Intramolecular Hydrogen Bond in Biologically Active o-Carbonyl Hydroquinones

 and
and
Abstract
:
1. Introduction

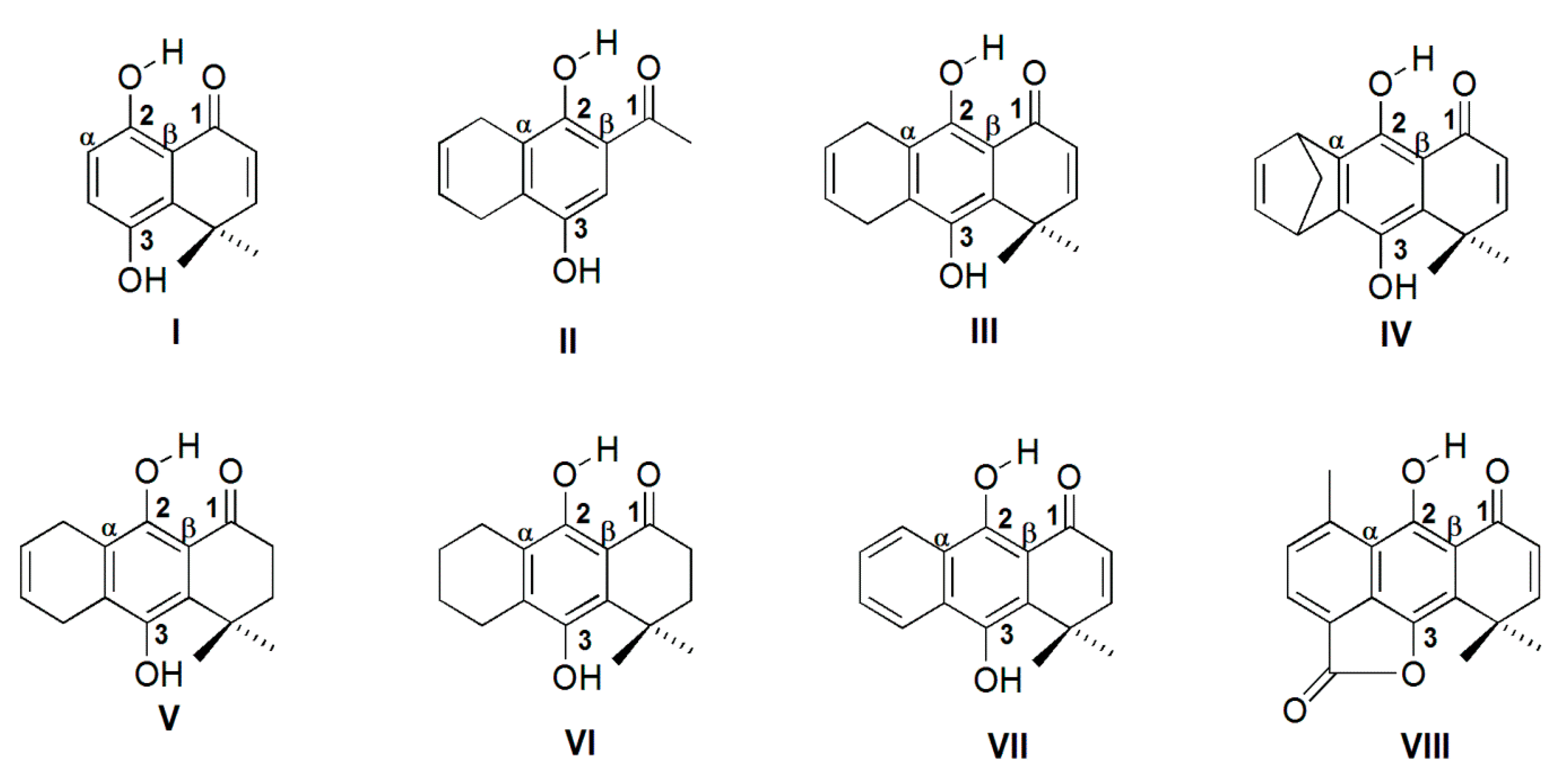
2. Results and Discussion
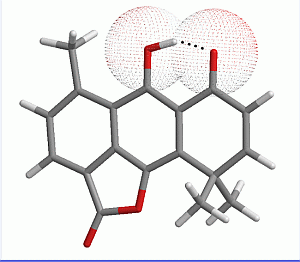
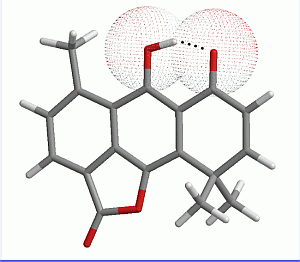
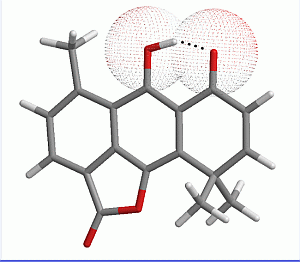
2.1. Geometry Optimization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | δH2 | B3LYP/6-31++G(d,p) | MP2/6-31++G(d,p) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| O1…O2 | O2-H2 | O1…H2 | < O2-H2…O1 | O1…O2 | O2-H2 | O1…H2 | < O2-H2…O1 | ||
| I | 12.54 | 2.540 | 0.996 | 1.638 | 148 | 2.573 | 0.989 | 1.682 | 148 |
| II | 12.32 | 2.556 | 0.994 | 1.657 | 148 | 2.592 | 0.988 | 1.703 | 148 |
| III | 13.08 | 2.533 | 0.998 | 1.624 | 149 | 2.567 | 0.991 | 1.667 | 149 |
| IV | 12.70 | 2.538 | 0.996 | 1.634 | 149 | 2.571 | 0.990 | 1.679 | 148 |
| V | 12.95 | 2.525 | 0.997 | 1.617 | 149 | 2.570 | 0.989 | 1.676 | 148 |
| VI | 12.94 | 2.521 | 0.997 | 1.613 | 149 | 2.521 | 0.997 | 1.613 | 149 |
| VII | 14.53 | 2.505 | 1.005 | 1.584 | 150 | 2.543 | 0.995 | 1.637 | 149 |
| VIII | 15.60 | 2.482 | 1.014 | 1.544 | 152 | 2.526 | 0.999 | 1.608 | 150 |
| R2 | 0.89 | 0.98 | 0.94 | 0.92 | 0.39 | 0.84 | 0.50 | 0.59 | |
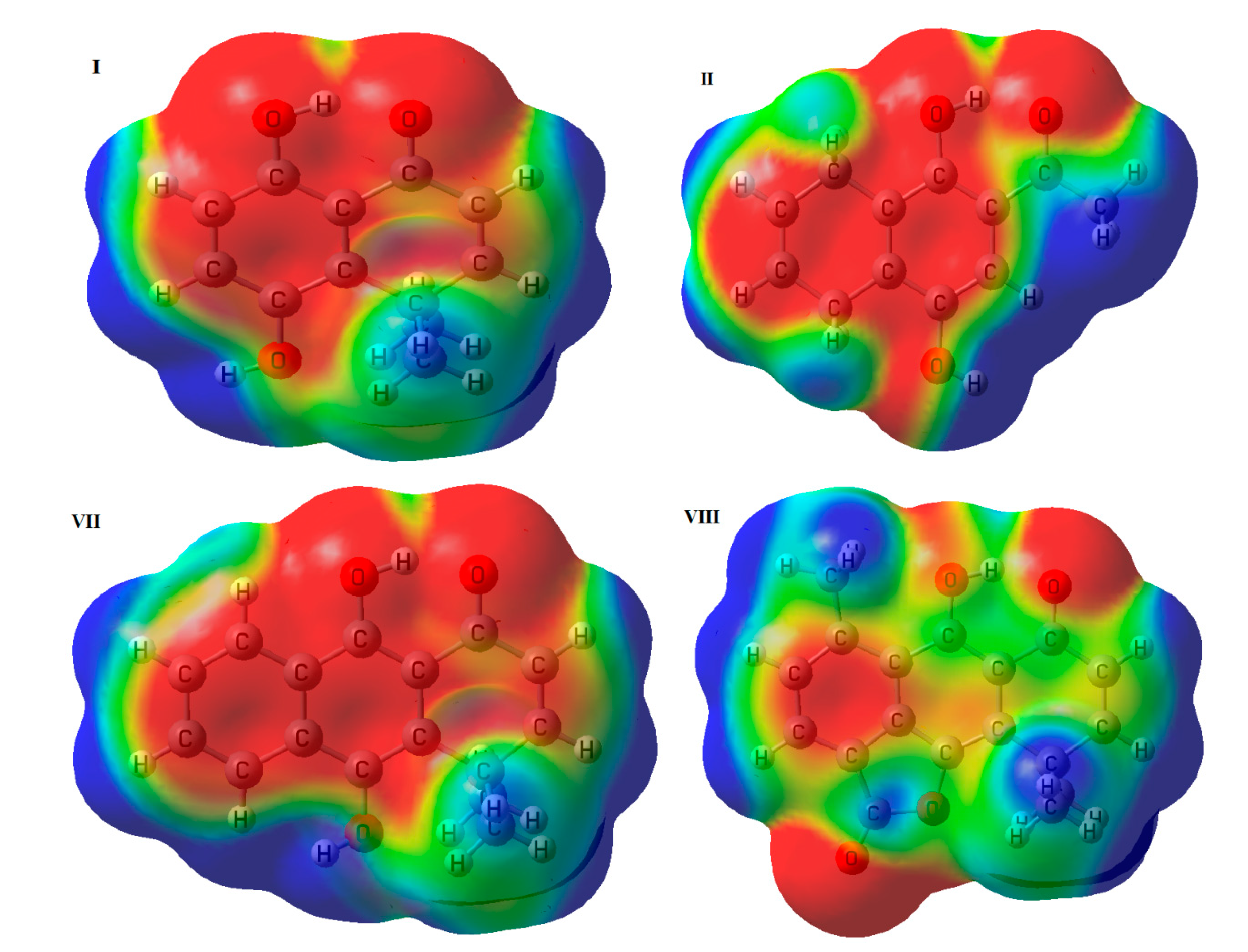
2.2. Molecular Electrostatic Potential

| Molecule | Vα(r) | Vmin(O1) |
|---|---|---|
| I | 165.0 | −48.9 |
| II | 169.2 | −45.0 |
| III | 163.2 | −50.5 |
| IV | 161.7 | −51.3 |
| V | 165.7 | −48.2 |
| VI | 164.1 | −49.2 |
| VII | 166.2 | −49.5 |
| VIII | 174.9 | −43.6 |
2.3. NBO Analysis
| Molecule | NC O1 | NC O2 | NC H2 | WBO O2-H2 | WBO H2…O1 |
|---|---|---|---|---|---|
| I | −0.721 | −0.753 | 0.522 | 0.6470 | 0.0699 |
| II | −0.717 | −0.759 | 0.524 | 0.6501 | 0.0647 |
| III | −0.726 | −0.765 | 0.525 | 0.6395 | 0.0747 |
| IV | −0.727 | −0.760 | 0.522 | 0.6460 | 0.0712 |
| V | −0.725 | −0.763 | 0.524 | 0.6393 | 0.0751 |
| VI | −0.726 | −0.765 | 0.523 | 0.6402 | 0.0757 |
| VII | −0.734 | −0.760 | 0.530 | 0.6197 | 0.0890 |
| VIII | −0.736 | −0.755 | 0.532 | 0.6004 | 0.1051 |
| Molecule | Φi | Φj | ∆Eij (2) | εj–εi/au | Fij/au | Φi | Φj | ∆Eij (2) | εj−εi/au | Fij/au |
|---|---|---|---|---|---|---|---|---|---|---|
| I | LP1 O1 | σ* O2-H2 | 4.04 | 1.58 | 0.072 | LP1 O2 | σ* C2-Cα | 10.15 | 1.60 | 0.114 |
| LP2 O1 | σ* O2-H2 | 28.33 | 1.18 | 0.165 | LP2 O2 | π* C2-Cα | 48.43 | 0.63 | 0.168 | |
| II | LP1 O1 | σ* O2-H2 | 3.67 | 1.58 | 0.068 | LP1 O2 | σ* C2-Cα | 9.19 | 1.61 | 0.109 |
| LP2 O1 | σ* O2-H2 | 26.02 | 1.18 | 1.159 | LP2 O2 | π* C2-Cα | 48.19 | 0.64 | 0.170 | |
| III | LP1 O1 | σ* O2-H2 | 4.08 | 1.57 | 0.072 | LP1 O2 | σ* C2-Cα | 9.42 | 1.61 | 0.110 |
| LP2 O1 | σ* O2-H2 | 30.58 | 1.18 | 0.171 | LP2 O2 | π* C2-Cα | 47.57 | 0.64 | 0.167 | |
| IV | LP1 O1 | σ* O2-H2 | 4.05 | 1.58 | 0.072 | LP1 O2 | σ* C2-Cα | 10.08 | 1.58 | 0.113 |
| LP2 O1 | σ* O2-H2 | 29.01 | 1.18 | 0.167 | LP2 O2 | π* C2-Cβ | 43.47 | 0.67 | 0.161 | |
| V | LP1 O1 | σ* O2-H2 | 4.10 | 1.57 | 0.072 | LP1 O2 | σ* C2-Cα | 9.46 | 1.61 | 0.110 |
| LP2 O1 | σ* O2-H2 | 30.88 | 1.18 | 0.173 | LP2 O2 | π* C2-Cα | 49.06 | 0.64 | 0.170 | |
| VI | LP1 O1 | σ* O2-H2 | 4.15 | 1.57 | 0.072 | LP1 O2 | σ* C2-Cα | 9.67 | 1.60 | 0.111 |
| LP2 O1 | σ* O2-H2 | 31.31 | 1.19 | 0.174 | LP2 O2 | π* C2-Cα | 48.22 | 0.63 | 0.169 | |
| VII | LP1 O1 | σ* O2-H2 | 4.36 | 1.54 | 0.074 | LP1 O2 | σ* C2-Cα | 9.70 | 1.63 | 0.112 |
| LP2 O1 | σ* O2-H2 | 37.44 | 1.17 | 0.189 | LP2 O2 | π* C2-Cα | 55.49 | 0.65 | 0.178 | |
| VIII | LP1 O1 | σ* O2-H2 | 4.64 | 1.51 | 0.076 | LP1 O2 | σ* C2-Cα | 9.82 | 1.60 | 0.112 |
| LP2 O1 | σ* O2-H2 | 45.67 | 1.16 | 0.208 | LP2 O2 | π* C2-Cα | 60.70 | 0.64 | 0.184 |
3. Experimental
3.1. General Information
3.2. Theoretical Methods

4. Conclusions
Supplementary Materials
Acknowlegments
Author Contributions
Conflicts of Interest
References
- Goulart, M.O.F.; Zani, C.L.; Tonholo, J.; Freitas, L.R.; deAbreu, F.C.; Oliveira, A.B.; Raslan, D.S.; Starling, S.; Chiari, E. Trypanocidal activity and redox potential of heterocyclic- and 2-hydroxy-naphthoquinones. Bioorg. Med. Chem. Lett. 1997, 7, 2043–2048. [Google Scholar] [CrossRef]
- Thomson, R.H. Naturally Occurring Quinones; Academic Press: New York, NY, USA, 1971; pp. 1–38. [Google Scholar]
- Monks, T.J.; Hanzlik, R.P.; Cohen, G.M.; Ross, D.; Graham, D.G. Quinone chemistry and toxicity. Toxicol. Appl. Pharm. 1992, 112, 2–16. [Google Scholar]
- De Abreu, F.C.; Ferraz, P.A.D.; Goulart, M.O.F. Some applications of electrochemistry in biomedical chemistry. Emphasis on the correlation of electrochemical and bioactive properties. J. Brazil. Chem. Soc. 2002, 13, 19–35. [Google Scholar] [CrossRef]
- Nyland, R.L.; Luo, M.H.; Kelley, M.R.; Borch, R.F. Design and synthesis of novel quinone inhibitors targeted to the redox function of apurinic/apyrimidinic endonuclease 1/redox enhancing factor-1 (Ape1/ref-1). J. Med. Chem. 2010, 53, 1200–1210. [Google Scholar] [CrossRef]
- Pickhardt, M.; Gazova, Z.; von Bergen, M.; Khlistunova, I.; Wang, Y.P.; Hascher, A.; Mandelkow, E.M.; Biernat, J.; Mandelkow, E. Anthraquinones inhibit tau aggregation and dissolve Alzheimer’s paired helical filaments in vitro and in cells. J. Biol. Chem. 2005, 280, 3628–3635. [Google Scholar] [CrossRef]
- Roginsky, V.; Barsukova, T.; Loshadkin, D.; Pliss, E. Substituted p-hydroquinones as inhibitors of lipid peroxidation. Chem. Phys. Lipids 2003, 125, 49–58. [Google Scholar]
- Inbaraj, J.J.; Chignell, C.F. Cytotoxic action of juglone and plumbagin: A mechanistic study using HaCaT keratinocytes. Chem. Res. Toxicol. 2004, 17, 55–62. [Google Scholar]
- Assimopoulu, A.N.; Boskou, D.; Papageorgiou, V.P. Antioxidant activities of alkannin, shikonin and alkanna tinctoria root extracts in oil substrates. Food Chem. 2004, 87, 433–438. [Google Scholar] [CrossRef]
- Schreiber, J.; Mottley, C.; Sinha, B.K.; Kalyanaraman, B.; Mason, R.P. One-electron reduction of daunomycin, daunomycinone, and 7-deoxydaunomycinone by the xanthine/xanthine oxidase system: Detection of semiquinone free radicals by electron spin resonance. J. Am. Chem. Soc. 1987, 109, 348–351. [Google Scholar] [CrossRef]
- Armendáriz-Vidales, G.; Martínez-González, E.; Cuevas-Fernández, H.J.; Fernández-Campos, D.O.; Burgos-Castillo, R.C.; Frontana, C. In situ characterization by cyclic voltammetry and conductance of composites based on polypyrrole, multi-walled carbon nanotubes and cobalt phthalocyanine. Electrochim. Acta 2013, 89, 840–847. [Google Scholar]
- Foti, M.C.; Johnson, E.R.; Vinqvist, M.R.; Wright, J.S.; Barclay, L.R.C.; Ingold, K.U. Naphthalene diols: A new class of antioxidants intramolecular hydrogen bonding in catechols, naphthalene diols, and their aryloxyl radicals. J. Org. Chem. 2002, 67, 5190–5196. [Google Scholar] [CrossRef]
- Foti, M.C.; Amorati, R.; Pedulli, G.F.; Daquino, C.; Pratt, D.A.; Ingold, K.U. Influence of "Remote" intramolecular hydrogen bonds on the stabilities of phenoxyl radicals and benzyl cations. J. Org. Chem. 2010, 75, 4434–4440. [Google Scholar] [CrossRef]
- Pihko, P.M. Hydrogen Bonding in Organic Synthesis, 1st ed.; Wiley-VHC: Weinhein, Germany, 2009; p. 383. [Google Scholar]
- Gilli, G.; Gilli, P. The Nature of the Hydrogen Bond: Outline of a Comprehensive Hydrogen Bond Theory, 1st ed.; Oxford University Press: Oxford, UK, 2009; p. 317. [Google Scholar]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Defining the hydrogen bond: An account (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 1619–1636. [Google Scholar]
- Lown, J.W. Molecular mechanisms of action of anticancer agents involving free radical intermediates. Adv. Free Radic. Biol. Med. 1985, 1, 225–264. [Google Scholar]
- Han, Y.; Jung, H.W.; Lee, J.Y.; Kim, J.S.; Kang, S.S.; Kim, Y.S.; Park, Y.K. 2,5-Dihydroxyacetophenone isolated from rehmanniae radix preparata inhibits inflammatory responses in lipopolysaccharide-stimulated RAW264.7 macrophages. J. Med. Food 2012, 15, 505–510. [Google Scholar]
- Talpir, R.; Rudi, A.; Kashman, Y.; Loya, Y.; Hizi, A. Three new sesquiterpene hydroquinones from marine origin. Tetrahedron 1994, 50, 4179–4184. [Google Scholar]
- Loya, S.; Bakhanashvili, M.; Kashman, Y.; Hizi, A. Peyssonol-a and peyssonal-b, 2 novel inhibitors of the reverse transcriptases of human-immunodeficiency-virus type-1 and type-2. Arch. Biochem. Biophys. 1995, 316, 789–796. [Google Scholar] [CrossRef]
- Maruyama, K.; Naruta, Y. A new method for the synthesis of β-glucosides using 2-chloro-3,5-dinitropyridine. Chem. Lett. 1979, 8, 847–848. [Google Scholar]
- Brimble, M.A.; Lynds, S.M. A short synthesis of deoxyfrenolicin. J. Chem. Soc. Perk. Trans. 1 1994, 5, 493–496. [Google Scholar] [CrossRef]
- Kraus, G.A.; Maeda, H. A direct preparation of 1,4-benzodiazepines - the synthesis of medazepam and related-compounds via a common intermediate. Tetrahedron Lett. 1994, 35, 9189–9190. [Google Scholar]
- Valderrama, J.A.; Benites, J.; Cortes, M.; Pessoa-Mahana, D.; Prina, E.; Fournet, A. Studies on quinones. Part 35: Access to antiprotozoal active euryfurylquinones and hydroquinones. Tetrahedron 2002, 58, 881–886. [Google Scholar]
- Valderrama, J.A.; Zamorano, C.; Gonzalez, M.F.; Prina, E.; Fournet, A. Studies on quinones. Part 39: Synthesis and leishmanicidal activity of acylchloroquinones and hydroquinones. Bioorg. Med. Chem. 2005, 13, 4153–4159. [Google Scholar] [CrossRef]
- Rios, D.; Benites, J.; Valderrama, J.A.; Farias, M.; Pedrosa, R.C.; Verrax, J.; Calderon, P.B. Biological evaluation of 3-acyl-2-arylamino-1,4-naphthoquinones as inhibitors of Hsp90 chaperoning function. Curr. Top. Med. Chem. 2012, 12, 2094–2102. [Google Scholar]
- Araya-Maturana, R.; Delgado-Castro, T.; Garate, M.; Ferreira, J.; Pavani, M.; Pessoa-Mahana, H.; Cassels, B.K. Effects of 4,4-dimethyl-5,8-dihydroxynaphtalene-1-one and 4,4-dimethyl-5,8-dihydroxytetralone derivatives on tumor cell respiration. Bioorg. Med. Chem. 2002, 10, 3057–3060. [Google Scholar]
- Araya-Maturana, R.; Cardona, W.; Cassels, B.K.; Delgado-Castro, T.; Ferreira, J.; Miranda, D.; Pavani, M.; Pessoa-Mahana, H.; Soto-Delgado, J.; Weiss-Lopez, B. Effects of 9,10-dihydroxy-4,4-dimethyl-5,8-dihydro-1(4H)-anthracenone derivatives on tumor cell respiration. Bioorg. Med. Chem. 2006, 14, 4664–4669. [Google Scholar]
- Urra, F.A.; Martínez-Cifuentes, M.; Pavani, M.; Lapier, M.; Jana-Prado, F.; Parra, E.; Maya, J.D.; Pessoa-Mahana, H.; Ferreira, J.; Araya-Maturana, R. An ortho-carbonyl substituted hydroquinone derivative is an anticancer agent that acts by inhibiting mitochondrial bioenergetics and by inducing G(2)/M-phase arrest in mammary adenocarcinoma TA3. Toxicol. Appl. Pharm. 2013, 267, 218–227. [Google Scholar]
- Dobado, J.A.; Gómez-Tamayo, J.C.; Calvo-Flores, F.G.; Martínez-García, H.; Cardona, W.; Weiss-López, B.; Ramírez-Rodríguez, O.; Pessoa-Mahana, H.; Araya-Maturana, R. NMR assignment in regioisomeric hydroquinones. Magn. Reson. Chem. 2011, 49, 358–365. [Google Scholar]
- Rodriguez, J.; Olea-Azar, C.; Cavieres, C.; Norambuena, E.; Delgado-Castro, T.; Soto-Delgado, J.; Araya-Maturana, R. Antioxidant properties and free radical-scavenging reactivity of a family of hydroxynaphthalenones and dihydroxyanthracenones. Bioorg. Med. Chem. 2007, 15, 7058–7065. [Google Scholar] [CrossRef]
- Soto-Delgado, J.; Bahamonde-Padilla, V.; Araya-Maturana, R.; Weiss-Lopez, B.E. On the mechanism of biological activity of hydroquinone derivatives that inhibit tumor cell respiration. A theoretical study. Comput. Theor. Chem. 2013, 1013, 97–101. [Google Scholar] [CrossRef]
- Graton, J.; Wang, Z.; Brossard, A.M.; Monteiro, D.G.; le Questel, J.Y.; Linclau, B. An Unexpected and Significantly Lower Hydrogen-Bond-Donating Capacity of Fluorohydrins Compared to Nonfluorinated Alcohols. Angew. Chem. Int. Ed. 2012, 51, 6176–6180. [Google Scholar]
- Afonin, A.V.; Vashchenko, A.V. Theoretical study of bifurcated hydrogen bonding effects on the 1J(N,H), 1hJ(N,H), 2hJ(N,N) couplings and 1H, 15N shieldings in model pyrroles. Magn. Reson. Chem. 2010, 48, 309–317. [Google Scholar]
- Luque, F.J.; Lopez, J.M.; Orozco, M. Perspective on “Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects”. Theor. Chem. Acc. 2000, 103, 343–345. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. The fundamental nature and role of the electrostatic potential in atoms and molecules. Theor. Chem. Acc. 2002, 108, 134–142. [Google Scholar] [CrossRef]
- Paul, B.K.; Guchhait, N. Geometrical criteria versus quantum chemical criteria for assessment of intramolecular hydrogen bond (IMHB) interaction: A computational comparison into the effect of chlorine substitution on IMHB of salicylic acid in its lowest energy ground state conformer. Chem. Phys. 2013, 412, 58–67. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Gilli, G.; Bellucci, F.; Ferretti, V.; Bertolasi, V. Evidence for resonance-assisted hydrogen-bonding from crystal-structure correlations on the enol form of the beta-diketone fragment. J. Am. Chem. Soc. 1989, 111, 1023–1028. [Google Scholar] [CrossRef]
- Sobczyk, L.; Grabowski, S.J.; Krygowski, T.M. Interrelation between H-bond and Pi-electron delocalization. Chem. Rev. 2005, 105, 3513–3560. [Google Scholar] [CrossRef]
- Jablonski, M.; Kaczmarek, A.; Sadlej, A.J. Estimates of the energy of intramolecular hydrogen bonds. J. Phys. Chem. A 2006, 110, 10890–10898. [Google Scholar] [CrossRef]
- Woodford, J.N. Density functional theory and atoms-in-molecules investigation of intramolecular hydrogen bonding in derivatives of malonaldehyde and implications for resonance-assisted hydrogen bonding. J. Phys. Chem. A 2007, 111, 8519–8530. [Google Scholar]
- Sanz, P.; Mo, O.; Yanez, M.; Elguero, J. Non-resonance-assisted hydrogen bonding in hydroxymethylene and aminomethylene cyclobutanones and cyclobutenones and their nitrogen counterparts. ChemPhysChem 2007, 8, 1950–1958. [Google Scholar]
- Trujillo, C.; Sanchez-Sanz, G.; Alkorta, I.; Elguero, J.; Mo, O.; Yanez, M. Resonance assisted hydrogen bonds in open-chain and cyclic structures of malonaldehyde enol: A theoretical study. J. Mol. Struct. 2013, 1048, 138–151. [Google Scholar]
- Abraham, M.H. Scales of solute hydrogen-bonding-their construction and application to physicochemical and biochemical processes. Chem. Soc. Rev. 1993, 22, 73–83. [Google Scholar] [CrossRef]
- Devereux, M.; Popelier, P.L.A.; McLay, I.M. A refined model for prediction of hydrogen bond acidity and basicity parameters from quantum chemical molecular descriptors. Phys. Chem. Chem. Phys. 2009, 11, 1595–1603. [Google Scholar]
- Schwobel, J.; Ebert, R.U.; Kuhne, R.; Schuurmann, G. Prediction of the intrinsic hydrogen bond acceptor strength of chemical substances from molecular structure. J. Phys. Chem. A 2009, 113, 10104–10112. [Google Scholar]
- Lamarche, O.; Platts, J.A. Complementary nature of hydrogen bond basicity and acidity scales from electrostatic and atoms in molecules properties. Phys. Chem. Chem. Phys. 2003, 5, 677–684. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Peralta-Inga, Z. Molecular surface electrostatic potentials in relation to noncovalent interactions in biological systems. Int. J. Quantum Chem. 2001, 85, 676–684. [Google Scholar]
- Atkinson, A.P.; Baguet, E.; Galland, N.; le Questel, J.Y.; Planchat, A.; Graton, J. Structural features and hydrogen-bond properties of galanthamine and codeine: An experimental and theoretical study. Chem. Eur. J. 2011, 17, 11637–11649. [Google Scholar]
- Tabatchnik, A.; Blot, V.; Pipelier, M.; Dubreuil, D.; Renault, E.; le Questel, J.Y. Theoretical study of the structures and hydrogen-bond properties of new alternated heterocyclic compounds. J. Phys. Chem. A 2010, 114, 6413–6422. [Google Scholar] [CrossRef]
- Parafiniuk, M.; Mitoraj, M.P. Origin of binding of ammonia-borane to transition-metal-based catalysts: An insight from the charge and energy decomposition method ETS-NOCV. Organometallics 2013, 32, 4103–4113. [Google Scholar] [CrossRef]
- Murray, J.S.; Ranganathan, S.; Politzer, P. Correlations between the solvent hydrogen-bond acceptor parameter beta and the calculated molecular electrostatic potential. J. Org. Chem. 1991, 56, 3734–3737. [Google Scholar]
- Kenny, P.W. Prediction of hydrogen-bond basicity from computed molecular electrostatic properties-implications for comparative molecular-field analysis. J. Chem. Soc. Perkin Trans. 2 1994, 199–202. [Google Scholar] [CrossRef]
- Kenny, P.W. Hydrogen Bonding, Electrostatic Potential, and Molecular Design. J. Chem. Inf. Model. 2009, 49, 1234–1244. [Google Scholar] [CrossRef]
- Saeed, A.; Khurshid, A.; Jasinski, J.P.; Pozzi, C.G.; Fantoni, A.C.; Erben, M.F. Competing intramolecular N-H…O=C hydrogen bonds and extended intermolecular network in 1-(4-chlorobenzoyl)-3-(2-methyl-4-oxopentan-2-yl) thiourea analyzed by experimental and theoretical methods. Chem. Phys. 2014, 431–432, 39–46. [Google Scholar] [CrossRef]
- Cooper, S.C.; Sammes, P.G. (1,5)-Acetyl shifts in cycloadducts derived from 2-acetyl-1,4-benzoquinones. J. Chem. Soc. Chem. Comm. 1980, 13, 633–634. [Google Scholar]
- Castro, C.G.; Santos, J.G.; Valcarcel, J.C.; Valderrama, J.A. Kinetic study of the acid-catalyzed rearrangement of 4-acetyl-3,3-dimethyl-5-hydroxy-2-morpholino-2,3-dihydrobenzo[b]furan. J. Org. Chem. 1983, 48, 3026–3029. [Google Scholar]
- Valderrama, J.A.; Araya-Maturana, R.; Zuloaga, F. Studies on quinones. Part 27. Diels-Alder reaction of 8,8-dimethylnaphthalene-1,4,5(8H)-trione. J. Chem. Soc. Perkin Trans. 1 1993, 1103–1107. [Google Scholar]
- Araya-Maturana, R.; Cassels, B.K.; Delgado-Castro, T.; Valderrama, J.A.; Weiss-Lopez, B.E. Regioselectivity in the Diels-Alder reaction of 8,8-dimethylnaphthalene-1,4,5(8H)-trione with 2,4-hexadien-1-ol. Tetrahedron 1999, 55, 637–648. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03; Revision E. 01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 5. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry . 3. the role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Siani, G.; Angelini, G.; de Maria, P.; Fontana, A.; Pierini, M. Solvent effects on the rate of the keto-enol interconversion of 2-nitrocyclohexanone. Org. Biomol. Chem. 2008, 6, 4236–4241. [Google Scholar] [CrossRef]
- Wilmot, N.; Marsella, M.J. Visualization method to predict the nucleophilic asymmetric induction of prochiral electrophiles. Org. Lett. 2006, 8, 3109–3112. [Google Scholar]
- Prasad, V.; Birzin, E.T.; McVaugh, C.T.; van Rijn, R.D.; Rohrer, S.P.; Chicchi, G.; Underwood, D.J.; Thornton, E.R.; Smith, A.B.; Hirschmann, R. Effects of heterocyclic aromatic substituents on binding affinities at two distinct sites of somatostatin receptors. Correlation with the electrostatic potential of the substituents. J. Med. Chem. 2003, 46, 1858–1869. [Google Scholar]
- Moro, S.; Bacilieri, M.; Cacciari, B.; Spalluto, G. Autocorrelation of molecular electrostatic potential surface properties combined with partial least squares analysis as new strategy for the prediction of the activity of human A(3) adenosine receptor antagonists. J. Med. Chem. 2005, 48, 5698–5704. [Google Scholar]
- Lämmermann, A.; Szatmari, I.; Fulop, F.; Kleinpeter, E. Inter- or Intramolecular N center dot center dot center dot H-O or N-H center dot center dot center dot O hydrogen bonding in 1,3-amino-alpha/beta-naphthols: An experimental NMR and computational study. J. Phys. Chem. A 2009, 113, 6197–6205. [Google Scholar] [CrossRef]
- Shchavlev, A.E.; Pankratov, A.N.; Enchev, V. Intramolecular hydrogen-bonding interactions in 2-nitrosophenol and nitrosonaphthols: Ab initio, density functional, and nuclear magnetic resonance theoretical study. J. Phys. Chem. A 2007, 111, 7112–7123. [Google Scholar]
- Weinhold, F. Chemistry—A new twist on molecular shape. Nature 2001, 411, 539–541. [Google Scholar] [CrossRef]
- Schreiner, P.R. Teaching the right reasons: Lessons from the mistaken origin of the rotational barrier in ethane. Angew. Chem. Int. Ed. 2002, 41, 3579–3581. [Google Scholar]
- Li, X.Y.; Wang, Y.; Zheng, S.J.; Meng, L.P. Substituent effects on the intramolecular hydrogen bond in 1-hydroxyanthraquinone: AIM and NBO analyses. Struct. Chem. 2012, 23, 1233–1240. [Google Scholar] [CrossRef]
- Haghdadi, M. DFT molecular orbital calculations and natural bond orbital analysis of 1,2,7-thiadiazepane conformers. Monatsh. Chem. 2013, 144, 1653–1661. [Google Scholar] [CrossRef]
- Sample Availability: Samples of all compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Cifuentes, M.; Weiss-López, B.E.; Santos, L.S.; Araya-Maturana, R. Intramolecular Hydrogen Bond in Biologically Active o-Carbonyl Hydroquinones. Molecules 2014, 19, 9354-9368. https://doi.org/10.3390/molecules19079354
Martínez-Cifuentes M, Weiss-López BE, Santos LS, Araya-Maturana R. Intramolecular Hydrogen Bond in Biologically Active o-Carbonyl Hydroquinones. Molecules. 2014; 19(7):9354-9368. https://doi.org/10.3390/molecules19079354
Chicago/Turabian StyleMartínez-Cifuentes, Maximiliano, Boris E. Weiss-López, Leonardo S. Santos, and Ramiro Araya-Maturana. 2014. "Intramolecular Hydrogen Bond in Biologically Active o-Carbonyl Hydroquinones" Molecules 19, no. 7: 9354-9368. https://doi.org/10.3390/molecules19079354