Synthesis and Biological Activities of Some New (Nα-Dinicotinoyl)- bis-L-Leucyl Linear and Macrocyclic Peptides

Abstract
:1. Introduction
2. Results and Discussion
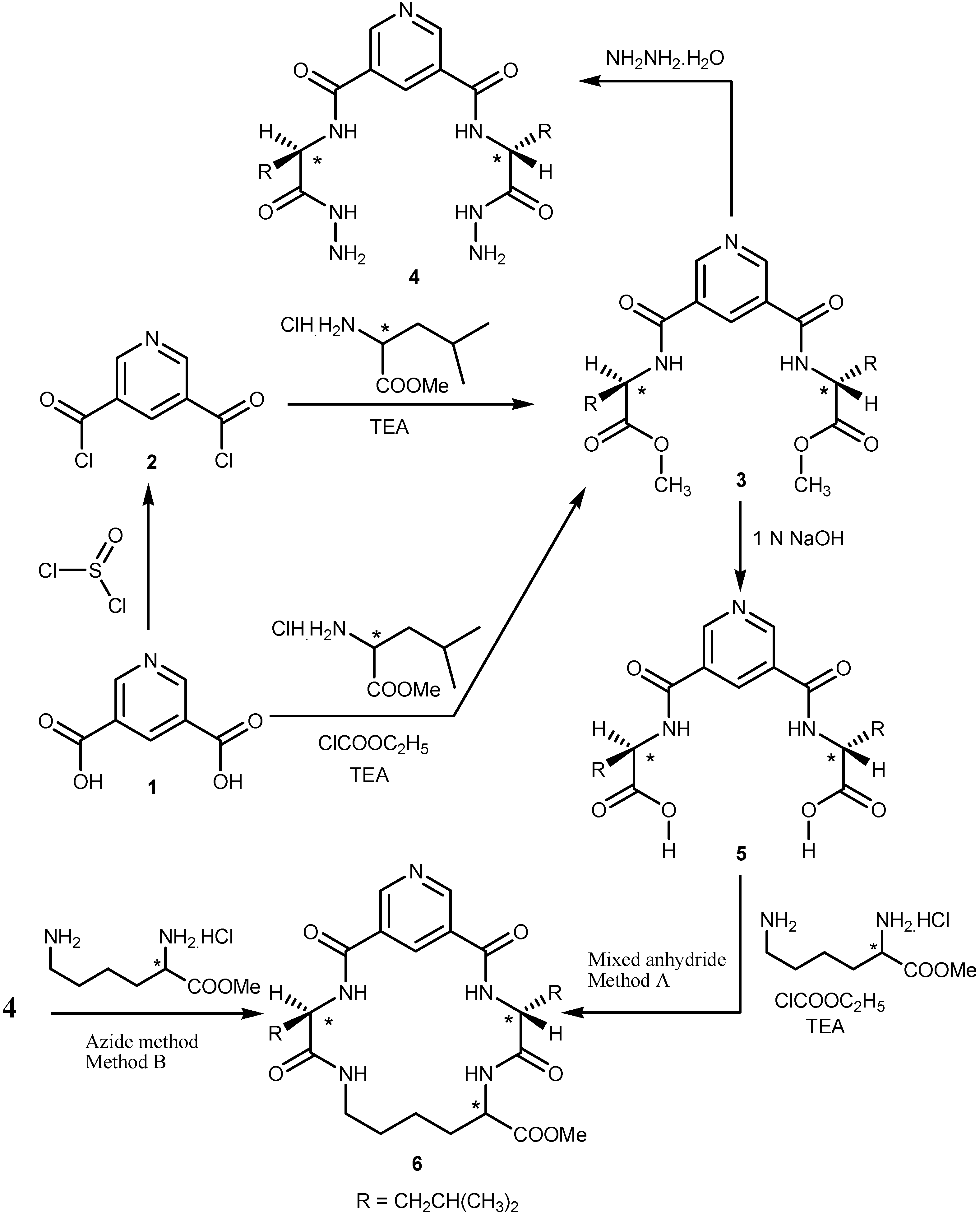
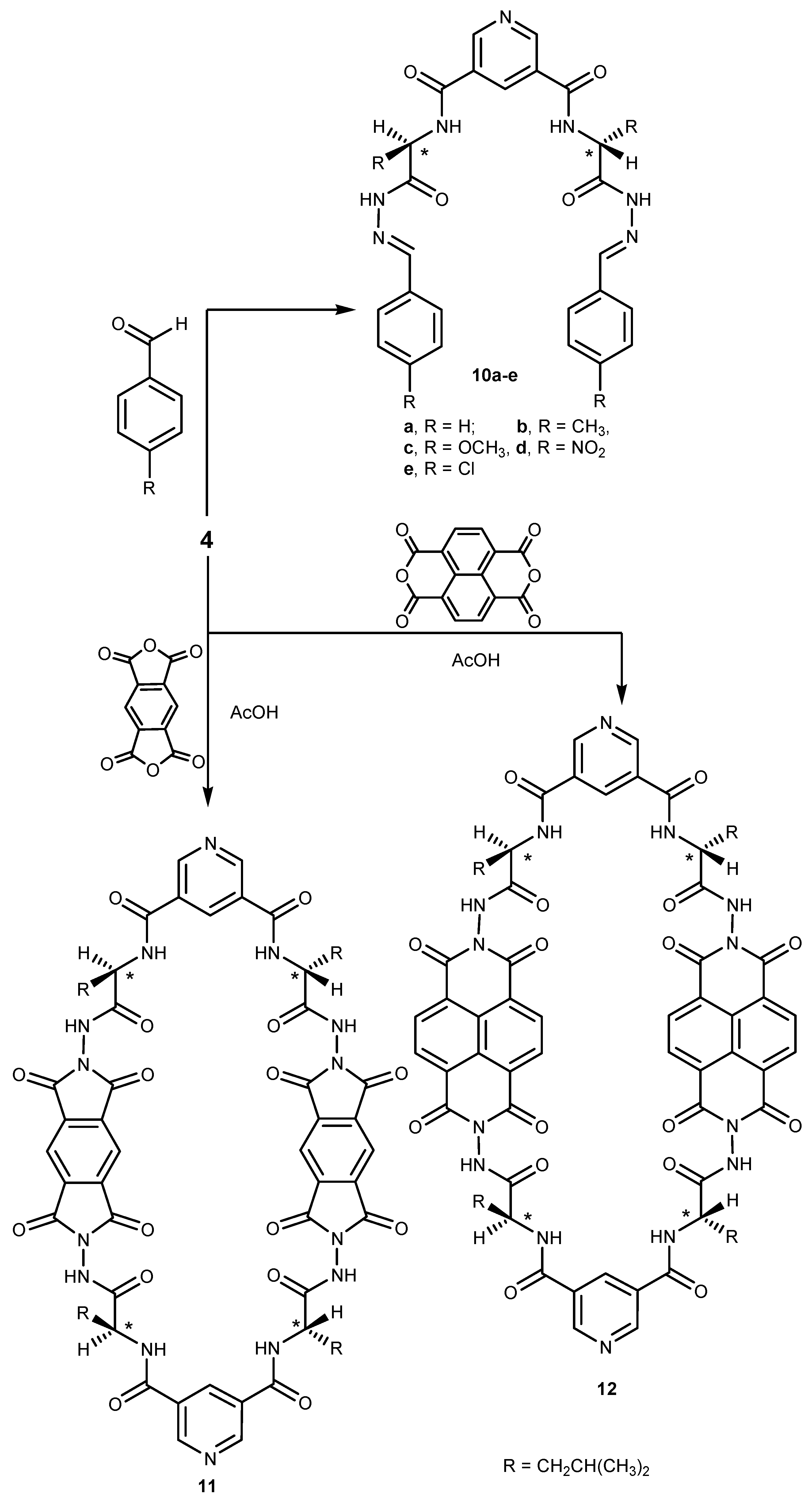
2.1. Chemistry



2.2. Pharmacological Screening
2.2.1. Antimicrobial Activity

{kind=link}
{kind=link}
{kind=link}
| Compound No. | Inhibition Zome (cm) | |||||
|---|---|---|---|---|---|---|
| Gram+ ve | Gram− ve | Yeast | Fungi | |||
| Bacillus subtilis | Bacillus aureus | Staphylococcus aureus | Escherichia coli | Candida albicans | Aspergillus niger | |
| 3 | 1.46 | 1.65 | 1.76 | 0.62 | - | 1.78 |
| 4 | 1.85 | 1.92 | 1.80 | 0.80 | 0.92 | 1.56 |
| 5 | 1.68 | 1.14 | 1.76 | 0.64 | - | 1.72 |
| 6 | 1.82 | 1.50 | 1.54 | 0.65 | - | 1.75 |
| 7a | 1.55 | 1.84 | 1.58 | 0.75 | 1.02 | 1.64 |
| 7b | 1.46 | 1.80 | 1.45 | 0.66 | - | 1.95 |
| 7c | 1.72 | 1.56 | 1.76 | 0.74 | 0.95 | 1.88 |
| 8 | 1.90 | 1.94 | 1.95 | 0.90 | 0.98 | 2.00 |
| 9a | 1.84 | 1.88 | 1.76 | 0.92 | 1.10 | 2.01 |
| 9b | 1.60 | 1.74 | 1.72 | 0.75 | 1.00 | 1.88 |
| 9c | 1.72 | 1.22 | 1.66 | 0.60 | - | 1.76 |
| 10a | 1.56 | 1.45 | 1.56 | 0.66 | - | 1.68 |
| 10b | 1.80 | 1.95 | 1.85 | 0.78 | 0.95 | 1.56 |
| 10c | 1.76 | 1.56 | 1.64 | 0.80 | 1.00 | 1.95 |
| 10d | 1.85 | 2.00 | 1.92 | 0.90 | 0.96 | 2.00 |
| 10e | 1.65 | 1.96 | 1.80 | 0.78 | 0.94 | 1.48 |
| 11 | 1.60 | 1.17 | 1.98 | 0.64 | - | 1.75 |
| 12 | 1.65 | 1.83 | 1.65 | 0.64 | - | 1.58 |
| Chloramphenicol | 2.00 | 2.10 | 2.00 | 0.95 | - | - |
| Fusidic Acid | - | - | - | - | 1.9 | 1.9 |
2.2.2. Anti-Inflammatory Activity
Purpose and Rational
| Compound No. | Dose mg/kg | % Protection against Edema | % Inhibition of Plasma PGE2 |
|---|---|---|---|
| 3 | 2.5 | 86.16 ± 0.052 | 59.45 ± 0.050 |
| 5.0 | 98.18 ± 0.053 | 81.10 ± 0.056 | |
| 4 | 2.5 | 93.45 ± 0.074 | 75.66 ± 0.040 |
| 5.0 | 98.56 ± 0.060 | 80.01 ± 0.058 | |
| 5 | 2.5 | 85.14 ± 0.056 | 61.16 ± 0.041 |
| 5.0 | 96.18 ± 0.067 | 77.14 ± 0.036 | |
| 6 | 2.5 | 92.14 ± 0.076 | 84.18 ± 0.052 |
| 5.0 | 99.08 ± 0.062 | 86.01 ± 0.030 | |
| 7a | 2.5 | 97.45 ± 0.054 | 81.30 ± 0.044 |
| 5.0 | 98.86 ± 0.046 | 82.48 ± 0.052 | |
| 7c | 2.5 | 91.12 ± 0.064 | 73.48 ± 0.049 |
| 5.0 | 95.14 ± 0.075 | 78.55 ± 0.035 | |
| 8 | 2.5 | 78.88 ± 0.090 | 57.57 ± 0.045 |
| 5.0 | 92.00 ± 0.060 | 76.00 ± 0.035 | |
| 9a | 2.5 | 82.16 ± 0.076 | 58.96 ± 0.024 |
| 5.0 | 93.16 ± 0.064 | 74.58 ± 0.040 | |
| 9b | 2.5 | 79.34 ± 0.081 | 57.90 ± 0.040 |
| 5.0 | 92.92 ± 0.090 | 77.14 ± 0.052 | |
| 10a | 2.5 | 90.32 ± 0.035 | 62.12 ± 0.051 |
| 5.0 | 99.10 ± 0.036 | 83.01 ± 0.049 | |
| 10d | 2.5 | 81.00 ± 0.072 | 63.14 ± 0.023 |
| 5.0 | 95.82 ± 0.062 | 79.88 ± 0.031 | |
| 12 | 2.5 | 90.48 ± 0.090 | 63.16 ± 0.036 |
| 5.0 | 99.36 ± 0.080 | 86.18 ± 0.071 | |
| Diclofenac Potassium | 2.5 | 70.14 ± 0.064 | 54.00 ± 0.041 |
| 5.0 | 75.23 ± 0.083 | 70.00 ± 0.051 |
2.3. Anticancer Activity
In Vitro Anti-Breast Cancer Activities
| Compound No. | IC50 (μM ) for Tested Compounds against Breast Cell Lines | ||||||
|---|---|---|---|---|---|---|---|
| MCF7 | MDAMB231 | HS578T | MDAMB435 | MDN | BT549 | T47D | |
| 3 | 0.00029 | 3.23 | 3.34 | 56.38 | 0.00 | 0.00 | 0.00 |
| 4 | 0.00026 | 2.76 | 2.78 | 45.47 | 0.00 | 0.00 | 0.00 |
| 5 | 0.00017 | 2.34 | 2.56 | 33.66 | 0.00 | 0.00 | 0.00 |
| 6 | 0.00075 | 4.56 | 8.89 | 88.76 | 0.00 | 0.00 | 0.00 |
| 7a | 0.00086 | 7.89 | 9.75 | 89.87 | 0.00 | 0.00 | 0.00 |
| 7c | 0.00099 | 9.65 | 14.45 | 92.99 | 0.00 | 0.00 | 0.00 |
| 9a | 0.00097 | 8.65 | 11.43 | 90.98 | 0.00 | 0.00 | 0.00 |
| 9b | 0.00064 | 3.56 | 7.47 | 78.65 | 0.00 | 0.00 | 0.00 |
| 10a | 0.00014 | 1.23 | 1.54 | 10.55 | 0.00 | 0.00 | 0.00 |
| 10d | 0.00035 | 3.67 | 5.67 | 57.47 | 0.00 | 0.00 | 0.00 |
| 12 | 0.00056 | 3.56 | 6.65 | 68.56 | 0.00 | 0.00 | 0.00 |
| Compound No. | Tumor Growth Vt/Vo for Compounds after Times in Days | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 2 | 4 | 6 | 8 | 10 | 12 | 14 | 16 | 18 | 20 | |
| Control | 1.00 | 1.57 | 2.11 | 4.87 | 9.67 | 12.55 | 24.34 | 26.45 | 28.30 | 39.90 | 41.12 |
| 3 | 1.00 | 1.40 | 1.56 | 1.87 | 2.01 | 2.89 | 3.54 | 5.10 | 6.12 | 7.14 | 8.12 |
| 4 | 1.00 | 1.34 | 1.45 | 1.77 | 1.91 | 2.77 | 3.44 | 4.89 | 5.56 | 6.12 | 7.14 |
| 5 | 1.00 | 1.21 | 1.28 | 1.65 | 1.81 | 2.67 | 3.21 | 4.45 | 5.22 | 5.77 | 6.23 |
| 6 | 1.00 | 1.40 | 1.67 | 2.45 | 2.79 | 3.56 | 4.57 | 5.98 | 7.01 | 8.88 | 9.56 |
| 7a | 1.00 | 1.41 | 1.71 | 2.78 | 3.20 | 3.78 | 4.78 | 6.11 | 7.16 | 8.90 | 9.77 |
| 7c | 1.00 | 1.42 | 1.72 | 2.98 | 3.40 | 4.00 | 5.01 | 6.18 | 7.28 | 8.99 | 9.99 |
| 9a | 1.00 | 1.41 | 1.70 | 2.89 | 3.30 | 3.90 | 4.90 | 6.13 | 7.13 | 8.79 | 9.88 |
| 9b | 1.00 | 1.42 | 1.78 | 2.34 | 2.67 | 3.45 | 4.56 | 5.90 | 6.88 | 8.89 | 9.45 |
| 10a | 1.00 | 1.14 | 1.24 | 1.56 | 1.76 | 2.54 | 3.11 | 4.32 | 5.12 | 5.89 | 6.12 |
| 10d | 1.00 | 1.40 | 1.6 | 1.99 | 2.34 | 3.23 | 4.34 | 5.55 | 6.66 | 8.45 | 9.00 |
| 12 | 1.00 | 1.41 | 1.65 | 2.14 | 2.56 | 3.25 | 4.39 | 5.78 | 6.79 | 8.68 | 9.14 |
3. Experimental Section
3.1. General Information

 :−132.5 (c = 0.5, DMF). IR (KBr): ν = 3370 (NH str.), 3045 (CH-Ar), 29525 (CH-aliph.), 1752 (C=O, ester), 1657, 1532, 1253 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 0.95–0.88 (d, 12H, 4CH3), 1.80–1.73 (m, 6H, 2CH2 + 2CH), 3.81 (s, 6H, 2OCH3), 4.84 (t, 2H, 2CHNH), 8.48 (s, 2H, 2NH, exchangeable with D2O), 8.40, 9.01 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.35 (2C, 2CH), 22.82 (4C, 4CH3), 41.04 (2C, 2CH2), 49.56 (2C, 2CH), 52.08 (2C, 2CH3), 131.56, 140.03, 152.10 (5C, Pyr-C), 167.45 (2C, 2C=O), 172.08 (2C, 2C=O). MS (EI, 70 eV): m/z (%) = 421 [M+, 22], 278 (32), 247 (100), 144 (16). C21H31N3O6 (421.49): calcd. C 59.84, H 7.41, N 9.97; found C 59.78, H 7.34, N 9.90.: −98.8 (c = 0.5, DMF). IR (KBr): ν = 3400–3210 (NH, NH2), 3050 (CH-Ar), 2954 (CH-aliph.), 1662, 1530, 1245 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 0.98–0.86 (m, 12H, 4CH3), 1.82–1.73 (m, 6H, 2CH2 + 2CH), 3.45 (s, 4H, 2 NH2, exchangeable with D2O), 4.64 (t, 2H, 2CHNH), 8.58, 8.98 (2s, 4H, 4NH, exchangeable with D2O), 8.48, 9.22 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.84 (4C, 4CH3), 22.95 (2C, 2CH), 40.98 (2C, 2CH2), 42.86 (2C, 2CH), 132.06, 140.10, 152.25 (5C, Pyr-C), 167.53 (2C, 2C=O), 170.16 (2C, 2C=O). MS (EI, 70 eV): m/z (%) = 422 [M++1, 8], 374 (12), 247 (100), 134 (86). C19H31N7O4 (421.49): calcd. C 54.14, H 7.41, N 23.26; found C 54.05, H 7.35, N 23.20.: −104.5 (c = 0.5, DMF). IR (KBr): ν = 3578–3385 (OH, NH), 3065 (CH-Ar), 2958 (CH-aliph.), 1732 (C=O, acid), 1664, 1536, 1248 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.05–0.96 (m, 12H, 4CH3), 1.80–1.71 (m, 6H, 2CH2 + 2CH), 4.60 (t, 2H, 2CHNH), 8.68 (s, 2H, 2NH, exchangeable with D2O), 8.42, 9.18 (2s, 3H, Pyr-H), 12.24 (s, 2H, 2OH, exchangeable with D2O). 13C-NMR: δ = 22.12 (2C, 2CH), 22.92 (4C, 4CH3), 39.95 (2C, 2CH2), 52.35 (2C, 2CH), 132.12, 139.88, 152.18 (5C, Pyr-C), 167.42 (2C, 2C=O), 174.56 (2C, 2C=O). MS (EI, 70 eV): m/z (%) = 393 [M+, 16], 360 (32), 246 (24), 134 (100). C19H27N3O6 (393.43): calcd. C 58.00, H 6.92, N 10.68; found C 57.90, H 6.87, N 10.62.: = −178.6 (c = 0.5, DMF). IR (KBr): ν = 3410–3335 (NH), 3062 (CH-Ar), 2955 (CH-aliph.), 1747 (C=O, ester), 1660, 1528, 1341 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.05–0.96 (m, 12H, 4CH3), 1.34–1.24 (m, 2H, CH2), 1.56–1.53 (m, 2H, CH2), 1.86–1.75 (m, 6H, 2CH2 + 2CH), 2.45–2.36 (m, 2H, CH2), 3.30–3.20 (m, 2H, CH2), 3.64 (s, 3H, OCH3), 4.42 (t, 1H, CHNH), 4.58 (t, 2H, 2CHNH), 8.62, 8.98 (2s, 4H, 4NH, exchangeable with D2O), 8.32, 9.15 (2s, 3H, Pyr-H). 13C-NMR: δ = 18.45, 30.60, 33.85, 41.45 (4C, 4CH2), 21.86 (2C, 2CH), 22.34 (4C, 4CH3), 40.92 (2C, 2CH2), 51.45 (2C, 2CH), 51.14 (1C, CH3), 53.96 (1C, CH), 131.86, 139.84, 152.00 (5C, Pyr-C), 167.14 (2C, 2C=O), 169.62 (2C, 2C=O), 171.05 (1C, C=O, ester). MS (EI, 70 eV): m/z (%) = 518 [M+, 18], 486 (45), 403 (52), 246 (34), 188 (100). C26H39N5O6 (517.62): calcd. C 60.33, H 7.59, N 13.53; found C 60.26, H 7.52, N 13.45.: −84.4 (c = 0.5, DMF). IR (KBr): ν = 3590–3480 (NH), 3056 (CH-Ar), 2960 (CH-aliph), 1753 (C=O, imide), 1658, 1532, 1322 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.01–0.92 (m, 12H, 4CH3), 1.83–1.74 (m, 6H, 2CH2 + 2CH), 4.66 (t, 2H, 2CHNH), 7.45–8.10 (m, 8H, Ar-H), 8.62, 9.08 (2s, 4H, 4NH, exchangeable with D2O), 8.38, 9.22 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.56 (4C, 4CH3), 22.95 (2C, 2CH), 41.16 (2C, 2CH2), 52.86 (2C, 2CH), 131.48, 140.10, 152.12 (5C, Pyr-C), 167.42 (2C, 2C=O), 169.84 (2C, 2C=O), 163.56 (4C, 4C=O), 127.15, 132.02, 132.86 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 682 [M+, 14], 520 (21), 274 (86), 247 (53), 189 (100). C35H35N7O8 (681.69): calcd. C 61.67, H 5.18, N 14.38; found C 61.60, H 5.12, N 14.33.: −96.5 (c = 0.5, DMF). IR (KBr): ν = 3532–3450 (NH), 3058 (CH-Ar), 2961 (CH-aliph.), 1750 (C=O, imide), 1656, 1528, 1318 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.00–0.90 (m, 12H, 4CH3), 1.84–1.76 (m, 4H, 2CH2 + 2CH), 4.65 (t, 2H, 2CHNH), 8.60, 9.04 (2s, 4H, 4NH, exchangeable with D2O), 8.45, 9.24 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.62 (4C, 4CH3), 22.94 (2C, 2CH), 41.24 (2C, 2CH2), 52.88 (2C, 2CH), 131.44, 140.04, 152.10 (5C, Pyr-C), 167.36 (2C, 2C=O), 169.92 (2C, 2C=O), 164.04 (4C, 4C=O), 128.25, 132.95, 138.36 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 957 [M+, 8], 542 (14), 410 (16), 324 (45), 247 (45), 245 (100). C35H27Cl8N7O8 (957.25): calcd. C 43.91, H 2.84, Cl 29.63, N 10.24; found C 43.86, H 2.75, Cl 29.55, N 10.18.: −114.2 (c = 0.5, DMF). IR (KBr): ν = 3565–3328 (NH), 3048 (CH-Ar), 2962 (CH-aliph.), 1748 (C=O, imide), 1655, 1536, 1320 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.00–0.94 (m, 12H, 4 CH3), 1.85–1.69 (m, 6H, 2 CH2 + 2CH), 4.65 (t, 2H, 2CHNH), 8.72, 9.04 (2d, 4H, Pyr-H), 8.02 (t, 2H, Pyr-H), 9.18, 8.64 (2s, 4H, 4NH, exchangeable with D2O), 8.36, 9.10 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.56 (4C, 4CH3), 22.92 (2C, 2CH), 41.25 (2C, 2CH2), 52.95 (2C, 2CH), 131.46, 140.10, 151.98 (5C, Pyr-C), 167.22 (2C, 2C=O), 170.02 (2C, 2C=O), 164.12, 165.05 (4C, 4C=O), 127.85, 128.15, 138.32, 146.10, 152.45 (10C, Pyr-C). MS (EI, 70 eV): m/z (%) = 684 [M+, 14], 494 (25), 304 (85), 247 (95), 190 (100). C33H33N9O8 (683.67): calcd. C 57.97, H 4.87, N 18.44; found C 57.90, H 4.80, N 18.38.: −108.8 (c = 0.5, DMF). IR (KBr): ν = 3548–3452 (NH), 3056 (CH-Ar), 2950 (CH-aliph.), 1746 (C=O, imide), 1656, 1532, 1328 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.00–0.95 (m, 12H, 4 CH3), 1.85–1.73 (m, 6H, 2 CH2 + 2CH), 4.62 (t, 2H, 2CHNH), 7.55–8.05 (m, 12H, Ar-H), 8.68, 9.12 (2s, 4H, 4NH, exchangeable with D2O), 8.35, 9.08 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.55 (4C, 4CH3), 22.98 (2C, 2CH), 41.25 (2C, 2CH2), 52.85 (2C, 2CH), 131.48, 140.10, 152.15 (5C, Pyr-C), 167.42 (2C, 2C=O), 169.88 (2C, 2C=O), 158.76 (4C, 4C=O), 123.45, 125.02, 128.20, 130.54, 137.05 137.65 (20C, Ar-C). MS (EI, 70 eV): m/z (%) = 782 [M+, 6], 570 (12), 324 (25), 246 (100), 134 (95). C43H39N7O8 (781.81): calcd. C 66.06, H 5.03, N 12.54; found C 66.00, H 4.96, N 12.50.: −106.6 (c = 0.5, DMF). IR (KBr): ν = 3510–3432 (NH), 3054 (CH-Ar), 2945 (CH-aliph.), 1660, 1526, 1316 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 0.85 (s, 6H, 2CH3), 0.98–0.92 (m, 12H, 4 CH3), 1.82–1.72 (m, 6H, 2 CH2 + 2CH), 4.64 (t, 2H, 2CHNH), 7.30–7.65 (m, 8H, Ar-H), 9.08, 8.35 (2s, 4H, 4NH, exchangeable with D2O), 8.42, 9.16 (2s, 3H, Pyr-H). 13C-NMR: δ = 14.05 (2C, 2CH3), 22.48 (4C, 4CH3), 22.95 (2C, 2CH), 41.35 (2C, 2CH2), 52.78 (2C, 2CH), 131.56, 140.18, 152.12 (5C, Pyr-C), 167.35 (2C, 2C=O), 172.84 (2C, 2C=O), 168.12 (2C, C=N), 128.45, 129.68, 131.89, 135.65 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 695 [M+, 10], 582 (8), 416 (24), 359 (100), 246 (95). C35H41Cl2N7O4 (694.65): calcd. C 60.52, H 5.95, Cl 10.21, N 14.11; found C 60.45, H 5.90, Cl 10.15, N 14.05.: −112.5 (c = 0.5, DMF). IR (KBr): ν = 3498–3386 (NH), 3065 (CH-Ar), 2943 (CH-aliph.), 1658, 1530, 1337 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 0.86 (s, 6H, 2CH3), 0.99–0.94 (m, 12H, 4 CH3), 1.80–1.74 (m, 6H, 2 CH2 + 2CH), 3.65 (s. 6H, 2 OCH3), 4.62 (t, 2H, 2CHNH), 7.02–7.60 (m, 8H, Ar-H), 7.85, 8.45 (2s, 4H, 4NH, exchangeable with D2O), 8.36, 9.12 (2s, 3H, Pyr-H). 13C-NMR: δ = 13.95 (2C, 2CH3), 22.56 (4C, 4CH3), 23.05 (2C, 2CH), 41.36 (2C, 2CH2), 52.84 (2C, 2CH), 56.04 (2C, 2OCH3), 131.58, 140.10, 152.10 (5C, Pyr-C), 167.28 (2C, 2C=O), 172.92 (2C, 2C=O), 168.15 (2C, C=N), 114.02, 125.65, 129.85, 162.15 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 686 [M+, 5], 522 (6), 276 (24), 246 (100). C37H47N7O6 (685.81): calcd. C 64.80, H 6.91, N 14.30; found C 64.72, H 6.83, N 14.24.: −122.4 (c = 0.5, DMF). IR (KBr): ν = 3495–3435 (NH), 3050 (CH-Ar), 2945 (CH-aliph.), 1655, 1531, 1324 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 0.88 (s, 6H, 2CH3), 1.00–0.96 (m, 12H, 4 CH3), 1.85–1.64 (m, 6H, 2 CH2 + 2CH), 4.65 (t, 2H, 2CHNH), 7.65–8.05 (m, 8H, Ar-H), 9.10, 8.36 (2s, 4H, 4NH, exchangeable with D2O), 8.38, 9.10 (2s, 3H, Pyr-H). 13C-NMR: δ = 13.98 (2C, 2CH3), 22.54 (4C, 4CH3), 22.98 (2C, 2CH), 41.32 (2C, 2CH2), 52.86 (2C, 2CH), 131.64, 140.08, 152.16 (5C, Pyr-C), 167.33 (2C, 2C=O), 173.05 (2C, 2C=O), 168.08 (2C, C=N), 119.84, 129.35, 139.88, 150.35 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 716 [M+, 16], 537 (22), 389 (18), 276 (32), 206 (100). C35H41N9O8 (715.76): calcd. C 58.73, H 5.77, N 17.61; found C 58.65, H 5.72, N 17.55.: −145.0 (c = 0.5, DMF). IR (KBr): ν = 3510–3447 (NH), 3038 (CH-Ar), 2955 (CH-aliph.), 1656, 1528, 1319 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.04–0.95 (m, 12H, 4 CH3), 1.86–1.70 (m, 6H, 2 CH2 + 2CH), 4.65 (t, 2H, 2CHNH), 5.60 (s, 2H, 2CH=N), 7.45–7.72 (m, 10H, Ar-H), 8.42, 9.10 (2s, 4H, 4NH, exchangeable with D2O), 8.35, 9.18 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.76 (4C, 4CH3), 23.01 (2C, 2CH), 41.16 (2C, 2CH2), 52.86 (2C, 2CH), 131.48, 140.32, 151.98 (5C, Pyr-C), 167.18 (2C, 2C=O), 173.05 (2C, 2C=O), 143.05 (2C, C=N), 128.68, 129.08, 130.85, 135.75 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 598 [M+, 24], 520 (15), 478 (18), 444 (10), 360 (100), 247 (32). C33H39N7O4 (597.71): calcd. C 66.31, H 6.58, N 16.40; found C 66.24, H 6.50, N 16.32.: −130.5 (c = 0.5, DMF). IR (KBr): ν = 3488–3432 (NH), 3065 (CH-Ar), 2952 (CH-aliph.), 1652, 1533, 1322 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.00–0.95 (m, 12H, 4 CH3), 1.78–1.72 (m, 6H, 2 CH2 + 2CH), 2.24 (s, 6H, 2CH3), 4.68 (t, 2H, 2CHNH), 5.58 (s, 2H, 2CH=N), 6.98–7.56 (m, 8H, Ar-H), 8.38, 9.12 (2s, 4H, 4NH, exchangeable with D2O), 8.26, 9.05 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.76 (4C, 4CH3), 22.95 (2C, 2CH), 24.12 (2C, 2CH3), 41.42 (2C, 2CH2), 52.76 (2C, 2CH), 131.46, 139.98, 152.15 (5C, Pyr-C), 166.95 (2C, 2C=O), 173.02 (2C, 2C=O), 142.55 (2C, C=N), 128.76, 129.01, 130.55, 139.85 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 626 [M+, 8], 534 (24), 402 (32), 360 (100), 246 (82). C35H43N7O4 (625.76): calcd. C 67.18, H 6.93, N 15.67; found C 67.09, H 6.84, N 15.60.: −98.5 (c = 0.5, DMF). IR (KBr): ν = 3490–3412 (NH), 3056 (CH-Ar), 2975 (CH-aliph.), 1657, 1532, 1336 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.10–0.94 (m, 12H, 4 CH3), 1.83–1.75 (m, 6H, 2 CH2 + 2CH), 3.68 (s, 6H, 2OCH3), 4.62 (t, 2H, 2CHNH), 5.62 (s, 2H, 2CH=N), 7.08–7.42 (m, 8H, Ar-H), 8.36, 9.08 (2s, 4H, 4NH, exchangeable with D2O), 8.24, 9.24 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.68 (4C, 4CH3), 23.00 (2C, 2CH), 41.32 (2C, 2CH2), 52.88 (2C, 2CH), 55.75 (2C, 2OCH3), 131.42, 140.05, 152.14 (5C, Pyr-C), 167.08 (2C, 2C=O), 172.85 (2C, 2C=O), 143.15 (2C, C=N), 113.95, 125.78, 130.02, 162.45 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 658 [M+, 6], 626 (12), 596 (15), 478 (56), 247 (100). C35H43N7O6 (657.76): calcd. C 63.91, H 6.59, N 14.91; found C 63.81, H 6.50, N 14.84.: −142.5 (c = 0.5, DMF). IR (KBr): ν = 3492–3398 (NH), 3045 (CH-Ar), 2965 (CH-aliph.), 1657, 1532, 1331 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.01–0.95 (m, 12H, 4 CH3), 1.85–1.64 (m, 6H, 2 CH2 + 2CH), 4.62 (t, 2H, 2CHNH), 5.65 (s, 2H, 2CH=N), 7.68–8.00 (m, 8H, Ar-H), 9.10, 8.36 (2s, 4H, 4NH, exchangeable with D2O), 8.56, 9.10 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.62 (4C, 4CH3), 22.95 (2C, 2CH), 41.40 (2C, 2CH2), 52.95 (2C, 2CH), 131.44, 140.10, 152.32 (5C, Pyr-C), 167.24 (2C, 2C=O), 172.88 (2C, 2C=O), 142.65 (2C, C=N), 120.05, 129.78, 139.65, 150.48 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 688 [M+, 16], 537 (22), 389 (18), 276 (32), 206 (100). C33H37N9O8 (687.70): calcd. C 57.63, H 5.42, N 18.33; found C 57.55, H 5.34, N 18.24.: −116.45 (c = 0.5, DMF). IR (KBr): ν = 3490–3444 (NH), 3065 (CH-Ar), 2958 (CH-aliph.), 1656, 1522, 1318 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.10–0.95 (m, 12H, 4 CH3), 1.80–1.70 (m, 6H, 2 CH2 + 2CH), 4.68 (t, 2H, 2CHNH), 5.48 (s, 2H, 2CH=N), 7.26–7.68 (m, 8H, Ar-H), 9.08, 8.35 (2s, 4H, 4NH, exchangeable with D2O), 8.45, 9.36 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.45 (4C, 4CH3), 22.90 (2C, 2CH), 41.33 (2C, 2CH2), 52.76 (2C, 2CH), 131.58, 140.22, 152.16 (5C, Pyr-C), 167.36 (2C, 2C=O), 172.91 (2C, 2C=O), 142.65 (2C, C=N), 128.50, 129.72, 131.94, 135.68 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 666 [M+, 6], 595 (12), 478 (45), 332(92), 260 (100). C33H37Cl2N7O4 (666.60): calcd. C 59.46, H 5.59, Cl 10.64, N 14.71; found C 59.40, H 5.53, Cl 10.60, N 14.65.: −112.6 (c = 0.5, DMF). IR (KBr): ν = 3478–3575 (NH), 3060 (CH-Ar), 2966 (CH-aliph.), 1753 (C=O, imide), 1656, 1530, 1320 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 0.98–0.90 (m, 24H, 8 CH3), 1.80–1.72 (m, 12H, 4 CH2 + 4 CH), 4.55 (t, 4H, 4CHNH), 9.08 (s, 4H, Ar-H), 8.78, 9.15 (2s, 8H, 8NH, exchangeable with D2O), 8.40, 9.20 (2s, 6H, Pyr-H). 13C-NMR: δ = 22.42 (8C, 8CH3), 22.98 (4C, 4CH), 41.18 (4C, 4CH2), 53.15 (4C, 4CH), 131.55, 140.08, 152.24 (10C, 2 Pyr-C), 167.16 (4C, 4C=O), 172.84 (4C, 4C=O), 164.16 (8C, 8C=O), 124.98, 134.85 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 1207 [M+, 4], 737 (5), 470 (32), 300 (100), 274 (72), 247 (85). C58H58N14O16 (1207.17): calcd. C 57.71, H 4.84, N 16.24; found C 57.65, H 4.78, N 16.20.: −124.5 (c = 0.5, DMF). IR (KBr): ν = 3544–3432 (NH), 3050 (CH-Ar), 2964 (CH-aliph.), 1742 (C=O, imide), 1657, 1530, 1326 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.05–0.98 (m, 24H, 8 CH3), 1.82–1.75 (m, 12H, 4 CH2 + 4 CH), 4.58 (t, 4H, 4 CHNH), 8.15–8.25 (m, 8H, Ar-H), 8.56, 9.16 (2s, 8H, 8NH, exchangeable with D2O), 8.40, 9.04 (2s, 6H, 2 Pyr-H). 13C-NMR: δ = 22.64 (8C, 8CH3), 22.95 (4C, 4CH), 41.35 (4C, 4CH2), 52.78 (4C, 4CH), 131.54, 140.12, 152.18 (10C, 2 Pyr-C), 167.36 (4C, 4C=O), 170.05 (4C, 4C=O), 158.68 (8C, 8 C=O), 120.25, 135.10, 139.73 (20C, Ar-C). MS (EI, 70 eV): m/z (%) = 1307 [M+, 4], 1014 (8), 787 (12), 654 (24), 294 (100), 246 (85). C66H62N14O16 (1307.28): calcd. C 60.64, H 4.78, N 15.00; found C 60.55, H 4.70, N 14.95.
:−132.5 (c = 0.5, DMF). IR (KBr): ν = 3370 (NH str.), 3045 (CH-Ar), 29525 (CH-aliph.), 1752 (C=O, ester), 1657, 1532, 1253 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 0.95–0.88 (d, 12H, 4CH3), 1.80–1.73 (m, 6H, 2CH2 + 2CH), 3.81 (s, 6H, 2OCH3), 4.84 (t, 2H, 2CHNH), 8.48 (s, 2H, 2NH, exchangeable with D2O), 8.40, 9.01 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.35 (2C, 2CH), 22.82 (4C, 4CH3), 41.04 (2C, 2CH2), 49.56 (2C, 2CH), 52.08 (2C, 2CH3), 131.56, 140.03, 152.10 (5C, Pyr-C), 167.45 (2C, 2C=O), 172.08 (2C, 2C=O). MS (EI, 70 eV): m/z (%) = 421 [M+, 22], 278 (32), 247 (100), 144 (16). C21H31N3O6 (421.49): calcd. C 59.84, H 7.41, N 9.97; found C 59.78, H 7.34, N 9.90.: −98.8 (c = 0.5, DMF). IR (KBr): ν = 3400–3210 (NH, NH2), 3050 (CH-Ar), 2954 (CH-aliph.), 1662, 1530, 1245 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 0.98–0.86 (m, 12H, 4CH3), 1.82–1.73 (m, 6H, 2CH2 + 2CH), 3.45 (s, 4H, 2 NH2, exchangeable with D2O), 4.64 (t, 2H, 2CHNH), 8.58, 8.98 (2s, 4H, 4NH, exchangeable with D2O), 8.48, 9.22 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.84 (4C, 4CH3), 22.95 (2C, 2CH), 40.98 (2C, 2CH2), 42.86 (2C, 2CH), 132.06, 140.10, 152.25 (5C, Pyr-C), 167.53 (2C, 2C=O), 170.16 (2C, 2C=O). MS (EI, 70 eV): m/z (%) = 422 [M++1, 8], 374 (12), 247 (100), 134 (86). C19H31N7O4 (421.49): calcd. C 54.14, H 7.41, N 23.26; found C 54.05, H 7.35, N 23.20.: −104.5 (c = 0.5, DMF). IR (KBr): ν = 3578–3385 (OH, NH), 3065 (CH-Ar), 2958 (CH-aliph.), 1732 (C=O, acid), 1664, 1536, 1248 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.05–0.96 (m, 12H, 4CH3), 1.80–1.71 (m, 6H, 2CH2 + 2CH), 4.60 (t, 2H, 2CHNH), 8.68 (s, 2H, 2NH, exchangeable with D2O), 8.42, 9.18 (2s, 3H, Pyr-H), 12.24 (s, 2H, 2OH, exchangeable with D2O). 13C-NMR: δ = 22.12 (2C, 2CH), 22.92 (4C, 4CH3), 39.95 (2C, 2CH2), 52.35 (2C, 2CH), 132.12, 139.88, 152.18 (5C, Pyr-C), 167.42 (2C, 2C=O), 174.56 (2C, 2C=O). MS (EI, 70 eV): m/z (%) = 393 [M+, 16], 360 (32), 246 (24), 134 (100). C19H27N3O6 (393.43): calcd. C 58.00, H 6.92, N 10.68; found C 57.90, H 6.87, N 10.62.: = −178.6 (c = 0.5, DMF). IR (KBr): ν = 3410–3335 (NH), 3062 (CH-Ar), 2955 (CH-aliph.), 1747 (C=O, ester), 1660, 1528, 1341 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.05–0.96 (m, 12H, 4CH3), 1.34–1.24 (m, 2H, CH2), 1.56–1.53 (m, 2H, CH2), 1.86–1.75 (m, 6H, 2CH2 + 2CH), 2.45–2.36 (m, 2H, CH2), 3.30–3.20 (m, 2H, CH2), 3.64 (s, 3H, OCH3), 4.42 (t, 1H, CHNH), 4.58 (t, 2H, 2CHNH), 8.62, 8.98 (2s, 4H, 4NH, exchangeable with D2O), 8.32, 9.15 (2s, 3H, Pyr-H). 13C-NMR: δ = 18.45, 30.60, 33.85, 41.45 (4C, 4CH2), 21.86 (2C, 2CH), 22.34 (4C, 4CH3), 40.92 (2C, 2CH2), 51.45 (2C, 2CH), 51.14 (1C, CH3), 53.96 (1C, CH), 131.86, 139.84, 152.00 (5C, Pyr-C), 167.14 (2C, 2C=O), 169.62 (2C, 2C=O), 171.05 (1C, C=O, ester). MS (EI, 70 eV): m/z (%) = 518 [M+, 18], 486 (45), 403 (52), 246 (34), 188 (100). C26H39N5O6 (517.62): calcd. C 60.33, H 7.59, N 13.53; found C 60.26, H 7.52, N 13.45.: −84.4 (c = 0.5, DMF). IR (KBr): ν = 3590–3480 (NH), 3056 (CH-Ar), 2960 (CH-aliph), 1753 (C=O, imide), 1658, 1532, 1322 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.01–0.92 (m, 12H, 4CH3), 1.83–1.74 (m, 6H, 2CH2 + 2CH), 4.66 (t, 2H, 2CHNH), 7.45–8.10 (m, 8H, Ar-H), 8.62, 9.08 (2s, 4H, 4NH, exchangeable with D2O), 8.38, 9.22 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.56 (4C, 4CH3), 22.95 (2C, 2CH), 41.16 (2C, 2CH2), 52.86 (2C, 2CH), 131.48, 140.10, 152.12 (5C, Pyr-C), 167.42 (2C, 2C=O), 169.84 (2C, 2C=O), 163.56 (4C, 4C=O), 127.15, 132.02, 132.86 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 682 [M+, 14], 520 (21), 274 (86), 247 (53), 189 (100). C35H35N7O8 (681.69): calcd. C 61.67, H 5.18, N 14.38; found C 61.60, H 5.12, N 14.33.: −96.5 (c = 0.5, DMF). IR (KBr): ν = 3532–3450 (NH), 3058 (CH-Ar), 2961 (CH-aliph.), 1750 (C=O, imide), 1656, 1528, 1318 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.00–0.90 (m, 12H, 4CH3), 1.84–1.76 (m, 4H, 2CH2 + 2CH), 4.65 (t, 2H, 2CHNH), 8.60, 9.04 (2s, 4H, 4NH, exchangeable with D2O), 8.45, 9.24 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.62 (4C, 4CH3), 22.94 (2C, 2CH), 41.24 (2C, 2CH2), 52.88 (2C, 2CH), 131.44, 140.04, 152.10 (5C, Pyr-C), 167.36 (2C, 2C=O), 169.92 (2C, 2C=O), 164.04 (4C, 4C=O), 128.25, 132.95, 138.36 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 957 [M+, 8], 542 (14), 410 (16), 324 (45), 247 (45), 245 (100). C35H27Cl8N7O8 (957.25): calcd. C 43.91, H 2.84, Cl 29.63, N 10.24; found C 43.86, H 2.75, Cl 29.55, N 10.18.: −114.2 (c = 0.5, DMF). IR (KBr): ν = 3565–3328 (NH), 3048 (CH-Ar), 2962 (CH-aliph.), 1748 (C=O, imide), 1655, 1536, 1320 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.00–0.94 (m, 12H, 4 CH3), 1.85–1.69 (m, 6H, 2 CH2 + 2CH), 4.65 (t, 2H, 2CHNH), 8.72, 9.04 (2d, 4H, Pyr-H), 8.02 (t, 2H, Pyr-H), 9.18, 8.64 (2s, 4H, 4NH, exchangeable with D2O), 8.36, 9.10 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.56 (4C, 4CH3), 22.92 (2C, 2CH), 41.25 (2C, 2CH2), 52.95 (2C, 2CH), 131.46, 140.10, 151.98 (5C, Pyr-C), 167.22 (2C, 2C=O), 170.02 (2C, 2C=O), 164.12, 165.05 (4C, 4C=O), 127.85, 128.15, 138.32, 146.10, 152.45 (10C, Pyr-C). MS (EI, 70 eV): m/z (%) = 684 [M+, 14], 494 (25), 304 (85), 247 (95), 190 (100). C33H33N9O8 (683.67): calcd. C 57.97, H 4.87, N 18.44; found C 57.90, H 4.80, N 18.38.: −108.8 (c = 0.5, DMF). IR (KBr): ν = 3548–3452 (NH), 3056 (CH-Ar), 2950 (CH-aliph.), 1746 (C=O, imide), 1656, 1532, 1328 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.00–0.95 (m, 12H, 4 CH3), 1.85–1.73 (m, 6H, 2 CH2 + 2CH), 4.62 (t, 2H, 2CHNH), 7.55–8.05 (m, 12H, Ar-H), 8.68, 9.12 (2s, 4H, 4NH, exchangeable with D2O), 8.35, 9.08 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.55 (4C, 4CH3), 22.98 (2C, 2CH), 41.25 (2C, 2CH2), 52.85 (2C, 2CH), 131.48, 140.10, 152.15 (5C, Pyr-C), 167.42 (2C, 2C=O), 169.88 (2C, 2C=O), 158.76 (4C, 4C=O), 123.45, 125.02, 128.20, 130.54, 137.05 137.65 (20C, Ar-C). MS (EI, 70 eV): m/z (%) = 782 [M+, 6], 570 (12), 324 (25), 246 (100), 134 (95). C43H39N7O8 (781.81): calcd. C 66.06, H 5.03, N 12.54; found C 66.00, H 4.96, N 12.50.: −106.6 (c = 0.5, DMF). IR (KBr): ν = 3510–3432 (NH), 3054 (CH-Ar), 2945 (CH-aliph.), 1660, 1526, 1316 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 0.85 (s, 6H, 2CH3), 0.98–0.92 (m, 12H, 4 CH3), 1.82–1.72 (m, 6H, 2 CH2 + 2CH), 4.64 (t, 2H, 2CHNH), 7.30–7.65 (m, 8H, Ar-H), 9.08, 8.35 (2s, 4H, 4NH, exchangeable with D2O), 8.42, 9.16 (2s, 3H, Pyr-H). 13C-NMR: δ = 14.05 (2C, 2CH3), 22.48 (4C, 4CH3), 22.95 (2C, 2CH), 41.35 (2C, 2CH2), 52.78 (2C, 2CH), 131.56, 140.18, 152.12 (5C, Pyr-C), 167.35 (2C, 2C=O), 172.84 (2C, 2C=O), 168.12 (2C, C=N), 128.45, 129.68, 131.89, 135.65 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 695 [M+, 10], 582 (8), 416 (24), 359 (100), 246 (95). C35H41Cl2N7O4 (694.65): calcd. C 60.52, H 5.95, Cl 10.21, N 14.11; found C 60.45, H 5.90, Cl 10.15, N 14.05.: −112.5 (c = 0.5, DMF). IR (KBr): ν = 3498–3386 (NH), 3065 (CH-Ar), 2943 (CH-aliph.), 1658, 1530, 1337 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 0.86 (s, 6H, 2CH3), 0.99–0.94 (m, 12H, 4 CH3), 1.80–1.74 (m, 6H, 2 CH2 + 2CH), 3.65 (s. 6H, 2 OCH3), 4.62 (t, 2H, 2CHNH), 7.02–7.60 (m, 8H, Ar-H), 7.85, 8.45 (2s, 4H, 4NH, exchangeable with D2O), 8.36, 9.12 (2s, 3H, Pyr-H). 13C-NMR: δ = 13.95 (2C, 2CH3), 22.56 (4C, 4CH3), 23.05 (2C, 2CH), 41.36 (2C, 2CH2), 52.84 (2C, 2CH), 56.04 (2C, 2OCH3), 131.58, 140.10, 152.10 (5C, Pyr-C), 167.28 (2C, 2C=O), 172.92 (2C, 2C=O), 168.15 (2C, C=N), 114.02, 125.65, 129.85, 162.15 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 686 [M+, 5], 522 (6), 276 (24), 246 (100). C37H47N7O6 (685.81): calcd. C 64.80, H 6.91, N 14.30; found C 64.72, H 6.83, N 14.24.: −122.4 (c = 0.5, DMF). IR (KBr): ν = 3495–3435 (NH), 3050 (CH-Ar), 2945 (CH-aliph.), 1655, 1531, 1324 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 0.88 (s, 6H, 2CH3), 1.00–0.96 (m, 12H, 4 CH3), 1.85–1.64 (m, 6H, 2 CH2 + 2CH), 4.65 (t, 2H, 2CHNH), 7.65–8.05 (m, 8H, Ar-H), 9.10, 8.36 (2s, 4H, 4NH, exchangeable with D2O), 8.38, 9.10 (2s, 3H, Pyr-H). 13C-NMR: δ = 13.98 (2C, 2CH3), 22.54 (4C, 4CH3), 22.98 (2C, 2CH), 41.32 (2C, 2CH2), 52.86 (2C, 2CH), 131.64, 140.08, 152.16 (5C, Pyr-C), 167.33 (2C, 2C=O), 173.05 (2C, 2C=O), 168.08 (2C, C=N), 119.84, 129.35, 139.88, 150.35 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 716 [M+, 16], 537 (22), 389 (18), 276 (32), 206 (100). C35H41N9O8 (715.76): calcd. C 58.73, H 5.77, N 17.61; found C 58.65, H 5.72, N 17.55.: −145.0 (c = 0.5, DMF). IR (KBr): ν = 3510–3447 (NH), 3038 (CH-Ar), 2955 (CH-aliph.), 1656, 1528, 1319 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.04–0.95 (m, 12H, 4 CH3), 1.86–1.70 (m, 6H, 2 CH2 + 2CH), 4.65 (t, 2H, 2CHNH), 5.60 (s, 2H, 2CH=N), 7.45–7.72 (m, 10H, Ar-H), 8.42, 9.10 (2s, 4H, 4NH, exchangeable with D2O), 8.35, 9.18 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.76 (4C, 4CH3), 23.01 (2C, 2CH), 41.16 (2C, 2CH2), 52.86 (2C, 2CH), 131.48, 140.32, 151.98 (5C, Pyr-C), 167.18 (2C, 2C=O), 173.05 (2C, 2C=O), 143.05 (2C, C=N), 128.68, 129.08, 130.85, 135.75 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 598 [M+, 24], 520 (15), 478 (18), 444 (10), 360 (100), 247 (32). C33H39N7O4 (597.71): calcd. C 66.31, H 6.58, N 16.40; found C 66.24, H 6.50, N 16.32.: −130.5 (c = 0.5, DMF). IR (KBr): ν = 3488–3432 (NH), 3065 (CH-Ar), 2952 (CH-aliph.), 1652, 1533, 1322 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.00–0.95 (m, 12H, 4 CH3), 1.78–1.72 (m, 6H, 2 CH2 + 2CH), 2.24 (s, 6H, 2CH3), 4.68 (t, 2H, 2CHNH), 5.58 (s, 2H, 2CH=N), 6.98–7.56 (m, 8H, Ar-H), 8.38, 9.12 (2s, 4H, 4NH, exchangeable with D2O), 8.26, 9.05 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.76 (4C, 4CH3), 22.95 (2C, 2CH), 24.12 (2C, 2CH3), 41.42 (2C, 2CH2), 52.76 (2C, 2CH), 131.46, 139.98, 152.15 (5C, Pyr-C), 166.95 (2C, 2C=O), 173.02 (2C, 2C=O), 142.55 (2C, C=N), 128.76, 129.01, 130.55, 139.85 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 626 [M+, 8], 534 (24), 402 (32), 360 (100), 246 (82). C35H43N7O4 (625.76): calcd. C 67.18, H 6.93, N 15.67; found C 67.09, H 6.84, N 15.60.: −98.5 (c = 0.5, DMF). IR (KBr): ν = 3490–3412 (NH), 3056 (CH-Ar), 2975 (CH-aliph.), 1657, 1532, 1336 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.10–0.94 (m, 12H, 4 CH3), 1.83–1.75 (m, 6H, 2 CH2 + 2CH), 3.68 (s, 6H, 2OCH3), 4.62 (t, 2H, 2CHNH), 5.62 (s, 2H, 2CH=N), 7.08–7.42 (m, 8H, Ar-H), 8.36, 9.08 (2s, 4H, 4NH, exchangeable with D2O), 8.24, 9.24 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.68 (4C, 4CH3), 23.00 (2C, 2CH), 41.32 (2C, 2CH2), 52.88 (2C, 2CH), 55.75 (2C, 2OCH3), 131.42, 140.05, 152.14 (5C, Pyr-C), 167.08 (2C, 2C=O), 172.85 (2C, 2C=O), 143.15 (2C, C=N), 113.95, 125.78, 130.02, 162.45 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 658 [M+, 6], 626 (12), 596 (15), 478 (56), 247 (100). C35H43N7O6 (657.76): calcd. C 63.91, H 6.59, N 14.91; found C 63.81, H 6.50, N 14.84.: −142.5 (c = 0.5, DMF). IR (KBr): ν = 3492–3398 (NH), 3045 (CH-Ar), 2965 (CH-aliph.), 1657, 1532, 1331 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.01–0.95 (m, 12H, 4 CH3), 1.85–1.64 (m, 6H, 2 CH2 + 2CH), 4.62 (t, 2H, 2CHNH), 5.65 (s, 2H, 2CH=N), 7.68–8.00 (m, 8H, Ar-H), 9.10, 8.36 (2s, 4H, 4NH, exchangeable with D2O), 8.56, 9.10 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.62 (4C, 4CH3), 22.95 (2C, 2CH), 41.40 (2C, 2CH2), 52.95 (2C, 2CH), 131.44, 140.10, 152.32 (5C, Pyr-C), 167.24 (2C, 2C=O), 172.88 (2C, 2C=O), 142.65 (2C, C=N), 120.05, 129.78, 139.65, 150.48 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 688 [M+, 16], 537 (22), 389 (18), 276 (32), 206 (100). C33H37N9O8 (687.70): calcd. C 57.63, H 5.42, N 18.33; found C 57.55, H 5.34, N 18.24.: −116.45 (c = 0.5, DMF). IR (KBr): ν = 3490–3444 (NH), 3065 (CH-Ar), 2958 (CH-aliph.), 1656, 1522, 1318 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.10–0.95 (m, 12H, 4 CH3), 1.80–1.70 (m, 6H, 2 CH2 + 2CH), 4.68 (t, 2H, 2CHNH), 5.48 (s, 2H, 2CH=N), 7.26–7.68 (m, 8H, Ar-H), 9.08, 8.35 (2s, 4H, 4NH, exchangeable with D2O), 8.45, 9.36 (2s, 3H, Pyr-H). 13C-NMR: δ = 22.45 (4C, 4CH3), 22.90 (2C, 2CH), 41.33 (2C, 2CH2), 52.76 (2C, 2CH), 131.58, 140.22, 152.16 (5C, Pyr-C), 167.36 (2C, 2C=O), 172.91 (2C, 2C=O), 142.65 (2C, C=N), 128.50, 129.72, 131.94, 135.68 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 666 [M+, 6], 595 (12), 478 (45), 332(92), 260 (100). C33H37Cl2N7O4 (666.60): calcd. C 59.46, H 5.59, Cl 10.64, N 14.71; found C 59.40, H 5.53, Cl 10.60, N 14.65.: −112.6 (c = 0.5, DMF). IR (KBr): ν = 3478–3575 (NH), 3060 (CH-Ar), 2966 (CH-aliph.), 1753 (C=O, imide), 1656, 1530, 1320 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 0.98–0.90 (m, 24H, 8 CH3), 1.80–1.72 (m, 12H, 4 CH2 + 4 CH), 4.55 (t, 4H, 4CHNH), 9.08 (s, 4H, Ar-H), 8.78, 9.15 (2s, 8H, 8NH, exchangeable with D2O), 8.40, 9.20 (2s, 6H, Pyr-H). 13C-NMR: δ = 22.42 (8C, 8CH3), 22.98 (4C, 4CH), 41.18 (4C, 4CH2), 53.15 (4C, 4CH), 131.55, 140.08, 152.24 (10C, 2 Pyr-C), 167.16 (4C, 4C=O), 172.84 (4C, 4C=O), 164.16 (8C, 8C=O), 124.98, 134.85 (12C, Ar-C). MS (EI, 70 eV): m/z (%) = 1207 [M+, 4], 737 (5), 470 (32), 300 (100), 274 (72), 247 (85). C58H58N14O16 (1207.17): calcd. C 57.71, H 4.84, N 16.24; found C 57.65, H 4.78, N 16.20.: −124.5 (c = 0.5, DMF). IR (KBr): ν = 3544–3432 (NH), 3050 (CH-Ar), 2964 (CH-aliph.), 1742 (C=O, imide), 1657, 1530, 1326 (C=O, amide I, II and III) cm−1. 1H-NMR: δ = 1.05–0.98 (m, 24H, 8 CH3), 1.82–1.75 (m, 12H, 4 CH2 + 4 CH), 4.58 (t, 4H, 4 CHNH), 8.15–8.25 (m, 8H, Ar-H), 8.56, 9.16 (2s, 8H, 8NH, exchangeable with D2O), 8.40, 9.04 (2s, 6H, 2 Pyr-H). 13C-NMR: δ = 22.64 (8C, 8CH3), 22.95 (4C, 4CH), 41.35 (4C, 4CH2), 52.78 (4C, 4CH), 131.54, 140.12, 152.18 (10C, 2 Pyr-C), 167.36 (4C, 4C=O), 170.05 (4C, 4C=O), 158.68 (8C, 8 C=O), 120.25, 135.10, 139.73 (20C, Ar-C). MS (EI, 70 eV): m/z (%) = 1307 [M+, 4], 1014 (8), 787 (12), 654 (24), 294 (100), 246 (85). C66H62N14O16 (1307.28): calcd. C 60.64, H 4.78, N 15.00; found C 60.55, H 4.70, N 14.95.3.2. Biological Activities
3.2.1. Antimicrobial Activity
3.2.2. Anti-Inflammatory Activity
3.2.2.1. Carrageenan-Induced Edema (Rat Paw Test)
3.2.2.2. Estimation of Plasma Prostaglandin E2 (PGE2)
3.3. Anticancer Activity
3.3.1. In Vitro Anti-Cancer Activities
3.3.2. Human Breast Cancer Xenograft Models and Animal Treatment
4. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Krakowiak, K.E.; Bradshaw, J.S.; Zamecka-Krakowiak, D.J. Synthesis of aza-crown ethers. Chem. Rev. 1989, 89, 929–972. [Google Scholar]
- Izatt, R.M.; Pawlak, K.; Bradshaw, J.S.; Bruening, R.L.; Tarbet, B.J. Thermodynamic and kinetic data for macrocycle interaction with neutral molecules. Chem. Rev. 1992, 92, 1261–1354. [Google Scholar] [CrossRef]
- Elwahy, A.H.M. New trends in the chemistry of condensed heteromacrocycles Part A: Condensed azacrown ethers and azathiacrown ethers. J. Heterocycl. Chem. 2003, 40, 1–23. [Google Scholar] [CrossRef]
- Hirschmann, R.; Smith, A.B.; Sprengeler, P.A. Some interactions of macromolecules with low molecular weight ligands. Recent advances in peptidomimetic research. In New Perspectives in Drug Design; Academic Press: San Diego, CA, USA, 1995; pp. 1–14. [Google Scholar]
- Bursavich, M.G.; West, C.W.; Rich, D.H. From peptides to non-peptide peptidomimetics: Design and synthesis of new piperidine inhibitors of aspartic peptidases. Org. Lett. 2001, 3, 2317–2320. [Google Scholar] [CrossRef]
- Amr, A.E.; Mohamed, A.M.; Ibrahim, A.A. Synthesis of some new chiral tricyclic and macrocyclic pyridine derivatives as antimicrobial agents. Z. Naturforsch. 2003, 58b, 861–868. [Google Scholar]
- Amr, A.E. Synthesis of some new linear and chiral macrocyclic pyridine carbazides as analgesic and anticonvulsant agents. Z. Naturforsch. 2005, 60b, 990–998. [Google Scholar]
- Abou-Ghalia, M.H.; Amr, A.E.; Abdalah, M.M. Synthesis of some new (Nα-dipicolinoyl)-bis-L-leucyl-DL-norvalyl linear tetra and cyclic octa bridged peptides as new anti-inflammatory agents. Z. Naturforsch. 2003, 58b, 903–910. [Google Scholar]
- Attia, A.; Abdel-Salam, O.I.; Amr, A.E.; Stibor, I.; Budesinsky, M. Synthesis and antimicrobial activity of some new chiral bridged macrocyclic pyridines. Egypt. J. Chem. 2000, 43, 187–201. [Google Scholar]
- Amr, A.E.; Abo-Ghalia, M.H.; Abdalah, M.M. Synthesis of novel macrocyclic peptide-calix[4]arenes and peptido-pyridines as precursors for potential molecular metallacages, chemo-sensors and biologically active candidates. Z. Naturforsch. 2006, 61b, 1335–1345. [Google Scholar]
- Amr, A.E.; Abo-Ghalia, M.H.; Abdalah, M.M. Synthesis of new (Nα-dipicolinoyl)-bis-L-valyl-L-phenylalanyl linear and macrocyclic bridged peptides as anti-inflammatory agents. Arch. Pharm. 2007, 340, 304–309. [Google Scholar] [CrossRef]
- Hassan, S.S.M.; Abo-Ghalia, M.H.; Amr, A.E.; Mohamed, A.H.K. Novel lead (II) selective membrane potentiometric sensors based on chiral 2,6-bis-pyridine-carboxamide derivatives. Talanta 2003, 60, 81–91. [Google Scholar] [CrossRef]
- Hassan, S.S.M.; Abo-Ghalia, M.H.; Amr, A.E.; Mohamed, A.H.K. Novel thiocyanate-selective membrane sensors based on di-, tetra-, and hexaimide pyridine ionophores. Anal. Chim. Acta 2003, 482, 9–18. [Google Scholar] [CrossRef]
- Abo-Ghalia, M.H.; Abd-El-Hamid, M.; Zweel, M.A.; Amr, A.E.; Moafi, S.A. Synthesis and reactions of new chiral linear and macrocyclic tetra and penta-peptide candidates. Z. Naturforsch. 2012, 67b, 806–818. [Google Scholar] [CrossRef]
- Khalifa, N.M.; Naglah, A.M.; Al-Omar, M.A.; Abo-Ghalia, M.H.; Amr, A.E. Synthesis and reactions of new chiral linear carboxamides with an incorporated peptide linkage using nalidixic acid and amino acids as starting materials. Z. Naturforsch. 2014, 69b, 351–361. [Google Scholar]
- Fakhr, I.M.; Amr, A.E.; Sabry, N.M.; Abdalah, M.M. Anti-inflammatory and analgesic activities of some newly synthesized chiral peptide derivatives using (3-benzoyl-4,5-dioxo-2-phenylpyrrolidin-1-yl)acetic acid ethyl ester as starting material. Arch. Pharm. Chem. Life Sci. 2008, 341, 174–180. [Google Scholar] [CrossRef]
- Abou-Ghalia, M.H.; Amr, A.E. Synthesis and investigation of a new cyclo-(Nα-dipicolinoyl)pentapeptide of a breast and CNS cytotoxic activity and an ionophoric specifity. Amino Acids 2004, 26, 283–289. [Google Scholar]
- Abd El-Salam, O.I.; Al-Omar, M.A.; Fayed, A.A.; Flefel, E.M.; Amr, A.E. Synthesis of new macrocyclic polyamides as antimicrobial agent candidates. Molecules 2012, 17, 14510–14521. [Google Scholar] [CrossRef]
- Ismail, O.A.; Al-Omar, M.A.; Amr, A.E. Facile synthesis of new chiral macrocyclic Schiff-base and tricyclopolyazacarboxamides candidates as antimicrobial agents. Curr. Org. Synth. 2012, 9, 406–412. [Google Scholar] [CrossRef]
- Neupert-Laves, K.; Dobler, M. The crystal structure of a K+ complex of valinomycin. Helv. Chim. Acta 1975, 58, 432–442. [Google Scholar] [CrossRef]
- Collison, M.E.; Aebli, G.V.; Petty, J.; Meyerhoff, M.E. Potentiometric combination ion/carbon dioxide sensors for in vitro and in vivo blood measurements. Anal. Chem. 1989, 61, 2365–2372. [Google Scholar] [CrossRef]
- Boitano, S.; Omoto, C.K. Membrane hyperpolarization activates trout sperm without an increase in intracellular pH. J. Cell. Sci. 1991, 98, 343–349. [Google Scholar]
- Talma, A.G.; Jouin, P.; Vries, J.G.; Troostwijk, C.B.; Buning, G.H. Reductions of activated carbonyl compounds with chiral bridged 1,4-dihydropyridines. An investigation of scope and structural effects. J. Am. Chem. Soc. 1985, 107, 3981–3997. [Google Scholar] [CrossRef]
- Amr, A.; Abd El-Salam, O.; Attia, A.; Stibor, I. Synthesis of new potential bis-intercallators based on chiral pyridine-2,6-dicarbox-amides. Collect. Czech Chem. Commun. 1999, 64, 288–298. [Google Scholar] [CrossRef]
- Roderick, A.B.; Henry, M.F. The Synthesis of the 3,9-diazabicyclo [3.3.1] nonane ring system. J. Am. Chem. Soc. 1953, 75, 975–977. [Google Scholar] [CrossRef]
- Winter, C.A.; Risley, E.A.; Nuss, G.W. Carrageenin-induced edema in hind paw of the rat as an assay for antiiflammatory drugs. Exp. Biol. Med. 1962, 111, 544–547. [Google Scholar] [CrossRef]
- Herrmann, F.; Lindemann, A.; Gamss, J.; Mertelsmann, R. Cytokine-stimulation of prostaglandin synthesis from endogenous and exogenous arachidonic acids in polymorphonuclear leukocytes involving activation and new synthesis of cyclooxygenase. Eur. J. Immunol. 1999, 20, 2513–2516. [Google Scholar]
- Furtada, G.L.; Medeiros, A.A. Single-disk diffusion testing (Kirby-Bauer) of susceptibility of Proteus mirabilis to chloramphenicol: Significance of the intermediate category. J. Clin. Microbiol. 1980, 12, 550–553. [Google Scholar]
- Jones, R.N.; Ballow, C.H.; Biedenbach, D.J. Multi-laboratory assessment of the linezolid spectrum of activity using the Kirby-Bauer disk diffusion method: Report of the Zyvox Antimicrobial Potency Study (ZAPS) in the United States. Diagn. Microbiol. Infect. Dis. 2001, 40, 59–66. [Google Scholar] [CrossRef]
- Wang, W.; Rayburn, E.R.; Velu, S.E.; Chen, D.; Nadkarni, D.H.; Murugesan, S.; Chen, D.; Zhang, R. A novel synthetic iminoquinone, BA-TPQ, as an anti-breast cancer agent: In vitro and in vivo activity and mechanisms of action. Breast Cancer Res. Treat. 2010, 123, 321–331. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Khayyat, S.; Amr, A.E.-G. Synthesis and Biological Activities of Some New (Nα-Dinicotinoyl)- bis-L-Leucyl Linear and Macrocyclic Peptides. Molecules 2014, 19, 10698-10716. https://doi.org/10.3390/molecules190810698
Khayyat S, Amr AE-G. Synthesis and Biological Activities of Some New (Nα-Dinicotinoyl)- bis-L-Leucyl Linear and Macrocyclic Peptides. Molecules. 2014; 19(8):10698-10716. https://doi.org/10.3390/molecules190810698
Chicago/Turabian StyleKhayyat, Suzan, and Abd El-Galil Amr. 2014. "Synthesis and Biological Activities of Some New (Nα-Dinicotinoyl)- bis-L-Leucyl Linear and Macrocyclic Peptides" Molecules 19, no. 8: 10698-10716. https://doi.org/10.3390/molecules190810698