Batch and Continuous Flow Preparation of Hantzsch 1,4-Dihydropyridines under Microwave Heating and Simultaneous Real-time Monitoring by Raman Spectroscopy. An Exploratory Study

Abstract
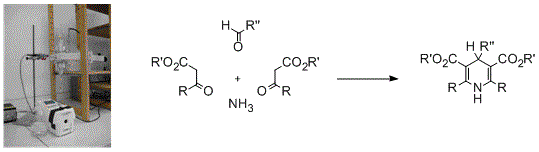
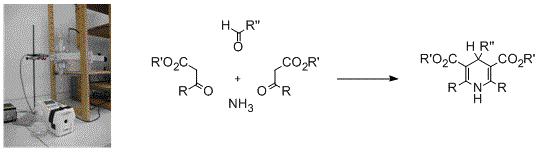
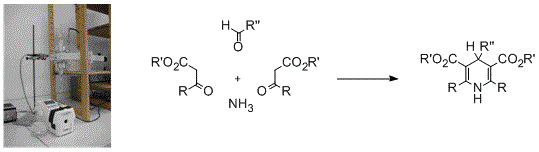
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
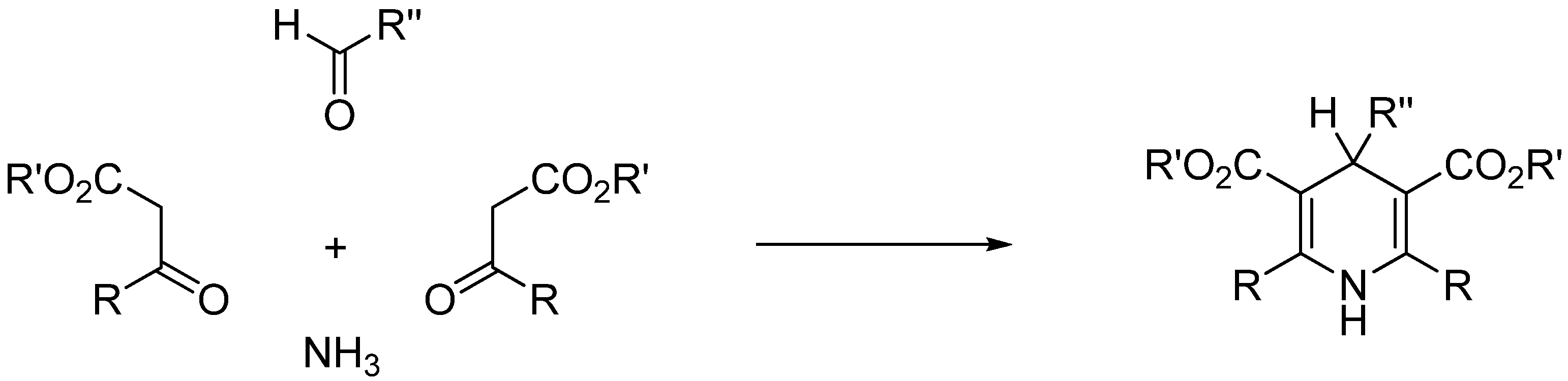{kind=link}
1. Introduction

- Infrared spectroscopy: in 1999 Te-As-Se based optical glass fibers were developed at the University of Rennes 1, France by Lucas et al. [14] and used as evanescent wave chemical sensors by Perio [15] in the Hamelin group. The fibers were directly introduced in the reaction media and infrared spectra were periodically recorded. In particular the disappearance of the carbonyl band due to the C=O vibration in 3-pentanone or cyclohexanone was monitored when those ketones were reacted with trimethyl orthoformate in the presence of Montmorillonite K10 clay or with mercaptoethanol in the presence of para-toluenesulfonic acid. Some patents [16,17,18] cover the subject, but for unknown reasons the technology has fallen into oblivion.
- Raman spectroscopy: the method does not require an immersion of the probe into the reactor but only a contact to the exterior wall. A first report [19] on the subject was published in 1995 and a full description of the instrumentation and its setup has been disclosed by Pivonka and Empfield [20] in 2004. These publications have been followed by the work of Leadbeater et al. [21,22,23,24,25,26,27], who used a CEM oven modified to allow introduction of a Raman probe into the microwave cavity.
2. Results and Discussion
2.1. Choice of Reaction

2.2. Materials and Methods
2.2.1. The Raman Spectrometer

2.2.2. The Conventional Heating System

2.2.3. The Microwave (Dielectric) Heating Method & Equipment

- 2.45 GHz microwaves are delivered in continuous wave (CW) by a solid state (transistor) microwave generator; the microwave power can be adjusted from 1 W to 200 W in steps of 1 W;
- The microwave power is transmitted from the solid state generator to the U-shaped waveguide using a coaxial cable 1 m long;
- The device enables measurement (via a built-in circulator) and adjustment (using the manual sliding short circuit and if necessary the manual impedance tuner) of the reflected microwave power so that maximum energy is available and transferred into the reaction medium with limited loss;
- The temperature inside the reactors was measured directly in the mixture with a fiber optic temperature sensor (Neoptix, Québec City, Québec, Canada) and after the heating ramp period, the power of the microwave irradiation was automatically adjusted in order to maintain the chosen final reaction temperature for a given period;
- A lateral chimney allows the introduction of a probe into the cavity of the U-shaped waveguide and more specifically the introduction of a quartz Raman probe;
- The same equipment can be used to perform reactions under both batch and continuous flow.
2.3. Preparation of a Reference Sample 1 under Conventional Heating
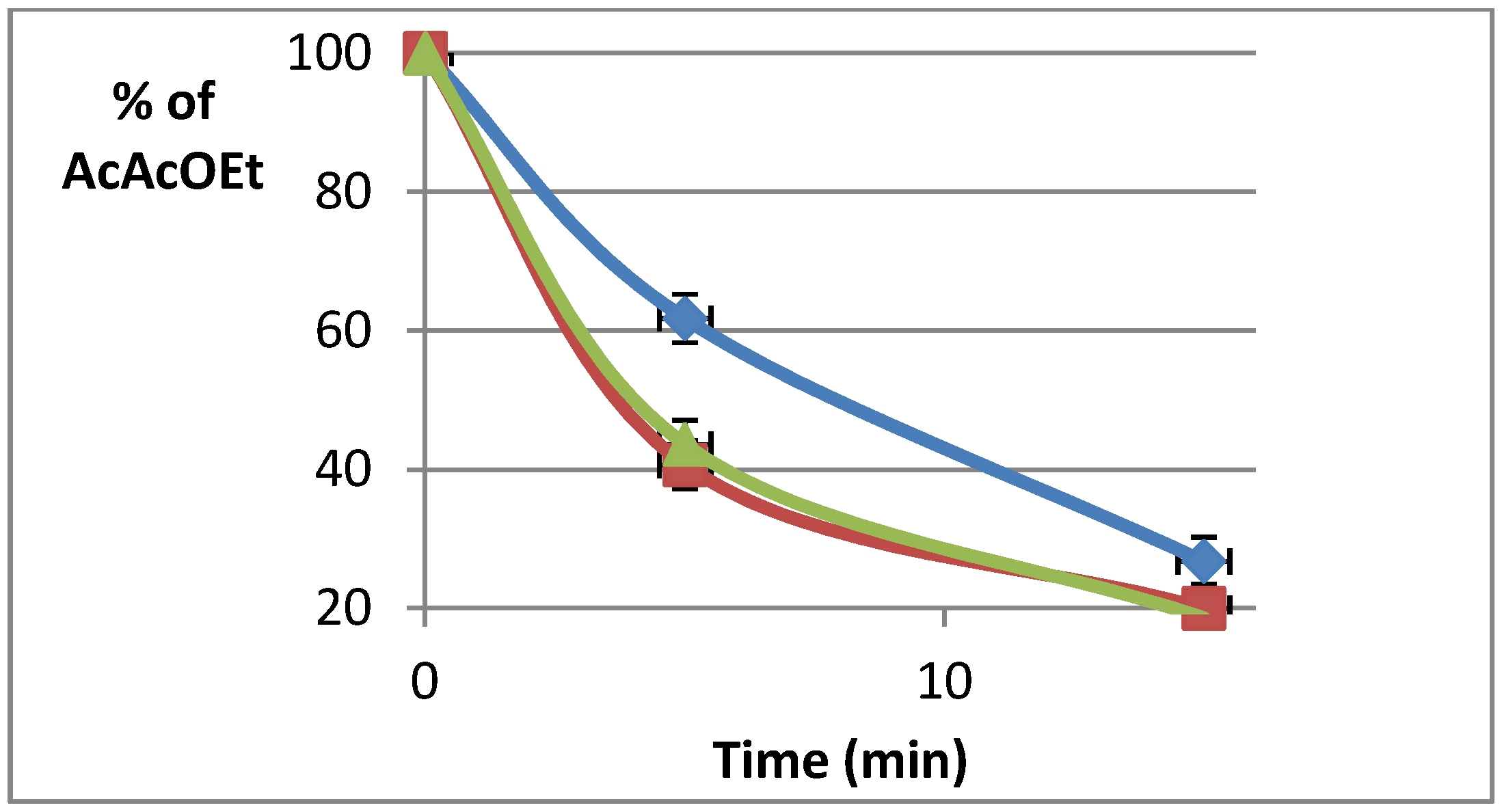
2.4. Preliminary Study by Raman Spectroscopy
2.5. Reactions in Batch Mode
2.5.1. Reaction in a Mixture of Methanol and Water

2.5.2. Reaction in a Mixture of Acetonitrile and Water
2.5.3. Extension to the Preparation of Other 1,4-DHPs
2.6. Reaction under Continuous Flow Conditions


3. Experimental Section
3.1. General
3.2. Chemistry
4. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References and Notes
- Leadbeater, N.E.; McGowan, C.B. Laboratory Experiments Using Microwave Heating; CRC Press: Boca Raton, FL, USA, 2013. [Google Scholar]
- Hassan, H.M.A.; Harakeh, S.; Sakkaf, K.A.; Denetiu, I. Progress in microwave-aided chemical synthesis. Aust. J. Chem. 2012, 65, 1647–1654. [Google Scholar] [CrossRef]
- Kruithof, A.; Ruijter, E.; Orru, R.V.A. Microwave-assisted multicomponent synthesis of heterocycles. Curr. Org. Chem. 2011, 15, 204–236. [Google Scholar] [CrossRef]
- Appukkuttan, P.; Mehta, V.P.; van Der Eycken, E.V. Microwave-assisted cycloaddition reactions. Chem. Soc. Rev. 2010, 39, 1467–1477. [Google Scholar] [CrossRef]
- Strauss, C.R. A strategic, “green” approach to organic chemistry with microwave assistance and predictive yield optimization as core, enabling technologies. Aust. J. Chem. 2009, 62, 3–15. [Google Scholar] [CrossRef]
- Bogdal, D.; Loupy, A. Application of microwave irradiation to phase-transfer catalyzed reactions. Org. Process Res. Dev. 2008, 12, 710–722. [Google Scholar] [CrossRef]
- Perio, B.; Dozias, M.-J.; Hamelin, J. Ecofriendly fast batch synthesis of dioxolanes, dithiolanes, and oxathiolanes without solvent under microwave irradiation. Org. Process Res. Dev. 1998, 2, 428–430. [Google Scholar] [CrossRef]
- Cleophax, J.; Liagre, M.; Loupy, A.; Petit, A. Application of focused microwaves to the scale-up of solvent-free organic reactions. Org. Process Res. Dev. 2000, 4, 498–504. [Google Scholar] [CrossRef]
- The Prolabo mark, popular in France in the 1990s, has now disappeared.
- Leadbeater, N.E.; Williams, V.A.; Barnard, T.M.; Collins, M.J., Jr. Open-vessel microwave-promoted Suzuki reactions using low levels of palladium catalyst: Optimization and scale-up. Org. Process Res. Dev. 2006, 10, 833–837. [Google Scholar] [CrossRef]
- Baxendale, I.R.; Hayward, J.J.; Ley, S.V. Microwave reactions under continuous flow conditions. Comb. Chem. High T. Scr. 2007, 10, 802–836. [Google Scholar]
- Barbry, D.; Vanden Eynde, J.J. Continuous flow organic synthesis under microwave heating. In Ultrasound and Microwaves: Recent Advances in Organic Chemistry; Bazureau, J.P., Draye, M., Eds.; Transworld Research Network: Trivandrum, India, 2011; pp. 2315–241. [Google Scholar]
- Strauss, C.R.; Trainor, R.W. Developments in microwave-assisted organic chemistry. Aust. J. Chem. 1995, 48, 1665–1692. [Google Scholar] [CrossRef]
- Hocde, S.; Boussard-Pledel, C.; le Coq, D.; Fonteneau, G.; Lucas, J. Remote analysis using IR glass fibers. In Infrared Optical Fibers and Their Applications, Proceedings of the Meeting of the Society of Photo-optical Instrumentation Engineers, Boston, MA, USA, 21–22 September 1999; Saad, M., Harrington, J.A., Eds.; SPIE The International Society for Optical Engineering: Ann Arbor, MI, USA; Volume 3849, pp. 50–59.
- Perio, B. Solvent-Free Protection of Carbonyl Groups under Microwave Irradiation: A Clean and Competitive Process. Ph.D. thesis, University of Rennes 1, Rennes, France, 6 October 1999. [Google Scholar]
- Brooks, H.J.; Mortenson, M.G.; Blum, B.J. Controlling Chemical Reactions by Spectral Chemistry and Spectral Conditioning. U.S. Patent 20050139485, 30 June 2005. [Google Scholar]
- McManus, M.E.; Collins, M.J., Sr.; Collins, M.J., Jr. Spectroscopy-Base Real-Time Control for Microwave-Assisted Chemistry. U.S. Patent 7141769, 28 November 2006. [Google Scholar]
- King, E.E. Real-Time Imaging and Spectroscopy during Microwave Assisted Chemistry. U.S. Patent 20070062934, 22 March 2007. [Google Scholar]
- Stellman, C.M.; Aust, J.F.; Myrick, M.L. In situ spectroscopic study of microwave polymerization. Appl. Spectrosc. 1995, 49, 392–394. [Google Scholar] [CrossRef]
- Pivonka, D.E.; Empfield, J.R. Real-time in situ Raman analysis of microwave-assisted organic reactions. Appl. Spectrosc. 2004, 58, 41–46. [Google Scholar] [CrossRef]
- Barnard, T.M.; Leadbeater, N.E. Real-time monitoring of microwave-promoted organometallic ligand-substitution reactions using in situ Raman spectroscopy. Chem. Commun. 2006, 2006, 3615–3616. [Google Scholar] [CrossRef]
- Leadbeater, N.E.; Smith, R.J. Real-time monitoring of microwave-promoted Suzuki coupling reaction using in situ Raman spectroscopy. Org. Lett. 2006, 8, 4589–4591. [Google Scholar] [CrossRef]
- Leadbeater, N.E.; Smith, R.J.; Barnard, T.M. Using in situ Raman monitoring as a tool for rapid optimization and scale-up of microwave-promoted organic synthesis: Esterification as an example. Org. Biomol. Chem. 2007, 5, 822–825. [Google Scholar] [CrossRef]
- Leadbeater, N.E.; Schmink, J.R. Use of Raman spectroscopy as a tool for in situ monitoring of microwave-promoted reactions. Nat. Protoc. 2008, 3, 1–7. [Google Scholar] [CrossRef]
- Schmink, J.R.; Holcomb, J.L.; Leadbeater, N.E. Use of Raman spectroscopy as an in situ tool to obtain kinetic data for organic transformations. Chem. Eur. J. 2008, 17, 9943–9950. [Google Scholar]
- Schmink, J.R.; Holcomb, J.L.; Leadbeater, N.E. Testing the validity of microwave-interfaced, in situ Raman spectroscopy as a tool for kinetic studies. Org. Lett. 2009, 11, 365–368. [Google Scholar] [CrossRef]
- Schmink, J.R.; Leadbeater, N.E. Probing “microwave effects” using Raman spectroscopy. Org. Biomol. Chem. 2009, 7, 3842–3846. [Google Scholar] [CrossRef]
- Hantzsch, A. Ueber die synthese pyridinartiger verbindungen aus acetessigäther und aldehydammoniak. Justus Liebigs Ann. Chem. 1882, 215, 1–82. [Google Scholar] [CrossRef]
- Vanden Eynde, J.J.; Mayence, A. 1,3-Dicarbonyl compound as third component (Hantzsch pyridine synthesis). In Science of Synthesis: Multicomponent Reactions 1; Müller, T.J.J., Ed.; Georg Thieme Verlag KG: Stuttgart, Germany, 2014; pp. 67–98. [Google Scholar]
- Bagley, M.C.; Jenkins, R.L.; Caterina Lubiou, M.; Mason, C.; Wood, R. A simple continuous flow microwave reactor. J. Org. Chem. 2005, 70, 7003–7006. [Google Scholar] [CrossRef]
- Baxendale, I.R.; Hornung, C.; Ley, S.V.; Molina, J.M.M.; Wikström, A. Flow microwave technology and microreactors in synthesis. Aust. J. Chem. 2013, 66, 131–144. [Google Scholar] [CrossRef]
- Huang, Y. Hantzsch 1,4-dihydropyridine—An effective and convenient reducing agent. Synlett 2007, 2007, 2304–2305. [Google Scholar] [CrossRef]
- Edraki, N.; Mehdipour, A.R.; Khoshneviszadeh, M.; Miri, R. Dihydropyridines: Evaluation of their current and future pharmacological applications. Drug Discov. Today 2009, 14, 1058–1066. [Google Scholar] [CrossRef]
- Norcross, B.E.; Clement, G.; Weinstein, M. The Hantzsch pyridine synthesis. A factorial design experiment for the introductory organic laboratory. J. Chem. Educ. 1969, 46, 694–695. [Google Scholar] [CrossRef]
- Lidström, P.; Tierney, J.; Wathey, B.; Westman, J. Microwave assisted organic synthesis—A Review. Tetrahedron 2001, 57, 9225–9283. [Google Scholar] [CrossRef]
- Vanden Eynde, J.J.; Delfosse, F.; Mayence, A.; van Haverbeke, Y. Old reagents, new results: Aromatization of Hantzsch 1,4-dihydropyridines with manganese dioxide and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone. Tetrahedron 1995, 51, 6511–6516. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the dihydropyridines are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christiaens, S.; Vantyghem, X.; Radoiu, M.; Vanden Eynde, J.J. Batch and Continuous Flow Preparation of Hantzsch 1,4-Dihydropyridines under Microwave Heating and Simultaneous Real-time Monitoring by Raman Spectroscopy. An Exploratory Study. Molecules 2014, 19, 9986-9998. https://doi.org/10.3390/molecules19079986
Christiaens S, Vantyghem X, Radoiu M, Vanden Eynde JJ. Batch and Continuous Flow Preparation of Hantzsch 1,4-Dihydropyridines under Microwave Heating and Simultaneous Real-time Monitoring by Raman Spectroscopy. An Exploratory Study. Molecules. 2014; 19(7):9986-9998. https://doi.org/10.3390/molecules19079986
Chicago/Turabian StyleChristiaens, Sylvain, Xavier Vantyghem, Marilena Radoiu, and Jean Jacques Vanden Eynde. 2014. "Batch and Continuous Flow Preparation of Hantzsch 1,4-Dihydropyridines under Microwave Heating and Simultaneous Real-time Monitoring by Raman Spectroscopy. An Exploratory Study" Molecules 19, no. 7: 9986-9998. https://doi.org/10.3390/molecules19079986