Determination of Phenol Compounds In Surface Water Matrices by Bar Adsorptive Microextraction-High Performance Liquid Chromatography-Diode Array Detection


Abstract
:1. Introduction
2. Results and Discussion
2.1. Instrumental Conditions
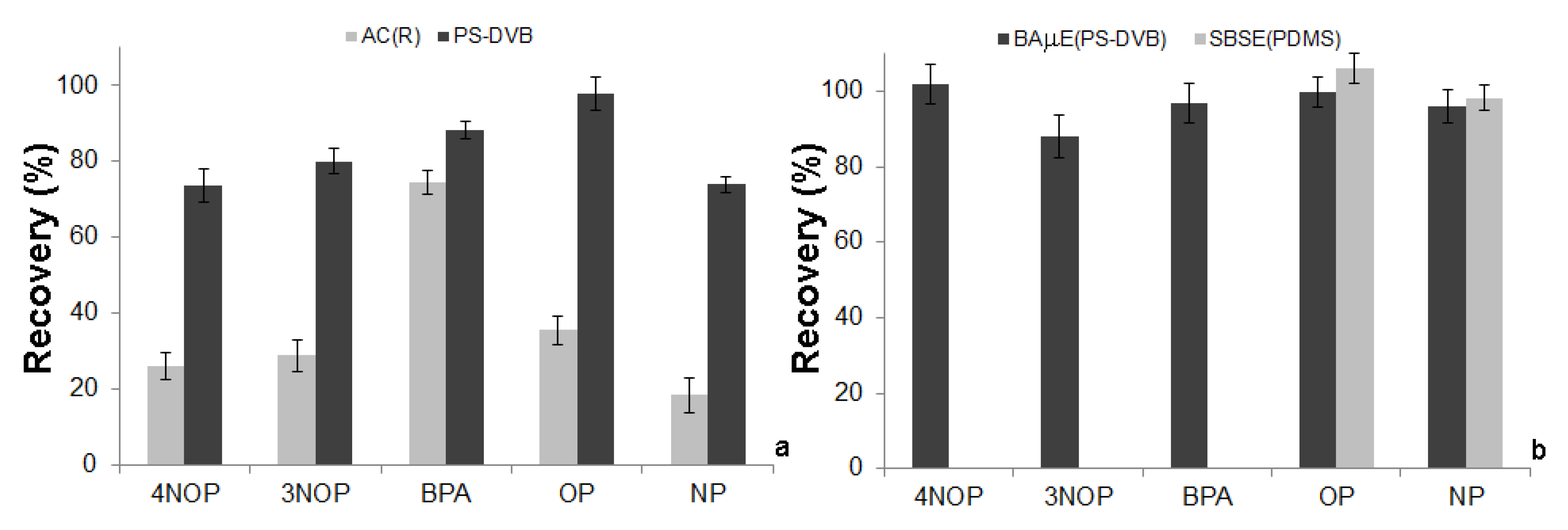
2.2. Selection of the Sorbent Phase

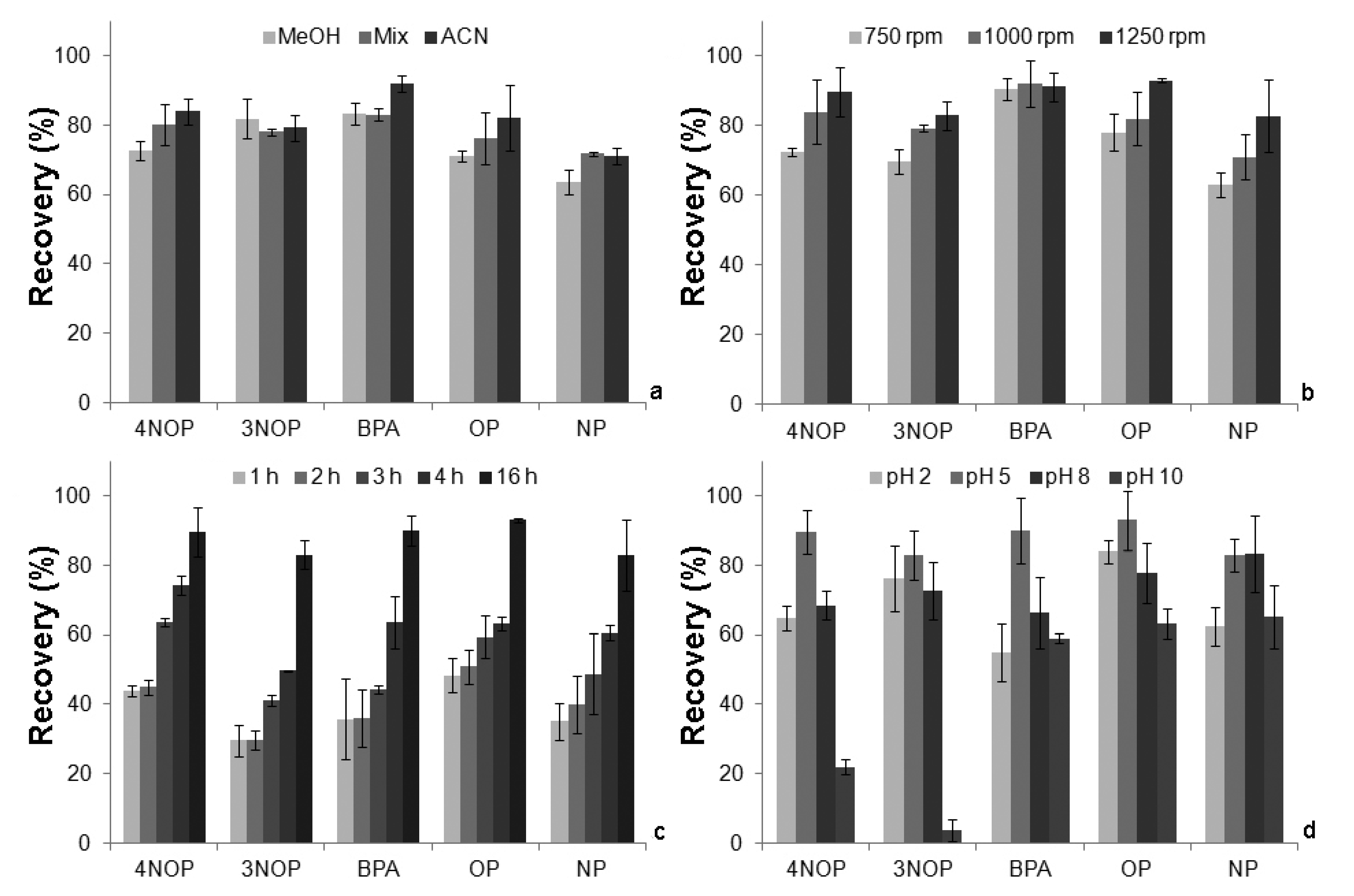
2.3. Optimization of the Recovery Assays

2.4. Validation of BAµE(PS-DVB)-LD/HPLC-DAD Methodology

{kind=link}
{kind=link}
{kind=link}
| Phenol Compounds | Recovery a (% ± RSD; n = 3) | r2 b | LOD (µg/L) | LOQ (µg/L) |
|---|---|---|---|---|
| 4NOP | 102.2 ± 5.2 | 0.9995 | 0.3 | 0.8 |
| 3NOP | 88.0 ± 5.7 | 0.9954 | 0.3 | 0.8 |
| BPA | 96.7 ± 5.3 | 0.9904 | 0.3 | 0.8 |
| OP | 99.8 ± 4.0 | 0.9946 | 0.3 | 0.8 |
| NP | 96.2 ± 4.5 | 0.9940 | 0.3 | 0.8 |
2.5. Comparison between BAµE(PS-DVB) and SBSE(PDMS)
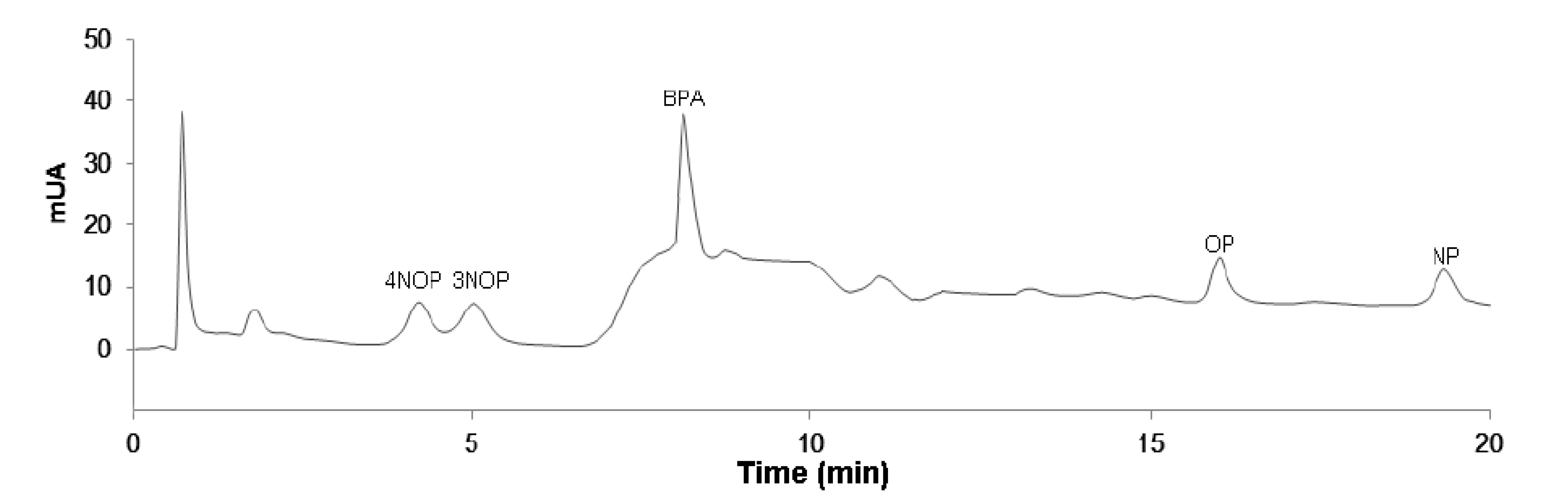
2.6. Application to Real Matrices

3. Experimental Section
3.1. Chemicals and Samples
3.2. Experimental Set-Up
3.2.1. Preparation of the Microextraction Bars
3.2.2. Recovery Assays and Method Validation
3.3. Instrumentation Settings
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Richardson, S.D.; Ternes, T.A. Water Analysis: Emerging Contaminants and Current Issues. Anal. Chem. 2011, 83, 4614–4648. [Google Scholar]
- Karim, K.; Gupta, S.K. Continuous biotransformation and removal of nitrophenols under denitrifying conditions. Water Res. 2003, 37, 2953–2959. [Google Scholar] [CrossRef]
- Ballesteros-Gómez, A.; Rubio, S.; Pérez-Bendito, D. Analytical methods for the determination of bisphenol A in food. J. Chromatogr. A 2009, 1216, 449–469. [Google Scholar] [CrossRef]
- Nakamura, S.; Daishima, S. Simultaneous determination of alkylphenols and bisphenol A in river water by stir bar sorptive extraction with in situ acetylation and thermal desorption-gas chromatography-mass spectrometry. J. Chromatogr. A 2004, 1038, 291–294. [Google Scholar]
- Huang, Y.Q.; Wong, C.K.C.; Zheng, J.S.; Bouwman, H.; Barra, R.; Wahlström, B.; Neretin, L.; Wong, M.H. Bisphenol A (BPA) in China: A review of sources, environmental levels, and potential human health impacts. Environ. Int. 2012, 42, 91–99. [Google Scholar] [CrossRef]
- Gassel, M.; Harwani, S.; Park, J.-S.; Jahn, A. Detection of nonylphenol and persistent organic pollutants in fish from the North Pacific Central Gyre. Mar. Pollut. Bull. 2013, 73, 231–242. [Google Scholar]
- Laws, S.C.; Carey, S.A.; Ferrell, J.M.; Bodman, G.J.; Cooper, R.L. Estrogenic Activity of Octylphenol, Nonylphenol, Bisphenol A and Methoxychlor in Rats. Toxicol. Sci. 2000, 54, 154–167. [Google Scholar] [CrossRef]
- Asimakopoulos, A.G.; Thomaidis, N.S.; Koupparis, M.A. Recent trends in biomonitoring of bisphenol A, 4-t-octylphenol, and 4-nonylphenol. Toxicol. Lett. 2012, 210, 141–154. [Google Scholar] [CrossRef]
- Cunha, S.C.; Fernandes, J.O. Assessment of bisphenol A and bisphenol B in canned vegetables and fruits by gas chromatography-mass spectrometry after QuEChERS and dispersive liquid-liquid microextraction. Food Control 2013, 33, 549–555. [Google Scholar] [CrossRef]
- Ballesteros, O.; Zafra, A.; Navalón, A.; Vílchez, J.L. Sensitive gas chromatographic-mass spectrometric method for the determination of phthalate esters, alkylphenols, bisphenol A and their chlorinated derivatives in wastewater samples. J. Chromatogr. A 2006, 1121, 154–162. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Ito, R.; Hayatsu, Y.; Nakata, H.; Sakui, N.; Okanouchi, N.; Saito, K.; Yokota, H.; Izumi, S.-I.; Makino, T.; et al. Stir bar sorptive extraction with in situ de-conjugation and thermal desorption gas chromatography-mass spectrometry for measurement of 4-nonylphenol glucuronide in human urine sample. J. Pharmaceut. Biomed. 2006, 40, 82–87. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Sakui, N.; Okanouchi, N.; Ito, R.; Saito, K.; Nakazawa, H. Stir bar sorptive extraction and trace analysis of alkylphenols in water samples by thermal desorption with in tube silylation and gas chromatography-mass spectrometry. J. Chromatogr. A 2005, 1062, 23–29. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Inoue, K.; Yoshimura, M.; Ito, R.; Sakui, N.; Nakazawa, H. Determination of 4-nonylphenol and 4-tert-octylphenol in water samples by stir bar sorptive extraction and thermal desorption-gas chromatography-mass spectrometry. Anal. Chim. Acta 2004, 505, 217–222. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Takahashi, S.; Seshimo, F.; Sakui, N.; Okanouchi, N.; Ito, R.; Inoue, K.; Yoshimura, Y.; Izumi, S.-I.; Makino, T.; et al. Determination of 4-tert-octylphenol and 4-nonylphenol in laboratory animal feed sample by stir bar sorptive extraction followed by liquid desorption and column-switching liquid chromatography–mass spectrometry with solid-phase extraction. J. Chromatogr. A 2004, 1046, 83–88. [Google Scholar] [CrossRef]
- Zhuang, Y.; Zhou, M.; Gu, J.; Li, X. Spectrophotometric and high performance liquid chromatographic methods for sensitive determination of bisphenol A. Spectrochim. Acta 2014, 122, 153–157. [Google Scholar] [CrossRef]
- Nogueira, J.M.F. Novel sorption-based methodologies for static microextraction analysis: A review on SBSE and related techniques. Anal. Chim. Acta 2012, 757, 1–10. [Google Scholar] [CrossRef]
- Neng, N.R.; Silva, A.R.M.; Nogueira, J.M.F. Adsorptive micro-extraction techniques—Novel analytical tools for trace levels of polar solutes in aqueous media. J. Chromatogr. A 2010, 1217, 7303–7310. [Google Scholar]
- Neng, N.R.; Nogueira, J.M.F. Development of a bar adsorptive micro-extraction-large-volume injection-gas chromatography-mass spectrometric method for pharmaceuticals and personal care products in environmental water matrices. Anal. Bioanal. Chem. 2012, 402, 1355–1364. [Google Scholar] [CrossRef]
- Almeida, C.; Nogueira, J.M.F. Comparison of the selectivity of different sorbent phases for bar adsorptive microextraction-Application to trace level analysis of fungicides in real matrices. J. Chromatogr. A 2012, 1265, 7–16. [Google Scholar]
- Neng, N.R.; Mestre, A.S.; Carvalho, A.P.; Nogueira, J.M.F. Powdered activated carbons as effective phases for bar adsorptive micro-extraction (BAμE) to monitor levels of triazinic herbicides in environmental water matrices. Talanta 2011, 83, 1643–1649. [Google Scholar] [CrossRef]
- Neng, N.R.; Mestre, A.S.; Carvalho, A.P.; Nogueira, J.M.F. Cork-based activated carbons as supported adsorbent materials for trace level analysis of ibuprofen and clofibric acid in environmental and biological matrices. J. Chromatogr. A 2011, 1218, 6263–6270. [Google Scholar]
- Baltussen, E.; Cramers, C.; Sandra, P. Sorptive sample preparation—A review. Anal. Bioanal. Chem. 2002, 373, 3–22. [Google Scholar]
- Sample Availability: All surface water samples were collected in the metropolitan area of Lisbon, Portugal.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neng, N.R.; Nogueira, J.M.F. Determination of Phenol Compounds In Surface Water Matrices by Bar Adsorptive Microextraction-High Performance Liquid Chromatography-Diode Array Detection. Molecules 2014, 19, 9369-9379. https://doi.org/10.3390/molecules19079369
Neng NR, Nogueira JMF. Determination of Phenol Compounds In Surface Water Matrices by Bar Adsorptive Microextraction-High Performance Liquid Chromatography-Diode Array Detection. Molecules. 2014; 19(7):9369-9379. https://doi.org/10.3390/molecules19079369
Chicago/Turabian StyleNeng, Nuno R., and José M. F. Nogueira. 2014. "Determination of Phenol Compounds In Surface Water Matrices by Bar Adsorptive Microextraction-High Performance Liquid Chromatography-Diode Array Detection" Molecules 19, no. 7: 9369-9379. https://doi.org/10.3390/molecules19079369