Flavonoids with M1 Muscarinic Acetylcholine Receptor Binding Activity

Abstract
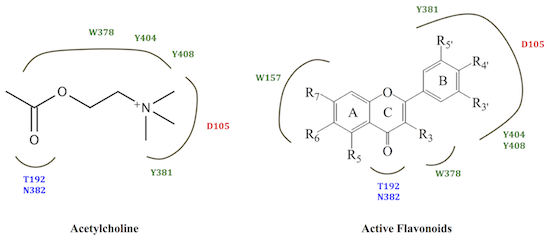
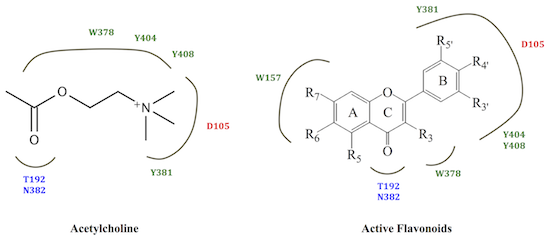
:
1. Introduction

2. Results and Discussion
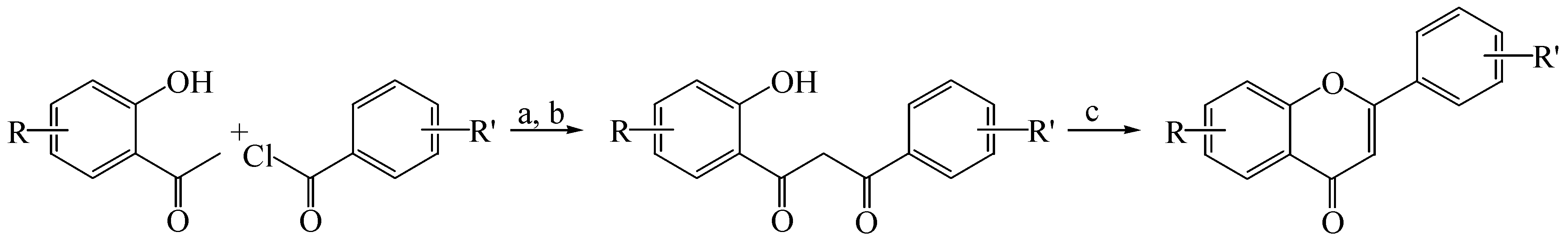
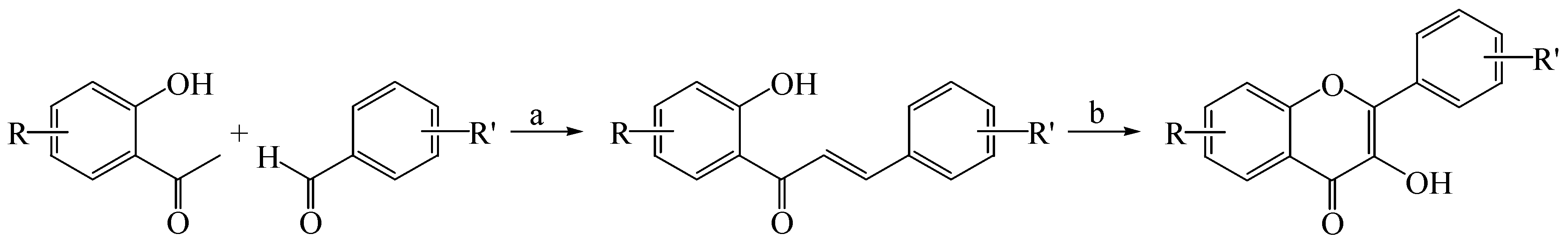
2.1. Synthesis of Flavones and Flavonols


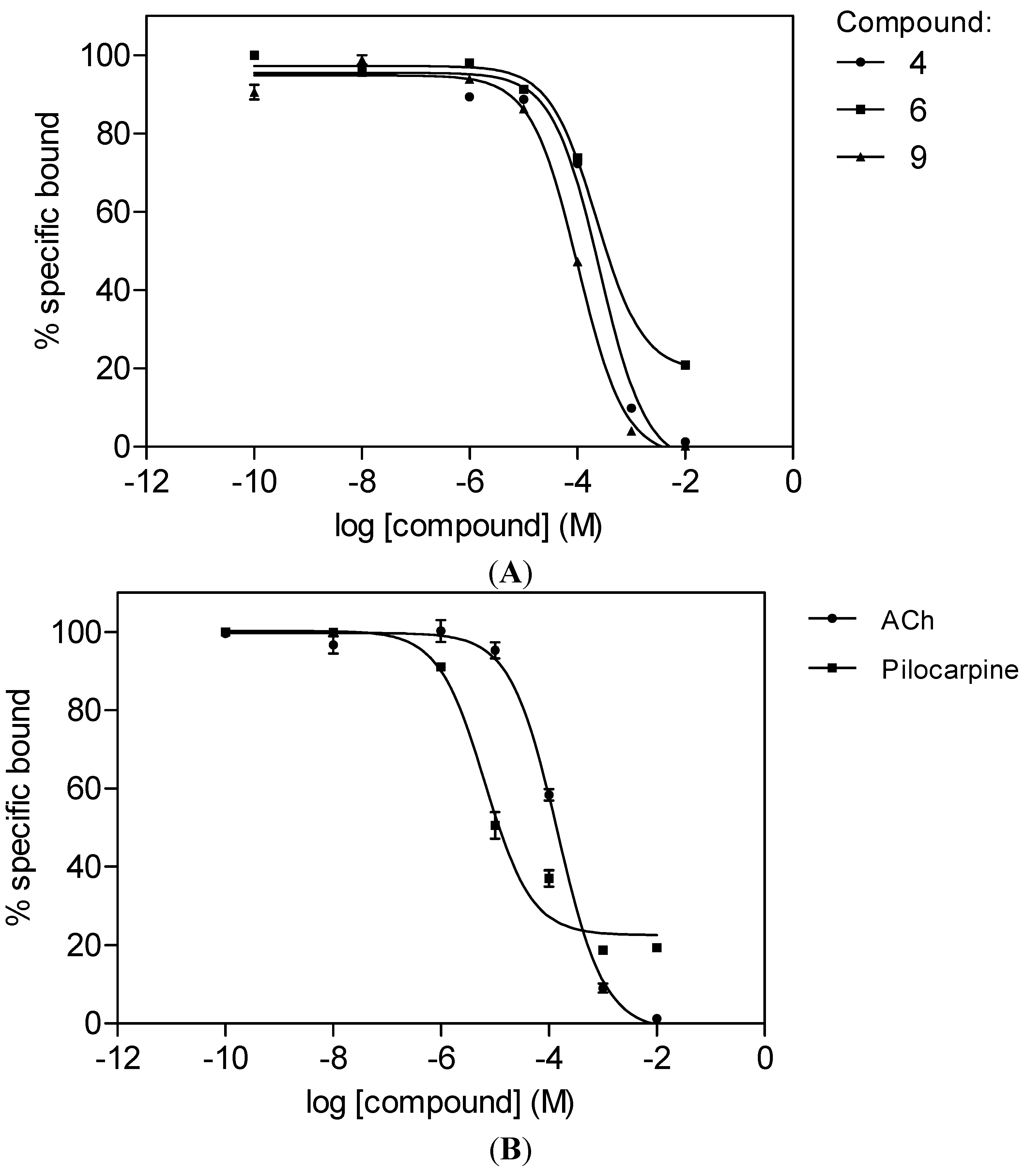
2.2. M1 mAChR Radioligand Binding Assay

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
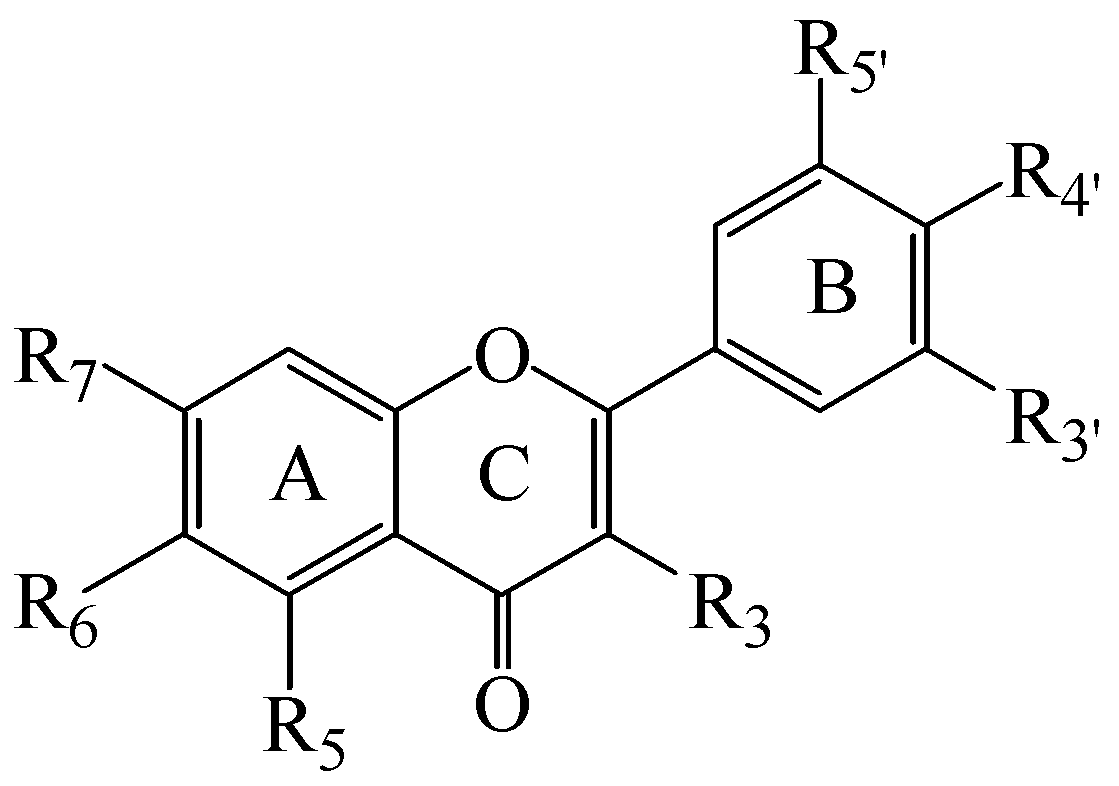
| Cpd | Trivial Name | R3 | R5 | R6 | R7 | R3' | R4' | R5' | IC50 µM | ( Ki µM) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | H | OMe | H | OMe | OMe | OMe | H | >10000 | ||
| 2 | H | OMe | H | OMe | OMe | OMe | OMe | >10000 | ||
| 3 | Sinensetin | H | OMe | OMe | OMe | OMe | OMe | H | >10000 | |
| 4 | H | OMe | OMe | OMe | OMe | OMe | OMe | 257 ± 27 | 107 ± 11 | |
| 5 | Apigenin | H | OH | H | OH | H | OH | H | >10000 | |
| 6 | Luteolin | H | OH | H | OH | OH | OH | H | 226 ± 14 | 94 ± 6 |
| 7 | Diosmetin | H | OH | H | OH | OH | OMe | H | >10000 | |
| 8 | Fisetin | OH | H | H | OH | OH | OH | H | >10000 | |
| 9 | Ombuin | OH | OH | H | OMe | OH | OMe | H | 101 ± 6 | 42 ± 3 |
| 10 | OH | H | OMe | OMe | OMe | OMe | H | >10000 | ||
| 11 | Kaempferol | OH | OH | H | OH | H | OH | H | >10000 | |
| 12 | Quercetin | OH | OH | H | OH | OH | OH | H | >10000 | |
| 13 | Myricetin | OH | OH | H | OH | OH | OH | OH | >10000 | |
| Acetylcholine | 142 ± 12 | 59 ± 5 | ||||||||
| Pilocarpine | 6.5 ± 0.1 | 2.7 ± 0.1 |

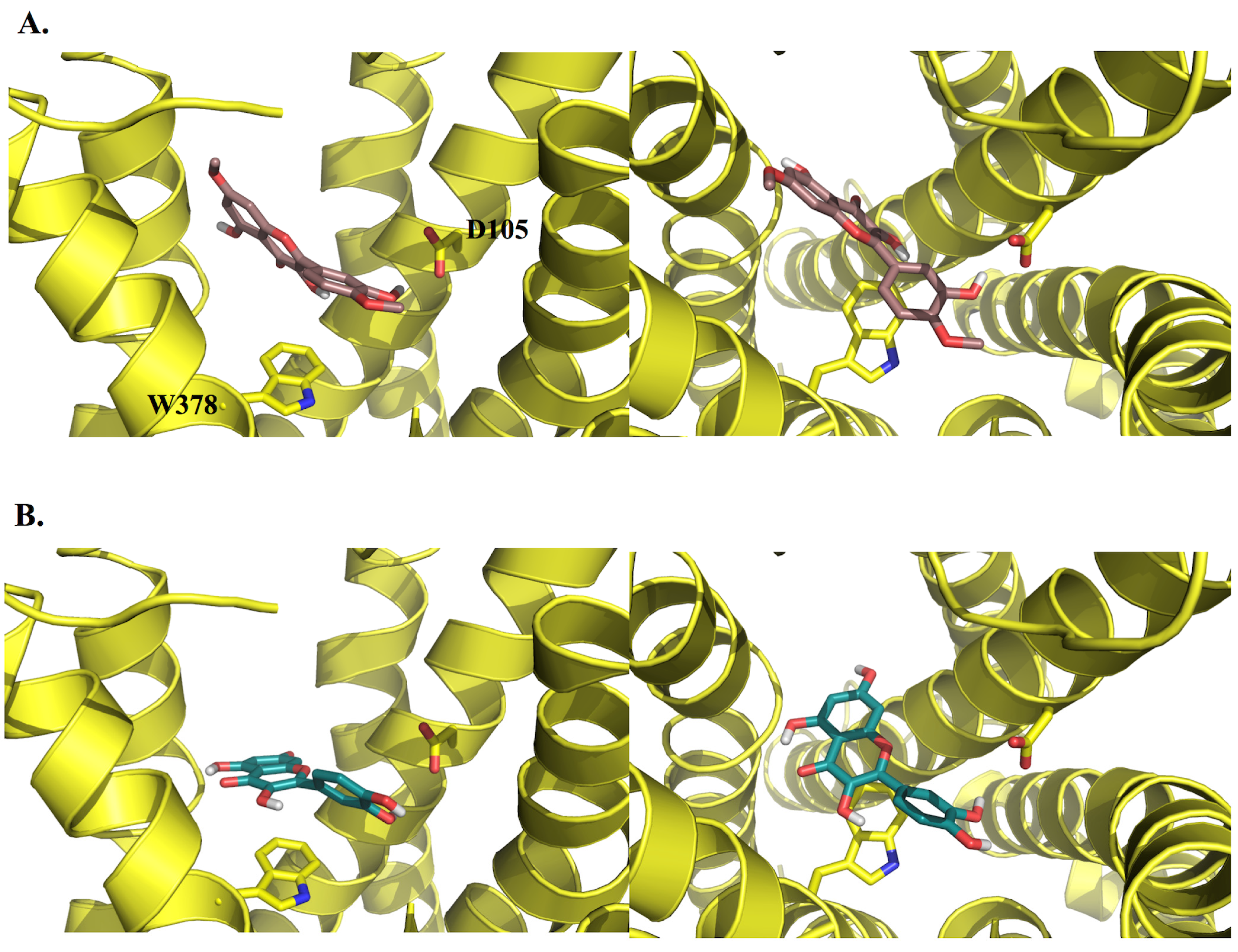
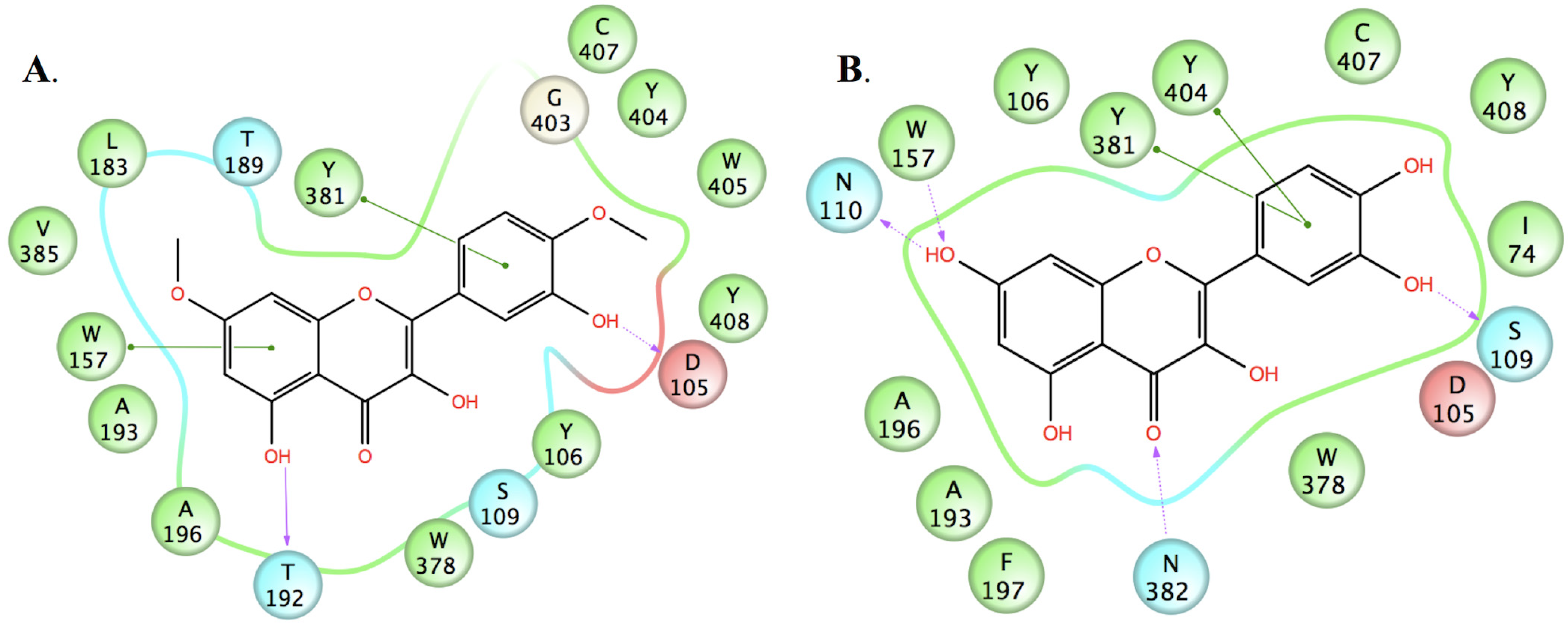
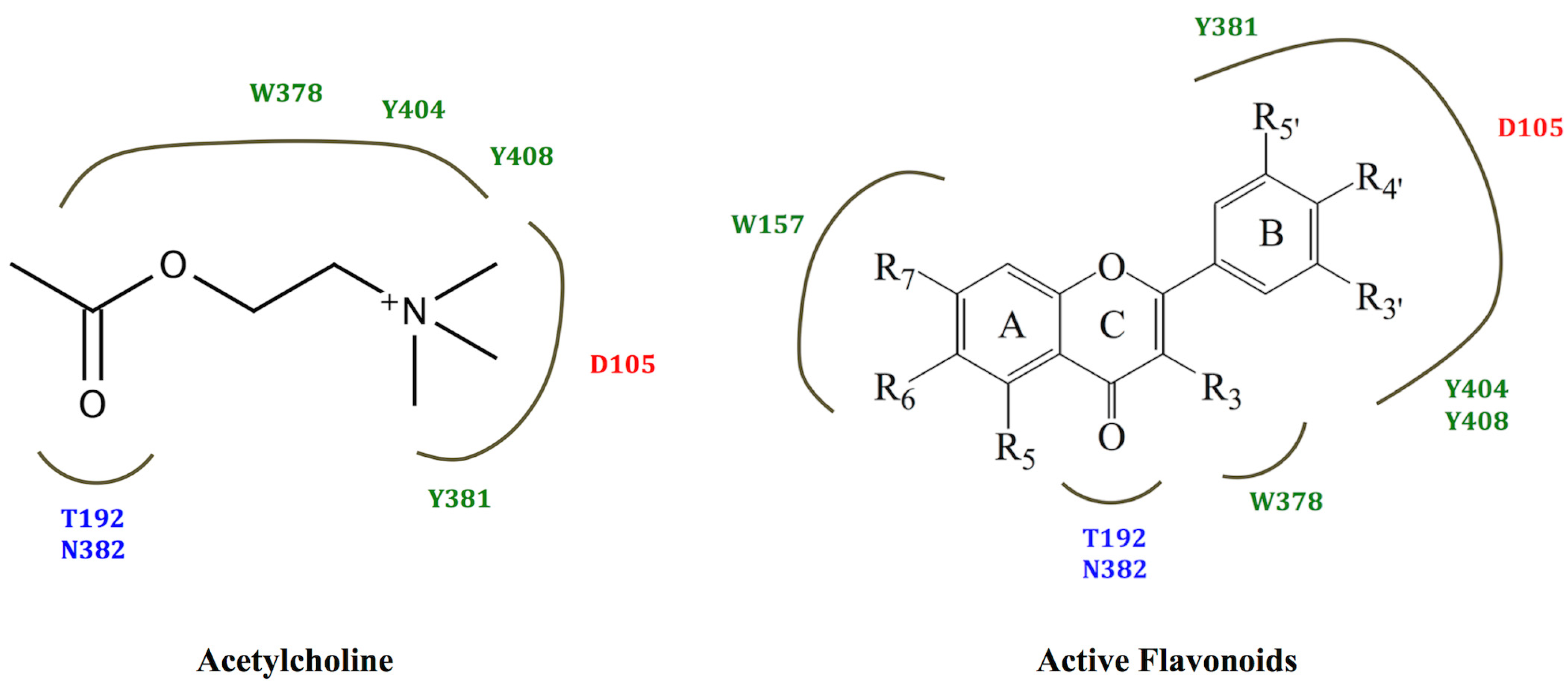
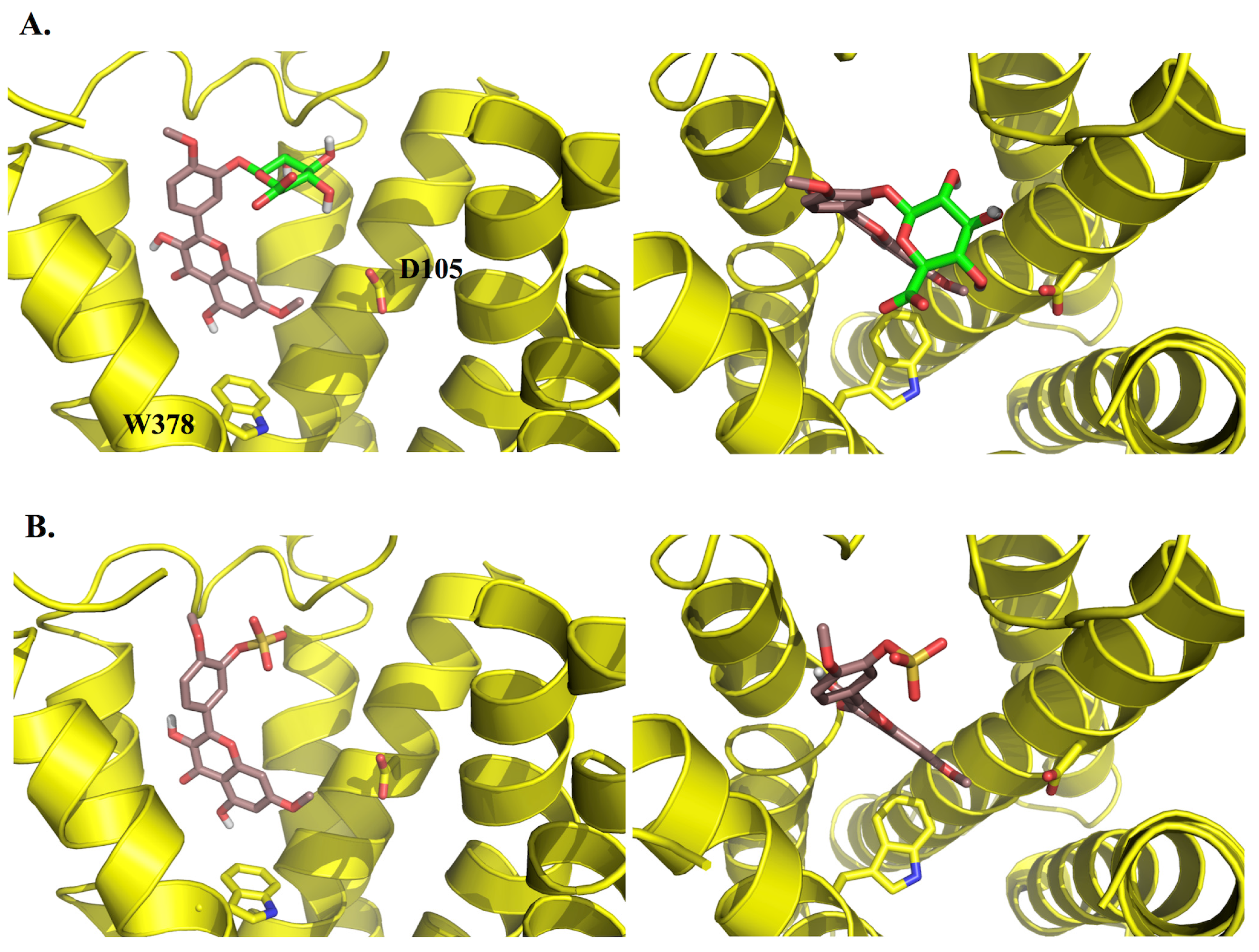
2.3. Molecular Modelling




3. Experimental Section
3.1. Chemistry
3.1.1. General Procedure for the Synthesis of Flavones
3.1.2. General Procedure for the Synthesis of Flavonols
3.2. M1 mAChR Radioligand Binding Assay
3.3. Molecular Modelling
3.3.1. Homology Modelling of the M1 mAChR
3.3.2. Docking Studies
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fisher, A. Cholinergic treatments with emphasis on M1 muscarinic agonists as potential disease-modifying agents for Alzheimer’s disease. Neurotherapeutics 2008, 5, 433–442. [Google Scholar]
- Langmead, C.J.; Watson, J.; Reavill, C. Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol. Ther. 2008, 117, 232–243. [Google Scholar]
- Davis, A.A.; Fritz, J.J.; Wess, J.; Lah, J.J.; Levey, A.I. Deletion of M1 muscarinic acetylcholine receptors increases amyloid pathology in vitro and in vivo. J. Neurosci. 2010, 30, 4190–4196. [Google Scholar]
- Manach, C.; Scalbert, A.; Morand, C.; Remesy, C.; Jimenez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar]
- Chun, O.K.; Chung, S.J.; Song, W.O. Estimated dietary flavonoid intake and major food sources of U.S. adults. J. Nutr. 2007, 137, 1244–1252. [Google Scholar]
- Vauzour, D.; Vafeiadou, K.; Rodriguez-Mateos, A.; Rendeiro, C.; Spencer, J.P.E. The neuroprotective potential of flavonoids: A multiplicity of effects. Genes Nutr. 2008, 3, 115–126. [Google Scholar] [CrossRef]
- Hwang, S.-L.; Shih, P.-H.; Yen, G.-C. Neuroprotective effects of citrus flavonoids. J. Agric. Food Chem. 2012, 60, 877–885. [Google Scholar] [CrossRef]
- Jäger, A.K.; Saaby, L. Flavonoids and the CNS. Molecules 2011, 16, 1471–1485. [Google Scholar] [CrossRef]
- Williams, R.J.; Spencer, J.P.E. Flavonoids, cognition, and dementia: Actions, mechanisms, and potential therapeutic utility for Alzheimer disease. Free Radic. Biol. Med. 2012, 52, 35–45. [Google Scholar] [CrossRef]
- Commenges, D.; Scotet, V.; Renaud, S.; Jacqmin-Gadda, H.; Barberger-Gateau, P.; Dartigues, J.-F. Intake of flavonoids and risk of dementia. Eur. J. Epidemiol. 2000, 16, 357–363. [Google Scholar] [CrossRef]
- Letenneur, L.; Proust-Lima, C.; le Gouge, A.; Dartigues, J.F.; Barberger-Gateau, P. Flavonoid intake and cognitive decline over a 10-year period. Am. J. Epidemiol. 2007, 165, 1364–1371. [Google Scholar] [CrossRef]
- Banister, J.; Whittaker, V.P. Pharmacological activity of the carbon analogue of acetylcholine. Nature 1951, 167, 605–606. [Google Scholar] [CrossRef]
- Barlow, R.B.; Tubby, J.H. Actions of some esters of 3,3-dimethylbutan-1-ol (the carbon analogue of choline) on the guinea-pig ileum. Br. J. Pharmacol. 1974, 51, 95–100. [Google Scholar] [CrossRef]
- Barlow, R.B.; Bond, S.; Holdup, D.W.; Howard, J.A.K.; McQueeen, D.S.; Paterson, A.; Veale, M.A. The contribution of charge to affinity at functional (M3) muscarinic receptors in guinea-pig ileum assessed from the effects of the carbon analogue of 4-DAMP methiodide. Br. J. Pharmacol. 1992, 106, 819–822. [Google Scholar]
- Waelbroeck, M.; Hou, X.; Wehrle, J.; Mutschler, E.; van Tilburg, E.; Menge, W.; Timmerman, H.; Lambrecht, G. Stereoselective interaction of uncharged esters at four muscarinic receptor subtypes muscarinic receptor subtypes. Eur. J. Pharmacol. 1996, 303, 221–226. [Google Scholar] [CrossRef]
- Chung, L.Y.; Yap, K.F.; Goh, S.H.; Mustafa, M.R.; Imiyabir, Z. Muscarinic receptor binding activity of polyoxygenated flavones from Melicope subunifoliolata. Phytochemistry 2008, 69, 1548–1554. [Google Scholar]
- Baker, W. Molecular rearrangement of some o-acyloxyacetophenones and the mechanism of the production of 3-acylchromones. J. Chem. Soc. 1933, 1381–1389. [Google Scholar] [CrossRef]
- Mahal, H.S.; Venkataraman, K. A synthesis of flavones at room temperature. Curr. Sci. 1933, 2, 214–215. [Google Scholar]
- Algar, J.; Flynn, J.P. New synthesis of flavonols. Proc. R. Irish Acad. 1934, 42B, 1–8. [Google Scholar]
- Oyamada, B. A new general method for the synthesis of the derivatives of flavonol. Bull. Chem. Soc. Jpn. 1935, 10, 182–186. [Google Scholar] [CrossRef]
- Kruse, A.C.; Hu, J.; Pan, A.C.; Arlow, D.H.; Rosenbaum, D.M.; Rosemond, E.; Green, H.F.; Liu, T.; Chae, P.S.; Dror, R.O.; et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 2012, 482, 552–556. [Google Scholar] [CrossRef]
- Chin, S.P.; Buckle, M.J.C.; Chalmers, D.K.; Yuriev, E; Doughty, S.W. Toward activated homology models of the human M1 muscarinic acetylcholine receptor. J. Mol. Graph. Model. 2014, 49, 91–98. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar]
- Hulme, E.C.; Lu, Z.L.; Bee, M.S. Scanning mutagenesis studies of the M1 muscarinic acetylcholine receptor. Recept. Channels 2003, 9, 215–228. [Google Scholar] [CrossRef]
- Goodwin, J.A.; Hulme, E.C.; Langmead, C.J.; Tehan, B.G. Roof and floor of the muscarinic binding pocket: Variations in the binding modes of orthosteric ligands. Mol. Pharmacol. 2007, 72, 1484–1496. [Google Scholar] [CrossRef]
- Haga, K.; Kruse, A.C.; Asada, H.; Yurugi-Kobayashi, T.; Shiroishi, M.; Zhang, C.; Weis, W.I.; Okada, T.; Kobilka, B.K.; Haga, T.; et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 2012, 482, 547–551. [Google Scholar]
- Gualtieri, F.; Romanelli, M.N.; Teodori, E. Eudismic analysis of a series of muscarinic ligands carrying a 1,3-oxathiolane nucleus. Chirality 1990, 2, 79–84. [Google Scholar] [CrossRef]
- Conforti, F.; Rigano, D.; Formisano, C.; Bruno, M.; Loizzo, M.R.; Menichini, F.; Senatore, F. Metabolic profile and and in vitro activities of Phagnalon saxatile (L.) Cass. relevant to treatment of Alzheimer’s disease. J. Enzym. Inhib. Med. Chem. 2010, 25, 97–104. [Google Scholar] [CrossRef]
- Kang, S.S.; Lee, J.Y.; Choi, Y.K.; Kim, G.S.; Han, B.H. Neuroprotective effects of flavones on hydrogen peroxide-induced apoptosis in SH-SY5Y neuroblostoma cells. Bioorg. Med. Chem. Lett. 2004, 14, 2261–2264. [Google Scholar]
- Liu, R.; Gao, M.; Qiang, G.F.; Zhang, T.T.; Lan, X.; Ying, J.; Du, G.H. The anti-amnesic effects of luteolin against amyloid β(25-35) peptide-induced toxicity in mice involve the protection of neurovascular unit. Neuroscience 2009, 162, 1232–1243. [Google Scholar] [CrossRef]
- Liu, Y.; Tian, X.; Gou, L.; Sun, L.; Ling, X.; Yin, X. Luteolin attenuates diabetes-associated cognitive decline in rats. Brain Res. Bull. 2013, 94, 23–29. [Google Scholar]
- Grabher, P.; Durieu, E.; Kouloura, E.; Halabalaki, M.; Skaltsounis, L.A.; Meijer, L.; Hamburger, M.; Potterat, O. Library-based discovery of DYRK1A/CLK1 inhibitors from natural product extracts. Planta Med. 2012, 78, 951–956. [Google Scholar] [CrossRef]
- Rangel-Ordóñez, L.; Nöldner, M.; Schubert-Zsilavecz, M.; Wurglics, M. Plasma levels and distribution of flavonoids in rat brain after single and repeated doses of standardized ginkgo biloba extract EGb 761®. Planta Med. 2010, 76, 1683–1690. [Google Scholar]
- Boersma, M.G.; van der Woude, H.; Bogaards, J.; Boeren, S.; Vervoort, J.; Cnubben, N.H.; van Iersel, M.L.; van Bladeren, P.J.; Rietjens, I.M. Regioselectivity of phase II metabolism of luteolin and quercetin by UDP-glucuronosyl transferases. Chem. Res. Toxicol. 2002, 15, 662–670. [Google Scholar]
- Day, A.J.; Bao, Y.; Morgan, M.R.; Williamson, G. Conjugation position of quercetin glucuronides and effect on biological activity. Free Radic. Biol. Med. 2000, 29, 1234–1243. [Google Scholar]
- Day, A.J.; Mellon, F.; Barron, D.; Sarrazin, G.; Morgan, M.R.; Williamson, G. Human metabolism of dietary flavonoids: Identification of plasma metabolites of quercetin. Free Radic. Res. 2001, 35, 941–952. [Google Scholar]
- O’Leary, K.A.; Day, A.J.; Needs, P.W.; Sly, W.S.; O’Brien, N.M.; Williamson, G. Flavonoid glucuronides are substrates for human liver β-glucuronidase. FEBS Lett. 2001, 503, 103–106. [Google Scholar] [CrossRef]
- Shimoi, K.; Saka, N.; Nozawa, R.; Sato, M.; Amano, I.; Nakayama, T.; Kinae, N. Deglucuronidation of a flavonoid, luteolin monoglucuronide, during inflammation. Drug Metab. Dispos. 2001, 29, 1521–1524. [Google Scholar]
- Bartholomé, R.; Haenen, G.; Hollman, C.H.; Bast, A.; Dagnelie, P.C.; Roos, D.; Keijer, J.; Kroon, P.A.; Needs, P.W.; Arts, I.C. Deconjugation kinetics of glucuronidated phase II flavonoid metabolites by β-glucuronidase from neutrophils. Drug Metab. Pharmacokinet. 2010, 25, 379–387. [Google Scholar] [CrossRef]
- Menendez, C.; Dueñas, M.; Galindo, P.; González-Manzano, S.; Jimenez, R.; Moreno, L.; Zarzuelo, M.J.; Rodríguez-Gómez, I.; Duarte, J.; Santos-Buelga, C.; et al. Vascular deconjugation of quercetin glucuronide: The flavonoid paradox revealed? Mol. Nutr. Food Res. 2011, 55, 1780–1790. [Google Scholar] [CrossRef]
- Sutthanut, K.; Sripanidkulchai, B.; Yenjai, C.; Jay, M. Simultaneous identification and quantitation of 11 flavonoid constituents in Kaempferia parviflora by gas chromatography. J. Chromatogr. A 2007, 1143, 227–233. [Google Scholar] [CrossRef]
- Mateeva, N.N.; Kode, R.N.; Redda, K.K. Synthesis of novel flavonoid derivatives as potential HIV-integrase inhibitors. J. Heterocycl. Chem. 2002, 39, 1251–1258. [Google Scholar] [CrossRef]
- Chu, H.W.; Wu, H.T.; Lee, Y.J. Regioselective hydroxylation of 2-hydroxychalcones by dimethyldioxirane towards polymethoxylated flavonoids. Tetrahedron 2004, 60, 2647–2655. [Google Scholar]
- Kong, C.; Liang, W.; Hu, F.; Xu, X.; Wang, P.; Jiang, Y.; Xing, B. Allelochemicals and their transformations in the Ageratum conyzoides intercropped citrus orchard soils. Plant Soil 2004, 264, 149–157. [Google Scholar] [CrossRef]
- Huong, D.T.; Luong, D.V.; Thao, T.T.; Sung, T.V. A new flavone and cytotoxic activity of flavonoid constituents isolated from Miliusa balansae (Annonaceae). Pharmazie 2005, 60, 627–629. [Google Scholar]
- King, F.E.; King, T.J.; Neill, K.G. The chemistry of extractives from hardwoods. Part XI. The isolation of a diterpene ester (methyl vinhaticoate), and of 6:7:3':4'-tetrahydroxyflavanone (plathymenin), and 2:4:5:3':4'-pentahydroxychalkone (neoplathymenin), from Plathymenia reticulate. J. Chem. Soc. 1953, 1055–1059. [Google Scholar]
- Cheng, Y.C.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The Swiss-Prot protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar]
- Armougom, F.; Moretti, S.; Poirot, O.; Audic, S.; Dumas, P.; Schaeli, B.; Keduas, V.; Notredame, C. Expresso: Automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res. 2006, 34, 604–608. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Sample Availability: Samples of compounds 1–4 and 9–10 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swaminathan, M.; Chee, C.F.; Chin, S.P.; Buckle, M.J.C.; Rahman, N.A.; Doughty, S.W.; Chung, L.Y. Flavonoids with M1 Muscarinic Acetylcholine Receptor Binding Activity. Molecules 2014, 19, 8933-8948. https://doi.org/10.3390/molecules19078933
Swaminathan M, Chee CF, Chin SP, Buckle MJC, Rahman NA, Doughty SW, Chung LY. Flavonoids with M1 Muscarinic Acetylcholine Receptor Binding Activity. Molecules. 2014; 19(7):8933-8948. https://doi.org/10.3390/molecules19078933
Chicago/Turabian StyleSwaminathan, Meyyammai, Chin Fei Chee, Sek Peng Chin, Michael J. C. Buckle, Noorsaadah Abd. Rahman, Stephen W. Doughty, and Lip Yong Chung. 2014. "Flavonoids with M1 Muscarinic Acetylcholine Receptor Binding Activity" Molecules 19, no. 7: 8933-8948. https://doi.org/10.3390/molecules19078933