, 8, 533–544. [Google Scholar] [CrossRef]
- Brana, M.F.; Ramos, A. Naphthalimides as anti-cancer agents: Synthesis and biological activity. Curr. Med. Chem. Anticancer Agents 2001, 1, 237–255. [Google Scholar] [CrossRef]
- Brana, M.F.; Castellano, J.M.; Moran, M.; Perez de Vega, M.J.; Perron, D.; Conlon, D.; Bousquet, P.F.; Romerdahl, C.A.; Robinson, S.P. Bis-naphthalimides 3: Synthesis and antitumor activity of N,N'-bis[2-(1,8-naphthalimido)-ethyl] alkanediamines. Anticancer Drug Des. 1996, 11, 297–309. [Google Scholar]
- Brana, M.F.; Castellano, J.M.; Moran, M.; Perez de Vega, M.J.; Romerdahl, C.R.; Qian, X.D.; Bousquet, P.; Emling, F.; Schlick, E.; Keilhauer, G. Bis-naphthalimides: A new class of antitumor agents. Anticancer Drug Des. 1993, 8, 257–268. [Google Scholar]
- Pain, A.; Samanta, S.; Dutta, S.; Saxena, A.K.; Shanmugavel, M.; Kampasi, H.; Qazi, G.N.; Sanyal, U. Evaluation of naphthalmustine, a nitrogen mustard derivative of naphthalimide as a rationally-designed anticancer agent. J. Exp. Clin. Cancer Res. 2003, 22, 411–418. [Google Scholar]
- Pain, A.; Samanta, S.; Dutta, S.; Saxena, A.K.; Shanmugavel, M.; Kampasi, H.; Qazi, G.N.; Sanyal, U. Evaluation of napromustine, a nitrogen mustard derivative of naphthalimide, as a rationally designed mixed-function anticancer agent. Exp. Oncol. 2002, 24, 173–179. [Google Scholar]
- Pain, A.; Samanta, S.; Dutta, S.; Saxena, A.K.; Shanmugavel, M.; Kampasi, H.; Quazi, G.N.; Sanyal, U. Synthesis and evaluation of substituted naphthalimide nitrogen mustards as rationally designed anticancer compounds. Acta. Pol. Pharm. 2003, 60, 285–291. [Google Scholar]
- Xu, F.; Gan, Y.; Zhang, Y.; Luo, W.; Ma, H.; Wang, C.; Zhao, J. Synthesis and biological activity of two kinds of nitrogen mustard derivatives. Chin. J. Org. Chem. 2011, 31, 1122–1126. [Google Scholar]
- Hossain, S.U.; Sengupta, S.; Bhattacharya, S. Synthesis and evaluation of antioxidative properties of a series of organoselenium compounds. Bioorg. Med. Chem. 2005, 13, 5750–5758. [Google Scholar] [CrossRef]
- Teiichi, M.; Kiyotaka, F. One-pot synthesis of aryl sulfones from alcohols. Synthesis 2002, 4, 479–482. [Google Scholar]
- Woo, L.W.; Bubert, C.; Sutcliffe, O.B.; Smith, A.; Chander, S.K.; Mahon, M.F.; Purohit, A.; Reed, M.J.; Potter, B.V. Dual aromatase-steroid sulfatase inhibitors. J. Med. Chem. 2007, 50, 3540–3560. [Google Scholar] [CrossRef]
- Hegde, R.; Thimmaiah, P.; Yerigeri, M.C.; Krishnegowda, G.; Thimmaiah, K.N.; Houghton, P.J. Anti-calmodulin acridone derivatives modulate vinblastine resistance in multidrug resistant (MDR) cancer cells. Eur. J. Med. Chem. 2004, 39, 161–177. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lou, Q.; Ji, L.; Zhong, W.; Li, S.; Yu, S.; Li, Z.; Meng, X. Synthesis and Cytotoxicity Evaluation of Naphthalimide Derived N-Mustards. Molecules 2014, 19, 8803-8819. https://doi.org/10.3390/molecules19078803
Lou Q, Ji L, Zhong W, Li S, Yu S, Li Z, Meng X. Synthesis and Cytotoxicity Evaluation of Naphthalimide Derived N-Mustards. Molecules. 2014; 19(7):8803-8819. https://doi.org/10.3390/molecules19078803
Chicago/Turabian StyleLou, Qinghua, Liyan Ji, Wenhe Zhong, Shasha Li, Siwang Yu, Zhongjun Li, and Xiangbao Meng. 2014. "Synthesis and Cytotoxicity Evaluation of Naphthalimide Derived N-Mustards" Molecules 19, no. 7: 8803-8819. https://doi.org/10.3390/molecules19078803